
Pathophysiological Molecular Mechanisms of Obesity: A Link between MAFLD and NASH with Cardiovascular Diseases


Abstract
:1. Introduction
2. Obesity
2.1. Determination of Obesity
2.2. Etiology of Obesity
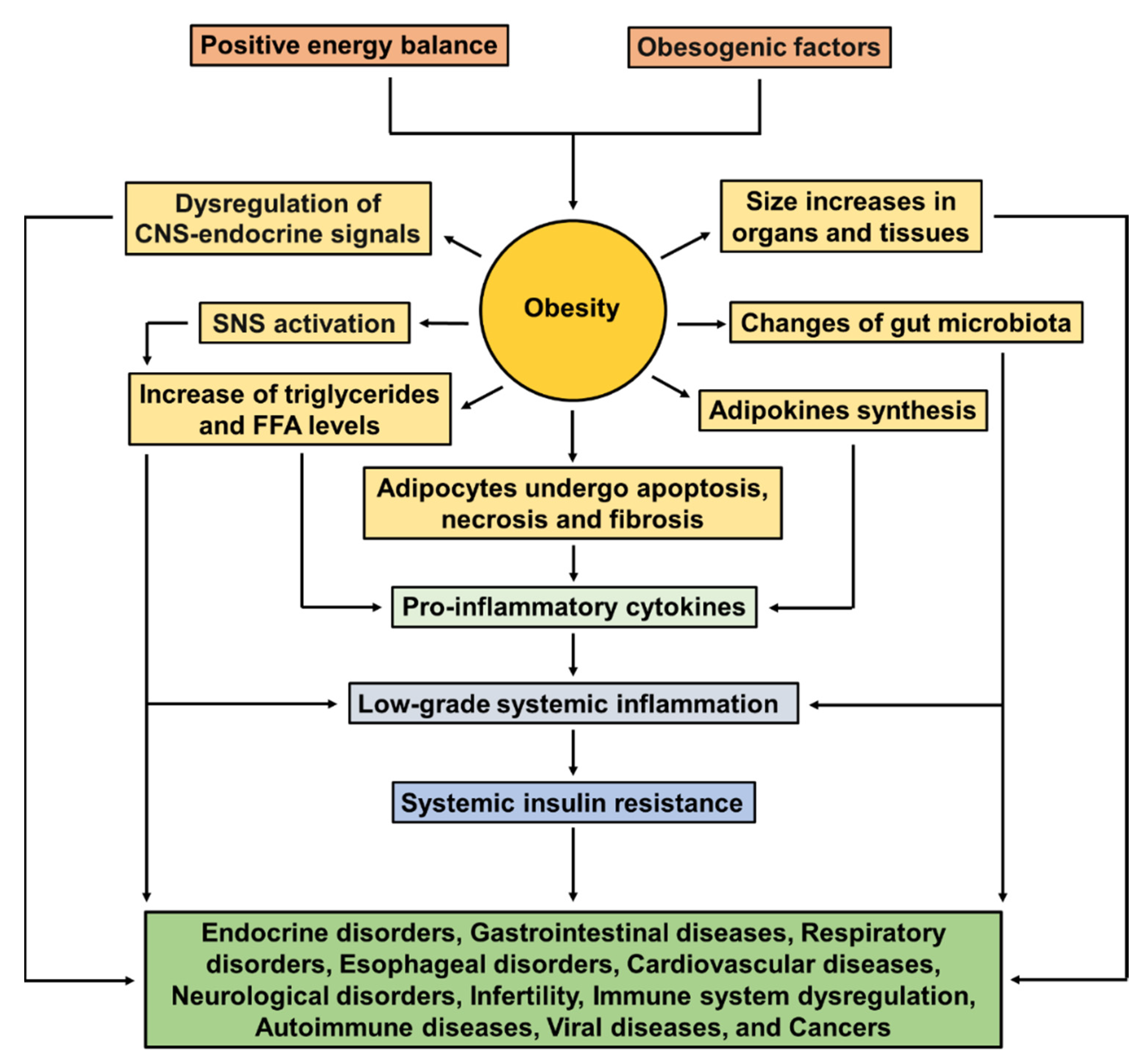
2.3. Pathophysiology of Obesity
2.4. Comorbidities Associated with Obesity
3. Features of MAFLD and NASH
Pathogenesis of MAFLD and NASH
4. Obesity and Cardiovascular Diseases
4.1. Pathogenesis of Obesity in Cardiovascular Diseases
4.2. Altered Lipid and Lipoprotein Metabolism in the Liver Contributes to CVD Risk
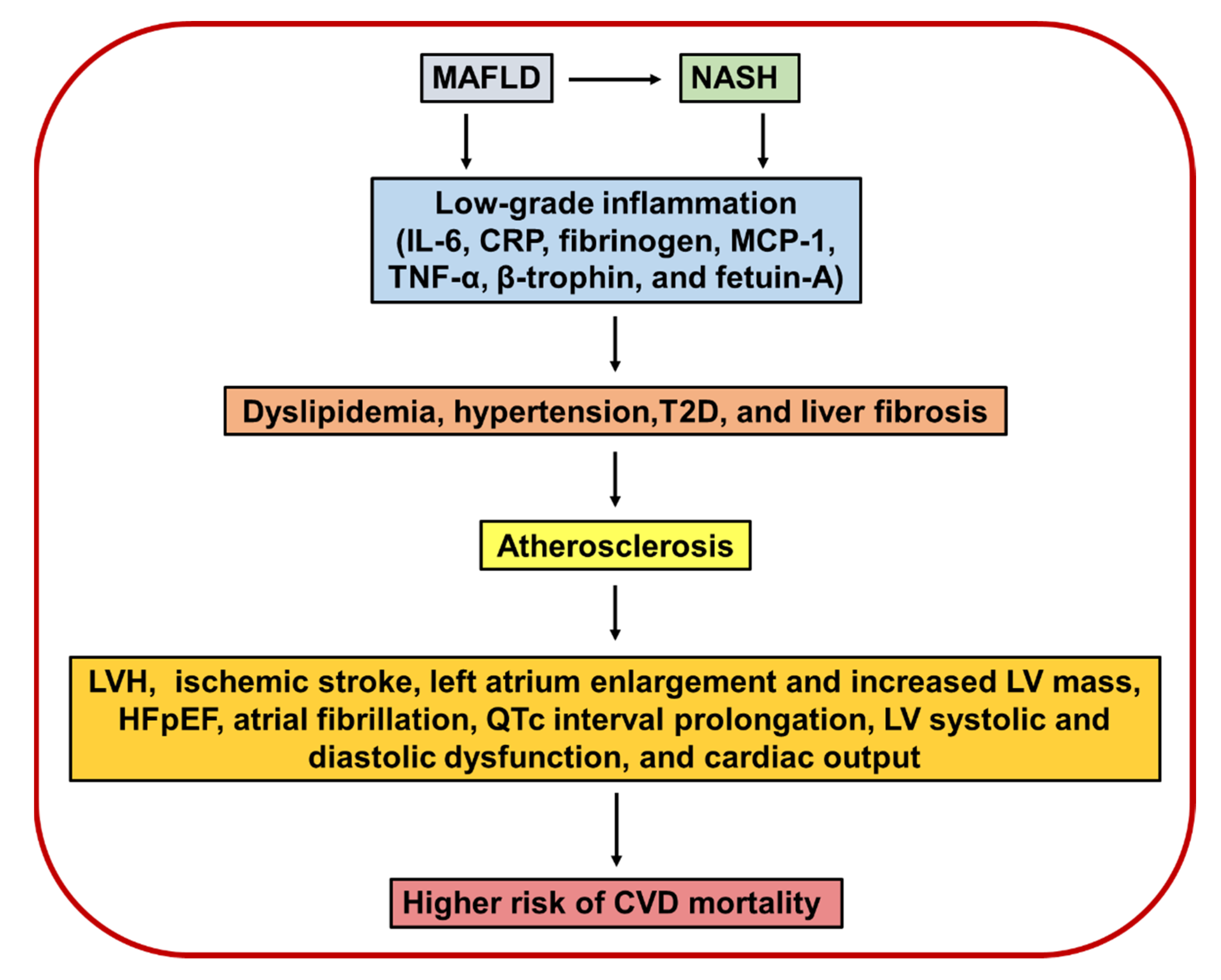
5. Association of MAFLD and NASH with Cardiovascular Diseases
5.1. Ischemic Stroke
5.2. Structural Cardiac Abnormalities
5.3. Cardiac Arrhythmias
6. Insulin Hormone
The Role of Insulin Resistance in MAFLD/NASH and in CVD
7. Strategies to Treat MAFLD and NASH and Cardiovascular Prevention
7.1. Lifestyle Modifications
7.2. Smoking Cessation
7.3. Weight Loss
7.4. Diet Modifications
7.5. Exercise and Physical Activity
7.6. Medical Therapy
7.6.1. Aspirin
7.6.2. Statins
7.6.3. Ezetimibe
7.6.4. PCSK9 Inhibitor
7.6.5. Fibroblast Growth Factor 21 Analogues
7.6.6. Farnesoid X Receptor Agonists
7.6.7. GLP1 Agonist
7.6.8. Bariatric Surgery
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eslam, M.; George, J. Refining the role of epicardial adipose tissue in non-alcoholic fatty liver disease. Hepatol. Int. 2019, 13, 662–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.J.; Rosenthal, M.D.; Miller, K.R.; Codner, P.; Kiraly, L. Martindale RG: The Critical Care Obesity Paradox and Implications for Nutrition Support. Curr. Gastroenterol. Rep. 2016, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Pigeyre, M.; Yazdi, F.T.; Kaur, Y.; Meyre, D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin. Sci. 2016, 130, 943–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchie, A.; Dallegri, F.; Carbone, F.; Bonaventura, A.; Liberale, L.; Portincasa, P.; Fruhbeck, G.; Montecucco, F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018, 48, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Redinger, R.N. The pathophysiology of obesity and its clinical manifestations. Gastroenterol. Hepatol. 2007, 3, 856–863. [Google Scholar]
- Souza-Mello, V. Peroxisome proliferator-activated receptors as targets to treat non-alcoholic fatty liver disease. World J. Hepatol. 2015, 7, 1012–1019. [Google Scholar] [CrossRef]
- Mathews, S.E.; Kumar, R.B.; Shukla, A.P. Nonalcoholic steatohepatitis, obesity, and cardiac dysfunction. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 315–320. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Shen, W.J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II: PPAR-beta/delta and PPAR-gamma. Future Cardiol. 2017, 13, 279–296. [Google Scholar] [CrossRef]
- Alkagiet, S.; Papagiannis, A.; Tziomalos, K. Associations between nonalcoholic fatty liver disease and ischemic stroke. World J. Hepatol. 2018, 10, 474–478. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, A.; Sookoian, S.; Pirola, C.J.; Targher, G. Non-alcoholic fatty liver disease and risk of cardiovascular disease. Metabolism 2016, 65, 1136–1150. [Google Scholar] [CrossRef]
- Kasper, P.; Martin, A.; Lang, S.; Kutting, F.; Goeser, T.; Demir, M.; Steffen, H.M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2021, 110, 921–937. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Mantovani, A.; Tilg, H.; Targher, G. Risk of cardiomyopathy and cardiac arrhythmias in patients with nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 425–439. [Google Scholar] [CrossRef]
- Stahl, E.P.; Dhindsa, D.S.; Lee, S.K.; Sandesara, P.B.; Chalasani, N.P.; Sperling, L.S. Nonalcoholic Fatty Liver Disease and the Heart: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 948–963. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Aubin, A.; Loomba, R. The Relationship Between Type 2 Diabetes, NAFLD, and Cardiovascular Risk. Curr. Diab. Rep. 2021, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Armandi, A.; Rosso, C.; Caviglia, G.P.; Bugianesi, E. Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 155. [Google Scholar] [CrossRef]
- Fujii, H.; Kawada, N.; Japan Study Group of Nafld J-N. The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Guo, P.; Guo, J.; Ares, I.; Lopez-Torres, B.; Martinez-Larranaga, M.R.; Wang, X.; Anadon, A.; Martinez, M.A. Targeting peroxisome proliferator-activated receptors: A new strategy for the treatment of cardiac fibrosis. Pharmacol. Ther. 2021, 219, 107702. [Google Scholar] [CrossRef]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuniga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Xu, H.M.; Yu, F.; Wang, M.; Li, M.Y.; Xu, T.; Gao, Y.Y.; Wang, J.X.; Li, P.F. Crosstalk between MicroRNAs and Peroxisome Proliferator-Activated Receptors and Their Emerging Regulatory Roles in Cardiovascular Pathophysiology. PPAR Res. 2018, 2018, 8530371. [Google Scholar] [CrossRef] [Green Version]
- Maclennan, W. On the Treatment of Obesity and Myxoedema by a New Preparation of Thyroid (“Thyroglandin”). Br. Med. J. 1898, 2, 79–80. [Google Scholar] [CrossRef]
- Oliver, T. Post-Mortem in a Case of Extreme Obesity. J. Anat. Physiol. 1880, 14 Pt 3, 345–347. [Google Scholar]
- Perry, A.W. Nature and Treatment of Obesity. Cal. State J. Med. 1903, 1, 356–359. [Google Scholar]
- Eknoyan, G. A history of obesity, or how what was good became ugly and then bad. Adv. Chronic Kidney Dis. 2006, 13, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Batsis, J.A.; Mackenzie, T.A.; Bartels, S.J.; Sahakyan, K.R.; Somers, V.K.; Lopez-Jimenez, F. Diagnostic accuracy of body mass index to identify obesity in older adults: NHANES 1999–2004. Int. J. Obes. 2016, 40, 761–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prado, C.M.; Gonzalez, M.C.; Heymsfield, S.B. Body composition phenotypes and obesity paradox. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 535–551. [Google Scholar] [CrossRef]
- Hu, F.B. Obesity and mortality: Watch your waist, not just your weight. Arch. Intern. Med. 2007, 167, 875–876. [Google Scholar] [CrossRef]
- Pischon, T.; Boeing, H.; Hoffmann, K.; Bergmann, M.; Schulze, M.B.; Overvad, K.; van der Schouw, Y.T.; Spencer, E.; Moons, K.G.; Tjonneland, A.; et al. General and abdominal adiposity and risk of death in Europe. N. Engl. J. Med. 2008, 359, 2105–2120. [Google Scholar] [CrossRef] [Green Version]
- Welborn, T.A.; Dhaliwal, S.S. Preferred clinical measures of central obesity for predicting mortality. Eur. J. Clin. Nutr. 2007, 61, 1373–1379. [Google Scholar] [CrossRef]
- Apovian, C.M. Obesity: Definition, comorbidities, causes, and burden. Am. J. Manag. Care 2016, 22 (Suppl. 7), s176–s185. [Google Scholar]
- Swinburn, B.; Sacks, G.; Ravussin, E. Increased food energy supply is more than sufficient to explain the US epidemic of obesity. Am. J. Clin. Nutr. 2009, 90, 1453–1456. [Google Scholar] [CrossRef] [PubMed]
- Akil, L.; Ahmad, H.A. Effects of socioeconomic factors on obesity rates in four southern states and Colorado. Ethn. Dis. 2011, 21, 58–62. [Google Scholar] [PubMed]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oussaada, S.M.; van Galen, K.A.; Cooiman, M.I.; Kleinendorst, L.; Hazebroek, E.J.; van Haelst, M.M.; Ter Horst, K.W.; Serlie, M.J. The pathogenesis of obesity. Metabolism 2019, 92, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Heymsfield, S.B.; Wadden, T.A. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 254–266. [Google Scholar] [CrossRef]
- Strable, M.S.; Ntambi, J.M. Genetic control of de novo lipogenesis: Role in diet-induced obesity. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 199–214. [Google Scholar] [CrossRef] [Green Version]
- Kinlen, D.; Cody, D.; O’Shea, D. Complications of obesity. QJM 2018, 111, 437–443. [Google Scholar] [CrossRef]
- Brochu, M.; Tchernof, A.; Dionne, I.J.; Sites, C.K.; Eltabbakh, G.H.; Sims, E.A.; Poehlman, E.T. What are the physical characteristics associated with a normal metabolic profile despite a high level of obesity in postmenopausal women? J. Clin. Endocrinol. Metab. 2001, 86, 1020–1025. [Google Scholar] [PubMed]
- Ortega, F.B.; Lavie, C.J.; Blair, S.N. Obesity and Cardiovascular Disease. Circ. Res. 2016, 118, 1752–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elagizi, A.; Kachur, S.; Lavie, C.J.; Carbone, S.; Pandey, A.; Ortega, F.B.; Milani, R.V. An Overview and Update on Obesity and the Obesity Paradox in Cardiovascular Diseases. Prog. Cardiovasc. Dis. 2018, 61, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, P.; Kowdley, K.V. The Metabolic Syndrome and Its Influence on Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2016, 20, 225–243. [Google Scholar] [CrossRef]
- D’Agati, V.D.; Chagnac, A.; de Vries, A.P.; Levi, M.; Porrini, E.; Herman-Edelstein, M.; Praga, M. Obesity-related glomerulopathy: Clinical and pathologic characteristics and pathogenesis. Nat. Rev. Nephrol. 2016, 12, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Anstey, K.J.; Cherbuin, N.; Budge, M.; Young, J. Body mass index in midlife and late-life as a risk factor for dementia: A meta-analysis of prospective studies. Obes. Rev. 2011, 12, e426–e437. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; De Schutter, A.; Parto, P.; Jahangir, E.; Kokkinos, P.; Ortega, F.B.; Arena, R.; Milani, R.V. Obesity and Prevalence of Cardiovascular Diseases and Prognosis-The Obesity Paradox Updated. Prog. Cardiovasc. Dis. 2016, 58, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Cuevas, J.; Sandoval-Rodriguez, A.; Meza-Rios, A.; Monroy-Ramirez, H.C.; Galicia-Moreno, M.; Garcia-Banuelos, J.; Santos, A.; Armendariz-Borunda, J. Molecular Mechanisms of Obesity-Linked Cardiac Dysfunction: An Up-Date on Current Knowledge. Cells 2021, 10, 629. [Google Scholar] [CrossRef]
- Louie, J.K.; Acosta, M.; Samuel, M.C.; Schechter, R.; Vugia, D.J.; Harriman, K.; Matyas, B.T.; California Pandemic Working Group. A novel risk factor for a novel virus: Obesity and 2009 pandemic influenza A (H1N1). Clin. Infect. Dis. 2011, 52, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Sanchis-Gomar, F.; Lavie, C.J.; Mehra, M.R.; Henry, B.M.; Lippi, G. Obesity and Outcomes in COVID-19: When an Epidemic and Pandemic Collide. Mayo Clin. Proc. 2020, 95, 1445–1453. [Google Scholar] [CrossRef]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M.; et al. High Prevalence of Obesity in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Requiring Invasive Mechanical Ventilation. Obesity 2020, 28, 1195–1199. [Google Scholar] [CrossRef]
- Petrakis, D.; Margina, D.; Tsarouhas, K.; Tekos, F.; Stan, M.; Nikitovic, D.; Kouretas, D.; Spandidos, D.A.; Tsatsakis, A. Obesity a risk factor for increased COVID19 prevalence, severity and lethality (Review). Mol. Med. Rep. 2020, 22, 9–19. [Google Scholar] [CrossRef]
- Bello-Chavolla, O.Y.; Bahena-Lopez, J.P.; Antonio-Villa, N.E.; Vargas-Vazquez, A.; Gonzalez-Diaz, A.; Marquez-Salinas, A.; Fermin-Martinez, C.A.; Naveja, J.J.; Aguilar-Salinas, C.A. Predicting Mortality Due to SARS-CoV-2: A Mechanistic Score Relating Obesity and Diabetes to COVID-19 Outcomes in Mexico. J. Clin. Endocrinol. Metab. 2020, 105, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Garduno, E. Obesity is the comorbidity more strongly associated for Covid-19 in Mexico. A case-control study. Obes. Res. Clin. Pract. 2020, 14, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Ismaiel, A.; Dumitrascu, D.L. Cardiovascular Risk in Fatty Liver Disease: The Liver-Heart Axis-Literature Review. Front. Med. 2019, 6, 202. [Google Scholar] [CrossRef] [Green Version]
- Adler, M.; Schaffner, F. Fatty liver hepatitis and cirrhosis in obese patients. Am. J. Med. 1979, 67, 811–816. [Google Scholar] [CrossRef]
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Ther. 2017, 179, 142–157. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Lau, J.K.; Zhang, X.; Yu, J. Animal models of non-alcoholic fatty liver disease: Current perspectives and recent advances. J. Pathol. 2017, 241, 36–44. [Google Scholar] [CrossRef]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Cardio-Metabolic Disorders in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2019, 20, 2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.D.; Valenti, L.; Romeo, S. Does nonalcoholic fatty liver disease cause cardiovascular disease? Current knowledge and gaps. Atherosclerosis 2019, 282, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Boeckmans, J.; Natale, A.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Human-based systems: Mechanistic NASH modelling just around the corner? Pharmacol. Res. 2018, 134, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballestri, S.; Lonardo, A.; Bonapace, S.; Byrne, C.D.; Loria, P.; Targher, G. Risk of cardiovascular, cardiac and arrhythmic complications in patients with non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1724–1745. [Google Scholar] [CrossRef]
- Leite, N.C.; Salles, G.F.; Cardoso, C.R.; Villela-Nogueira, C.A. Serum biomarkers in type 2 diabetic patients with non-alcoholic steatohepatitis and advanced fibrosis. Hepatol. Res. 2013, 43, 508–515. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Shang, L.; Hosseini, M.; Liu, X.; Kisseleva, T.; Brenner, D.A. Human hepatic stellate cell isolation and characterization. J. Gastroenterol. 2018, 53, 6–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Lanthier, N. Targeting Kupffer cells in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: Why and how? World J. Hepatol. 2015, 7, 2184–2188. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, W.R.; Kim, H.J.; Therneau, T.M. Association between noninvasive fibrosis markers and mortality among adults with nonalcoholic fatty liver disease in the United States. Hepatology 2013, 57, 1357–1365. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, 1–16. [Google Scholar] [CrossRef]
- Polotsky, V.Y.; Patil, S.P.; Savransky, V.; Laffan, A.; Fonti, S.; Frame, L.A.; Steele, K.E.; Schweizter, M.A.; Clark, J.M.; Torbenson, M.S.; et al. Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am. J. Respir. Crit. Care Med. 2009, 179, 228–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, S.S.; Halbower, A.; Pan, Z.; Robbins, K.; Capocelli, K.E.; Klawitter, J.; Shearn, C.T.; Sokol, R.J. Nocturnal hypoxia-induced oxidative stress promotes progression of pediatric non-alcoholic fatty liver disease. J. Hepatol. 2016, 65, 560–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papatheodoridi, A.M.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Nonalcoholic Fatty Liver Disease and Progression to Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Schnabl, B. Intestinal microbiota and nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol. 2017, 33, 128–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Bautista, L.; Franzosi, M.G.; Commerford, P.; Lang, C.C.; Rumboldt, Z.; Onen, C.L.; Lisheng, L.; et al. Obesity and the risk of myocardial infarction in 27,000 participants from 52 countries: A case-control study. Lancet 2005, 366, 1640–1649. [Google Scholar] [CrossRef]
- Gungor, N.; Thompson, T.; Sutton-Tyrrell, K.; Janosky, J.; Arslanian, S. Early signs of cardiovascular disease in youth with obesity and type 2 diabetes. Diabetes Care 2005, 28, 1219–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akil, L.; Ahmad, H.A. Relationships between obesity and cardiovascular diseases in four southern states and Colorado. J. Health Care Poor Underserved 2011, 22 (Suppl. S4), 61–72. [Google Scholar] [CrossRef] [Green Version]
- Csige, I.; Ujvarosy, D.; Szabo, Z.; Lorincz, I.; Paragh, G.; Harangi, M.; Somodi, S. The Impact of Obesity on the Cardiovascular System. J. Diabetes Res. 2018, 2018, 3407306. [Google Scholar] [CrossRef] [Green Version]
- Alpert, M.A.; Omran, J.; Bostick, B.P. Effects of Obesity on Cardiovascular Hemodynamics, Cardiac Morphology, and Ventricular Function. Curr. Obes. Rep. 2016, 5, 424–434. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Wong, C.; Marwick, T.H. Obesity cardiomyopathy: Pathogenesis and pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 436–443. [Google Scholar] [CrossRef]
- Mahajan, R.; Lau, D.H.; Sanders, P. Impact of obesity on cardiac metabolism, fibrosis, and function. Trends Cardiovasc. Med. 2015, 25, 119–126. [Google Scholar] [CrossRef]
- Turer, A.T.; Hill, J.A.; Elmquist, J.K.; Scherer, P.E. Adipose tissue biology and cardiomyopathy: Translational implications. Circ. Res. 2012, 111, 1565–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 2020, 598, 2977–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlobine, I.; Gopal, K.; Ussher, J.R. Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochim. Biophys. Acta 2016, 1861, 1555–1568. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Boutjdir, M.; Aromolaran, A.S. Cardiolipotoxicity, Inflammation, and Arrhythmias: Role for Interleukin-6 Molecular Mechanisms. Front. Physiol. 2018, 9, 1866. [Google Scholar] [CrossRef] [PubMed]
- Zamora, M.; Villena, J.A. Contribution of Impaired Insulin Signaling to the Pathogenesis of Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 2833. [Google Scholar] [CrossRef] [Green Version]
- Sletten, A.C.; Peterson, L.R.; Schaffer, J.E. Manifestations and mechanisms of myocardial lipotoxicity in obesity. J. Intern. Med. 2018, 284, 478–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinar, A.A.; Scott, T.E.; Huuskes, B.M.; Tapia Caceres, F.E.; Kemp-Harper, B.K.; Samuel, C.S. Targeting the NLRP3 inflammasome to treat cardiovascular fibrosis. Pharmacol. Ther. 2020, 209, 107511. [Google Scholar] [CrossRef]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef]
- Gutiérrez-Cuevas, J.; Sandoval-Rodriguez, A.; Monroy-Ramirez, H.C.; Vazquez-Del Mercado, M.; Santos-Garcia, A.; Armendariz-Borunda, J. Prolonged-release pirfenidone prevents obesity-induced cardiac steatosis and fibrosis in a mouse NASH model. Cardiovasc. Drugs Ther. 2020, 9, 980. [Google Scholar] [CrossRef]
- Niederseer, D.; Wernly, B.; Aigner, E.; Stickel, F.; Datz, C. NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations. J. Clin. Med. 2021, 10, 467. [Google Scholar] [CrossRef]
- Sonmez, A.; Nikolic, D.; Dogru, T.; Ercin, C.N.; Genc, H.; Cesur, M.; Tapan, S.; Karslioglu, Y.; Montalto, G.; Banach, M.; et al. Low- and high-density lipoprotein subclasses in subjects with nonalcoholic fatty liver disease. J. Clin. Lipidol. 2015, 9, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Corey, K.E.; Misdraji, J.; Gelrud, L.; Zheng, H.; Chung, R.T.; Krauss, R.M. Nonalcoholic steatohepatitis is associated with an atherogenic lipoprotein subfraction profile. Lipids Health Dis. 2014, 13, 100. [Google Scholar] [CrossRef] [Green Version]
- Tutunchi, H.; Naeini, F.; Ebrahimi-Mameghani, M.; Mobasseri, M.; Naghshi, S.; Ostadrahimi, A. The association of the steatosis severity, NAFLD fibrosis score and FIB-4 index with atherogenic dyslipidaemia in adult patients with NAFLD: A cross-sectional study. Int. J. Clin. Pract. 2021, 75, e14131. [Google Scholar] [CrossRef]
- Gottlieb, A.; Leven, A.S.; Sowa, J.P.; Borucki, K.; Link, A.; Yilmaz, E.; Aygen, S.; Canbay, A.; Porsch-Ozcurumez, M. Lipoprotein and Metabolic Profiles Indicate Similar Cardiovascular Risk of Liver Steatosis and NASH. Digestion 2021, 102, 671–681. [Google Scholar] [CrossRef]
- Deprince, A.; Haas, J.T.; Staels, B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol. Metab. 2020, 42, 101092. [Google Scholar] [CrossRef]
- Nass, K.J.; van den Berg, E.H.; Faber, K.N.; Schreuder, T.; Blokzijl, H.; Dullaart, R.P.F. High prevalence of apolipoprotein B dyslipoproteinemias in non-alcoholic fatty liver disease: The lifelines cohort study. Metabolism 2017, 72, 37–46. [Google Scholar] [CrossRef]
- Castillo-Leon, E.; Connelly, M.A.; Konomi, J.V.; Caltharp, S.; Cleeton, R.; Vos, M.B. Increased atherogenic lipoprotein profile in children with non-alcoholic steatohepatitis. Pediatr. Obes. 2020, 15, e12648. [Google Scholar] [CrossRef]
- Duong, M.; Uno, K.; Nankivell, V.; Bursill, C.; Nicholls, S.J. Induction of obesity impairs reverse cholesterol transport in ob/ob mice. PLoS ONE 2018, 13, e0202102. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Bezsonov, E.E.; Baig, M.S.; Popkova, T.V.; Orekhov, A.N. Mitochondrial Lipid Homeostasis at the Crossroads of Liver and Heart Diseases. Int. J. Mol. Sci. 2021, 22, 6949. [Google Scholar] [CrossRef]
- Brunner, K.T.; Pedley, A.; Massaro, J.M.; Hoffmann, U.; Benjamin, E.J.; Long, M.T. Increasing liver fat is associated with progression of cardiovascular risk factors. Liver Int. 2020, 40, 1339–1343. [Google Scholar] [CrossRef]
- Liu, B.; Li, Y.; Li, Y.; Liu, Y.; Yan, Y.; Luo, A.; Ren, H.; She, Q. Association of epicardial adipose tissue with non-alcoholic fatty liver disease: A meta-analysis. Hepatol. Int. 2019, 13, 757–765. [Google Scholar] [CrossRef]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.S.; Roh, J.H.; Lee, J.H.; Lee, H.; Min Kim, Y.; Yoon, Y.H.; Kim, M.; Kim, Y.G.; Park, G.M.; Park, J.H.; et al. Association between nonalcoholic fatty liver disease and cardiovascular disease revealed after comprehensive control of metabolic risk factors: A nationwide population-based study in Korea. Eur. J. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, G.; Di Marco, V.; Buscemi, C.; Mazzola, G.; Randazzo, C.; Spatola, F.; Craxi, A.; Buscemi, S.; Petta, S. Interplay between non-alcoholic fatty liver disease and cardiovascular risk in an asymptomatic general population. J. Gastroenterol. Hepatol. 2021, 36, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, G.T.S.; Longo, L.; Fonseca, M.A.; de Souza, V.E.G.; Alvares-da-Silva, M.R. Does the risk of cardiovascular events differ between biopsy-proven NAFLD and MAFLD? Hepatol. Int. 2021, 15, 380–391. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease-related risk of cardiovascular disease and other cardiac complications. Diabetes Obes. Metab. 2021. [Google Scholar] [CrossRef]
- Meyersohn, N.M.; Mayrhofer, T.; Corey, K.E.; Bittner, D.O.; Staziaki, P.V.; Szilveszter, B.; Hallett, T.; Lu, M.T.; Puchner, S.B.; Simon, T.G.; et al. Association of Hepatic Steatosis With Major Adverse Cardiovascular Events, Independent of Coronary Artery Disease. Clin. Gastroenterol. Hepatol. 2021, 19, 1480–1488.e14. [Google Scholar] [CrossRef]
- Henson, J.B.; Simon, T.G.; Kaplan, A.; Osganian, S.; Masia, R.; Corey, K.E. Advanced fibrosis is associated with incident cardiovascular disease in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2020, 51, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.F.; Wang, Y.W.; Lin, C.C.; Wang, Y.J.; Ding, Y.Z.; Liou, T.L.; Huang, S.S.; Lu, T.M.; Chan, W.L.; Lin, S.J.; et al. The association of the steatosis severity in fatty liver disease with coronary plaque pattern in general population. Liver Int. 2021, 41, 81–90. [Google Scholar] [CrossRef]
- Liu, Z.; Wei, R.; Li, Y. Coronary heart disease is associated with nonalcoholic fatty liver disease in patients without hypertension and diabetes. Medicine 2020, 99, e20898. [Google Scholar] [CrossRef]
- Labenz, C.; Huber, Y.; Michel, M.; Nagel, M.; Galle, P.R.; Kostev, K.; Schattenberg, J.M. Impact of NAFLD on the Incidence of Cardiovascular Diseases in a Primary Care Population in Germany. Dig. Dis. Sci. 2020, 65, 2112–2119. [Google Scholar] [CrossRef]
- Lee, H.; Lee, Y.H.; Kim, S.U.; Kim, H.C. Metabolic Dysfunction-Associated Fatty Liver Disease and Incident Cardiovascular Disease Risk: A Nationwide Cohort Study. Clin. Gastroenterol. Hepatol. 2020, S1542–S3565. [Google Scholar] [CrossRef]
- Xia, W.; Yang, N.; Li, Y. Analysis of Risk Factors for Adverse Cardiovascular Events in Elderly Patients with Acute Myocardial Infarction and Non-Alcoholic Fatty Liver Disease (NAFLD). Med. Sci. Monit. 2020, 26, e922913. [Google Scholar] [CrossRef] [PubMed]
- Tana, C.; Ballestri, S.; Ricci, F.; Di Vincenzo, A.; Ticinesi, A.; Gallina, S.; Giamberardino, M.A.; Cipollone, F.; Sutton, R.; Vettor, R.; et al. Cardiovascular Risk in Non-Alcoholic Fatty Liver Disease: Mechanisms and Therapeutic Implications. Int. J. Environ. Res. Public Health 2019, 16, 3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalic, A.J.; Satapathy, S.K. The Role of Nonalcoholic Fatty Liver Disease on Cardiovascular Manifestations and Outcomes. Clin. Liver Dis. 2018, 22, 141–174. [Google Scholar] [CrossRef] [PubMed]
- Golabi, P.; Fukui, N.; Paik, J.; Sayiner, M.; Mishra, A.; Younossi, Z.M. Mortality Risk Detected by Atherosclerotic Cardiovascular Disease Score in Patients with Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2019, 3, 1050–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Konyn, P.; Sandhu, K.K.; Dennis, B.B.; Cheung, A.C.; Ahmed, A. Metabolic dysfunction-associated fatty liver disease is associated with increased all-cause mortality in the United States. J. Hepatol. 2021. [Google Scholar] [CrossRef]
- Haring, R.; Wallaschofski, H.; Nauck, M.; Dorr, M.; Baumeister, S.E.; Volzke, H. Ultrasonographic hepatic steatosis increases prediction of mortality risk from elevated serum gamma-glutamyl transpeptidase levels. Hepatology 2009, 50, 1403–1411. [Google Scholar] [CrossRef]
- Konishi, K.; Miyake, T.; Furukawa, S.; Senba, H.; Kanzaki, S.; Nakaguchi, H.; Yukimoto, A.; Nakamura, Y.; Watanabe, T.; Koizumi, Y.; et al. Advanced fibrosis of non-alcoholic steatohepatitis affects the significance of lipoprotein(a) as a cardiovascular risk factor. Atherosclerosis 2020, 299, 32–37. [Google Scholar] [CrossRef]
- Baratta, F.; Pastori, D.; Angelico, F.; Balla, A.; Paganini, A.M.; Cocomello, N.; Ferro, D.; Violi, F.; Sanyal, A.J.; Del Ben, M. Nonalcoholic Fatty Liver Disease and Fibrosis Associated with Increased Risk of Cardiovascular Events in a Prospective Study. Clin. Gastroenterol. Hepatol. 2020, 18, 2324–2331.e4. [Google Scholar] [CrossRef]
- Dasarathy, S.; Dasarathy, J.; Khiyami, A.; Joseph, R.; Lopez, R.; McCullough, A.J. Validity of real time ultrasound in the diagnosis of hepatic steatosis: A prospective study. J. Hepatol. 2009, 51, 1061–1067. [Google Scholar] [CrossRef]
- Di Sessa, A.; Umano, G.R.; Miraglia Del Giudice, E.; Santoro, N. From the liver to the heart: Cardiac dysfunction in obese children with non-alcoholic fatty liver disease. World J. Hepatol. 2017, 9, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Azzam, H.; Malnick, S. Non-alcoholic fatty liver disease—The heart of the matter. World J. Hepatol. 2015, 7, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.C.; Wild, S.H.; Kwag, H.J.; Byrne, C.D. Fatty liver, insulin resistance, and features of metabolic syndrome: Relationships with coronary artery calcium in 10,153 people. Diabetes Care 2012, 35, 2359–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.G.; Jung, J.; Verma, K.K.; Kang, M.K.; Madamba, E.; Lopez, S.; Qas Yonan, A.; Liu, A.; Bettencourt, R.; Sirlin, C.; et al. Liver stiffness by magnetic resonance elastography is associated with increased risk of cardiovascular disease in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2021, 53, 1030–1037. [Google Scholar] [PubMed]
- Long, M.T.; Zhang, X.; Xu, H.; Liu, C.T.; Corey, K.E.; Chung, R.T.; Loomba, R.; Benjamin, E.J. Hepatic Fibrosis Associates with Multiple Cardiometabolic Disease Risk Factors: The Framingham Heart Study. Hepatology 2021, 73, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Pemmasani, G.; Yandrapalli, S.; Aronow, W. Sex differences in cardiovascular diseases and associated risk factors in non-alcoholic steatohepatitis. Am. J. Cardiovasc. Dis. 2020, 10, 362–366. [Google Scholar]
- Yang, Y.J.; Jung, M.H.; Jeong, S.H.; Hong, Y.P.; Kim, Y.I.; An, S.J. The Association between Nonalcoholic Fatty Liver Disease and Stroke: Results from the Korean Genome and Epidemiology Study (KoGES). Int. J. Environ. Res. Public Health 2020, 17, 9568. [Google Scholar] [CrossRef] [PubMed]
- Wijarnpreecha, K.; Panjawatanan, P.; Kroner, P.T.; Cheungpasitporn, W.; Ungprasert, P. Association between cardiac conduction defect and nonalcoholic fatty liver disease: A systematic review and meta-analysis. Ann. Gastroenterol. 2020, 33, 661–666. [Google Scholar] [CrossRef]
- Xu, J.; Dai, L.; Zhang, Y.; Wang, A.; Li, H.; Wang, Y.; Meng, X.; Wu, S.; Wang, Y. Severity of Nonalcoholic Fatty Liver Disease and Risk of Future Ischemic Stroke Events. Stroke 2021, 52, 103–110. [Google Scholar] [CrossRef]
- Parikh, N.S.; VanWagner, L.B.; Elkind, M.S.V.; Gutierrez, J. Association between nonalcoholic fatty liver disease with advanced fibrosis and stroke. J. Neurol. Sci. 2019, 407, 116524. [Google Scholar] [CrossRef]
- Parikh, N.S.; Koh, I.; VanWagner, L.B.; Elkind, M.S.V.; Zakai, N.A.; Cushman, M. Liver Fibrosis is Associated with Ischemic Stroke Risk in Women but not Men: The REGARDS Study. J. Stroke Cerebrovasc. Dis. 2021, 30, 105788. [Google Scholar] [CrossRef]
- Chiu, L.S.; Pedley, A.; Massaro, J.M.; Benjamin, E.J.; Mitchell, G.F.; McManus, D.D.; Aragam, J.; Vasan, R.S.; Cheng, S.; Long, M.T. The association of non-alcoholic fatty liver disease and cardiac structure and function-Framingham Heart Study. Liver Int. 2020, 40, 2445–2454. [Google Scholar] [CrossRef]
- VanWagner, L.B.; Wilcox, J.E.; Ning, H.; Lewis, C.E.; Carr, J.J.; Rinella, M.E.; Shah, S.J.; Lima, J.A.C.; Lloyd-Jones, D.M. Longitudinal Association of Non-Alcoholic Fatty Liver Disease with Changes in Myocardial Structure and Function: The CARDIA Study. J. Am. Heart Assoc. 2020, 9, e014279. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Miceli, S.; Succurro, E.; Sciacqua, A.; Andreozzi, F.; Sesti, G. Nonalcoholic fatty liver disease is associated with a decreased myocardial mechano-energetic efficiency. J. Intern. Med. 2021, 289, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Thanassoulis, G.; Massaro, J.M.; O’Donnell, C.J.; Hoffmann, U.; Levy, D.; Ellinor, P.T.; Wang, T.J.; Schnabel, R.B.; Vasan, R.S.; Fox, C.S.; et al. Pericardial fat is associated with prevalent atrial fibrillation: The Framingham Heart Study. Circ. Arrhythm. Electrophysiol. 2010, 3, 345–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnellan, E.; Cotter, T.G.; Wazni, O.M.; Elshazly, M.B.; Kochar, A.; Wilner, B.; Patel, D.; Kanj, M.; Hussein, A.; Baranowski, B.; et al. Impact of Nonalcoholic Fatty Liver Disease on Arrhythmia Recurrence Following Atrial Fibrillation Ablation. JACC Clin. Electrophysiol. 2020, 6, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Sciacqua, A.; Marcucci, R.; Farcomeni, A.; Perticone, F.; Del Ben, M.; Angelico, F.; Baratta, F.; Pignatelli, P.; Violi, F.; et al. Prevalence and Impact of Nonalcoholic Fatty Liver Disease in Atrial Fibrillation. Mayo Clin. Proc. 2020, 95, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zheng, S.; Liu, Y.; Zhang, Y.; Lu, J.; Huang, Y. Nonalcoholic fatty liver disease is associated with increased risk of atrial fibrillation. Liver Int. 2020, 40, 1594–1600. [Google Scholar] [CrossRef]
- Kang, M.K.; Park, J.G.; Kim, M.C. Association between Atrial Fibrillation and Advanced Liver Fibrosis in Patients with Non-Alcoholic Fatty Liver Disease. Yonsei Med. J. 2020, 61, 860–867. [Google Scholar] [CrossRef]
- Graner, M.; Nyman, K.; Siren, R.; Pentikainen, M.O.; Lundbom, J.; Hakkarainen, A.; Lauerma, K.; Lundbom, N.; Nieminen, M.S.; Taskinen, M.R. Ectopic fat depots and left ventricular function in nondiabetic men with nonalcoholic fatty liver disease. Circ. Cardiovasc. Imaging 2014, 8, e001979. [Google Scholar] [CrossRef] [Green Version]
- Gastaldelli, A.; Gaggini, M.; DeFronzo, R.A. Role of Adipose Tissue Insulin Resistance in the Natural History of Type 2 Diabetes: Results from the San Antonio Metabolism Study. Diabetes 2017, 66, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.F.; Lembo, E.; Mari, A.; Angelini, G.; Verrastro, O.; Nanni, G.; Pompili, M.; Raffaelli, M.; Vecchio, F.M.; Bornstein, S.R.; et al. Insulin Resistance Is Central to Long-Term Reversal of Histologic Nonalcoholic Steatohepatitis After Metabolic Surgery. J. Clin. Endocrinol. Metab. 2021, 106, 750–761. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Martinez-Rodriguez, J.; Gonzalez-Lucan, M.; Fernandez-Fernandez, C.; Castro-Quintela, E. Insulin resistance is a cardiovascular risk factor in humans. Diabetes Metab. Syndr. 2019, 13, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Perla, F.M.; Prelati, M.; Lavorato, M.; Visicchio, D.; Anania, C. The Role of Lipid and Lipoprotein Metabolism in Non-Alcoholic Fatty Liver Disease. Children 2017, 4, 46. [Google Scholar] [CrossRef] [Green Version]
- Pinto, X.; Fanlo-Maresma, M.; Corbella, E.; Corbella, X.; Mitjavila, M.T.; Moreno, J.J.; Casas, R.; Estruch, R.; Corella, D.; Bullo, M.; et al. A Mediterranean Diet Rich in Extra-Virgin Olive Oil Is Associated with a Reduced Prevalence of Nonalcoholic Fatty Liver Disease in Older Individuals at High Cardiovascular Risk. J. Nutr. 2019, 149, 1920–1929. [Google Scholar] [CrossRef]
- Seebacher, F.; Zeigerer, A.; Kory, N.; Krahmer, N. Hepatic lipid droplet homeostasis and fatty liver disease. Semin. Cell Dev. Biol. 2020, 108, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, J.L.; Laufs, U. New Insights in the Control of Low-Density Lipoprotein Cholesterol to Prevent Cardiovascular Disease. Curr. Cardiol. Rep. 2019, 21, 69. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 70, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Smeuninx, B.; Boslem, E.; Febbraio, M.A. Current and Future Treatments in the Fight against Non-Alcoholic Fatty Liver Disease. Cancers 2020, 12, 1714. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Types of Risk Factors | Specific Risk Factors |
|---|---|
| Genetics | Melanocortin-4 receptor mutation, leptin deficiency, pro-opiomelanocortin deficiency, variant rs9939609 of the FTO gene, parental obesity, and epigenetic modifications |
| Behavioral history | Nutrition, eating behavior, poor dietary choices, high calories, high-fat food, sugar-sweetened beverages, physical inactivity, sedentary lifestyle, insufficient sleep, stress, and smoking cessation |
| Socioeconomic | Low incomes, poverty, low education, unemployment, industrialization, mechanized transportation, urbanization, and socioeconomic status |
| Environmental | Cultural influences, television watching, fast food restaurants, culture, social bias, and environmental chemicals |
| Biological | Gut microbiome, viruses, brain-gut axis, prenatal determinants, pregnancy, gestational diabetes, menopause, neuroendocrine conditions, medications, and physical disability |
| Type of Comorbidity | Specific Comorbidity |
|---|---|
| Endocrine | Hyperleptinemia, hypothyroidism, hypercortisolism, Cushing’s syndrome, polycystic ovary syndrome, metabolic syndrome, and T2D |
| Gastrointestinal | Kidney stones, glomerulopathy, kidney dysfunction, urinary incontinence (in women), pancreatitis, gallbladder disease, and liver disease (MAFLD and NASH) |
| Respiratory | Obstructive sleep apnea and asthma |
| Esophageal | Gastroesophageal reflux disease and Barrett’s esophagus |
| Cardiovascular | Hypertension, CHD, AF, diastolic dysfunction, HF, ischemic stroke, and cardiac fibrosis |
| Neurological | Alzheimer’s disease, vascular dementia, any type of dementia, mood, anxiety, and other psychiatric disorders |
| Fertility | In women: preeclampsia, eclampsia of pregnancy, depression, amenorrhea, menorrhagia, and infertility In men: low sperm count and erectile dysfunction |
| Immune system dysregulation | Infections such as surgical-site, urinary tract, nosocomial, and skin |
| Autoimmune diseases | Rheumatoid arthritis, osteoarthritis, multiple sclerosis, psoriasis, and psoriatic arthritis |
| Viral | H1N1 influenza virus and SARS-CoV-2 (obesity increases severity of the disease) |
| Cancer | Esophageal, colon, pancreatic, endometrium, renal, gastric, uterine, gallbladder, cervical, thyroid, prostate, leukemia, liver, ovarian (epithelial), and breast (postmenopausal) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutiérrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Pathophysiological Molecular Mechanisms of Obesity: A Link between MAFLD and NASH with Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 11629. https://doi.org/10.3390/ijms222111629
Gutiérrez-Cuevas J, Santos A, Armendariz-Borunda J. Pathophysiological Molecular Mechanisms of Obesity: A Link between MAFLD and NASH with Cardiovascular Diseases. International Journal of Molecular Sciences. 2021; 22(21):11629. https://doi.org/10.3390/ijms222111629
Chicago/Turabian StyleGutiérrez-Cuevas, Jorge, Arturo Santos, and Juan Armendariz-Borunda. 2021. "Pathophysiological Molecular Mechanisms of Obesity: A Link between MAFLD and NASH with Cardiovascular Diseases" International Journal of Molecular Sciences 22, no. 21: 11629. https://doi.org/10.3390/ijms222111629
APA StyleGutiérrez-Cuevas, J., Santos, A., & Armendariz-Borunda, J. (2021). Pathophysiological Molecular Mechanisms of Obesity: A Link between MAFLD and NASH with Cardiovascular Diseases. International Journal of Molecular Sciences, 22(21), 11629. https://doi.org/10.3390/ijms222111629