V-Dock: Fast Generation of Novel Drug-like Molecules Using Machine-Learning-Based Docking Score and Molecular Optimization


Abstract
:1. Introduction
2. Results and Discussion
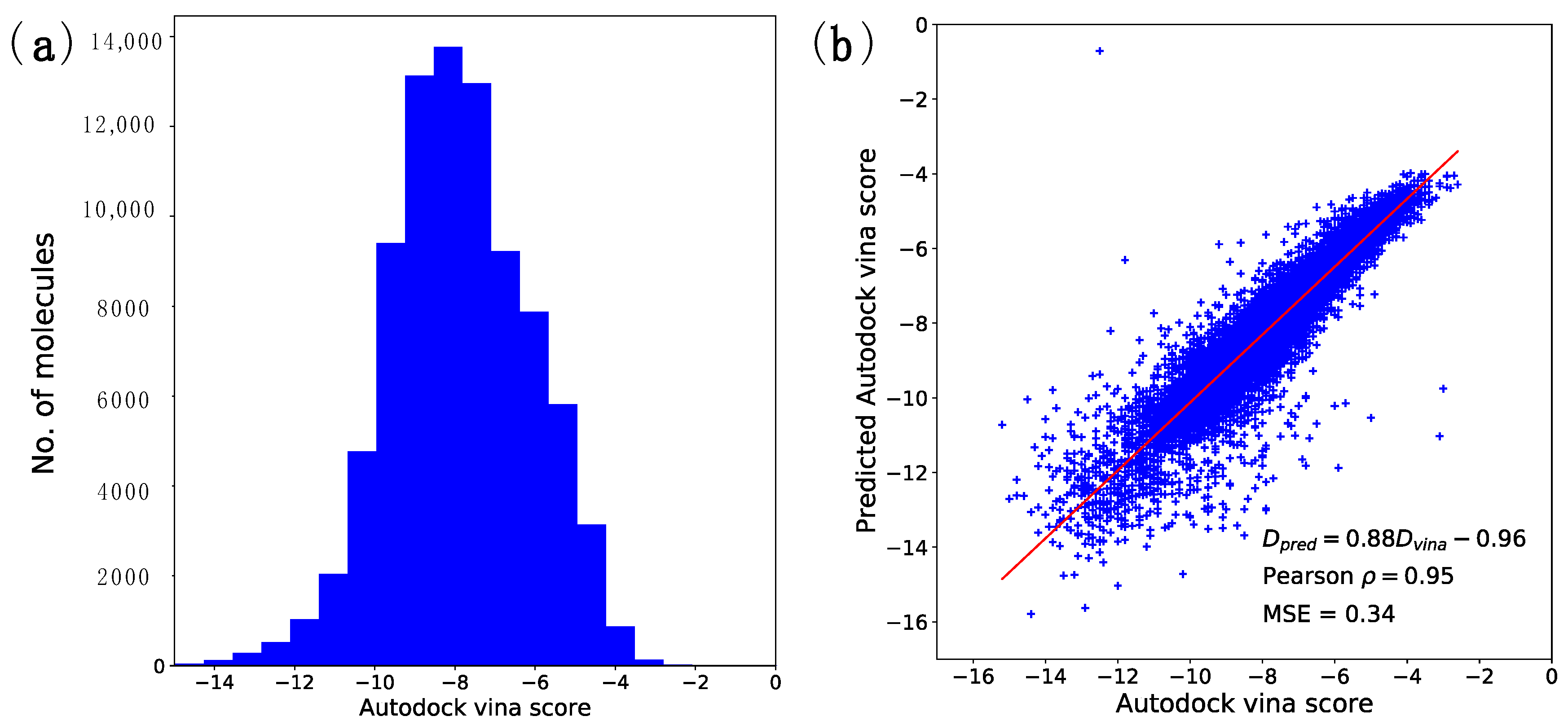
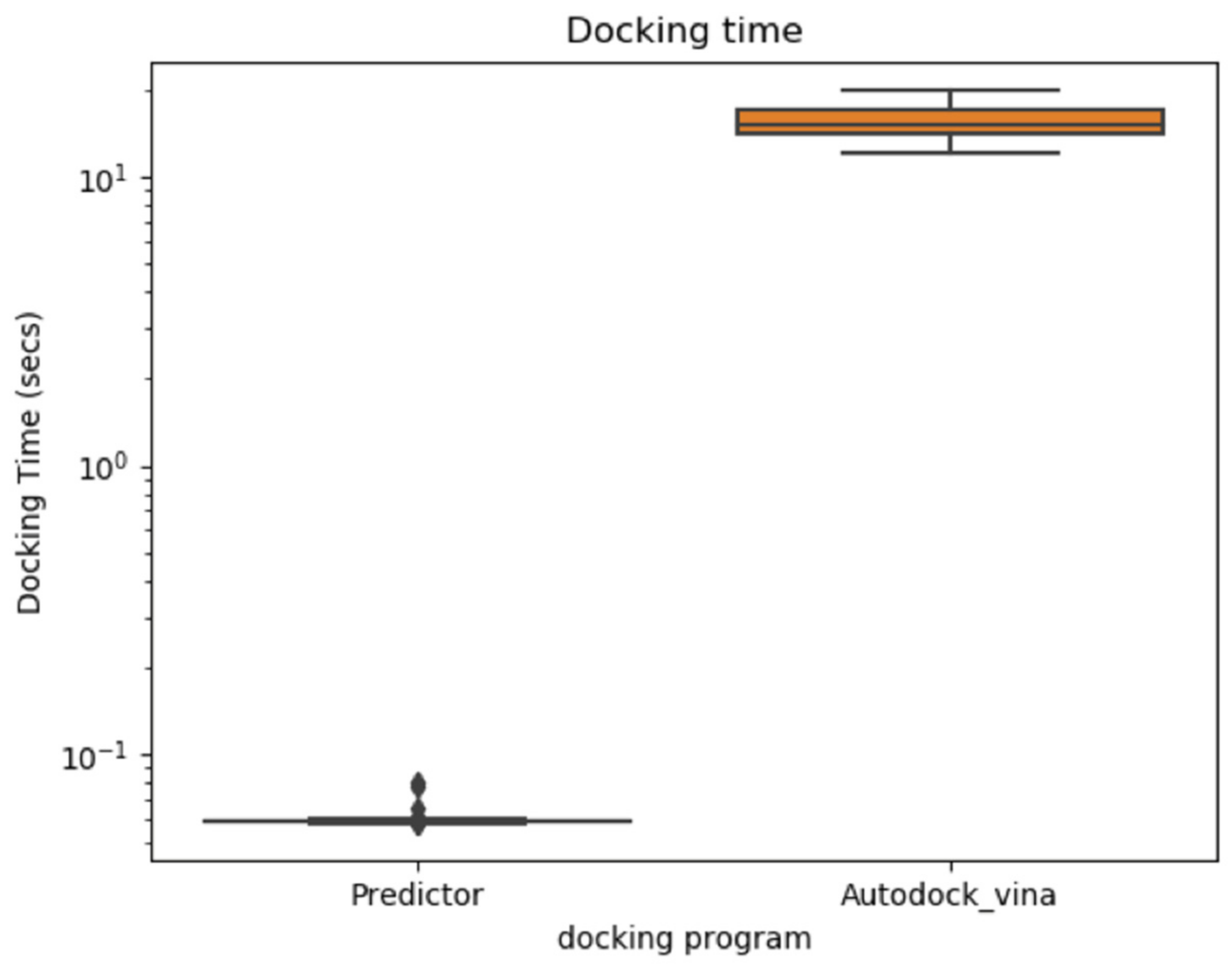
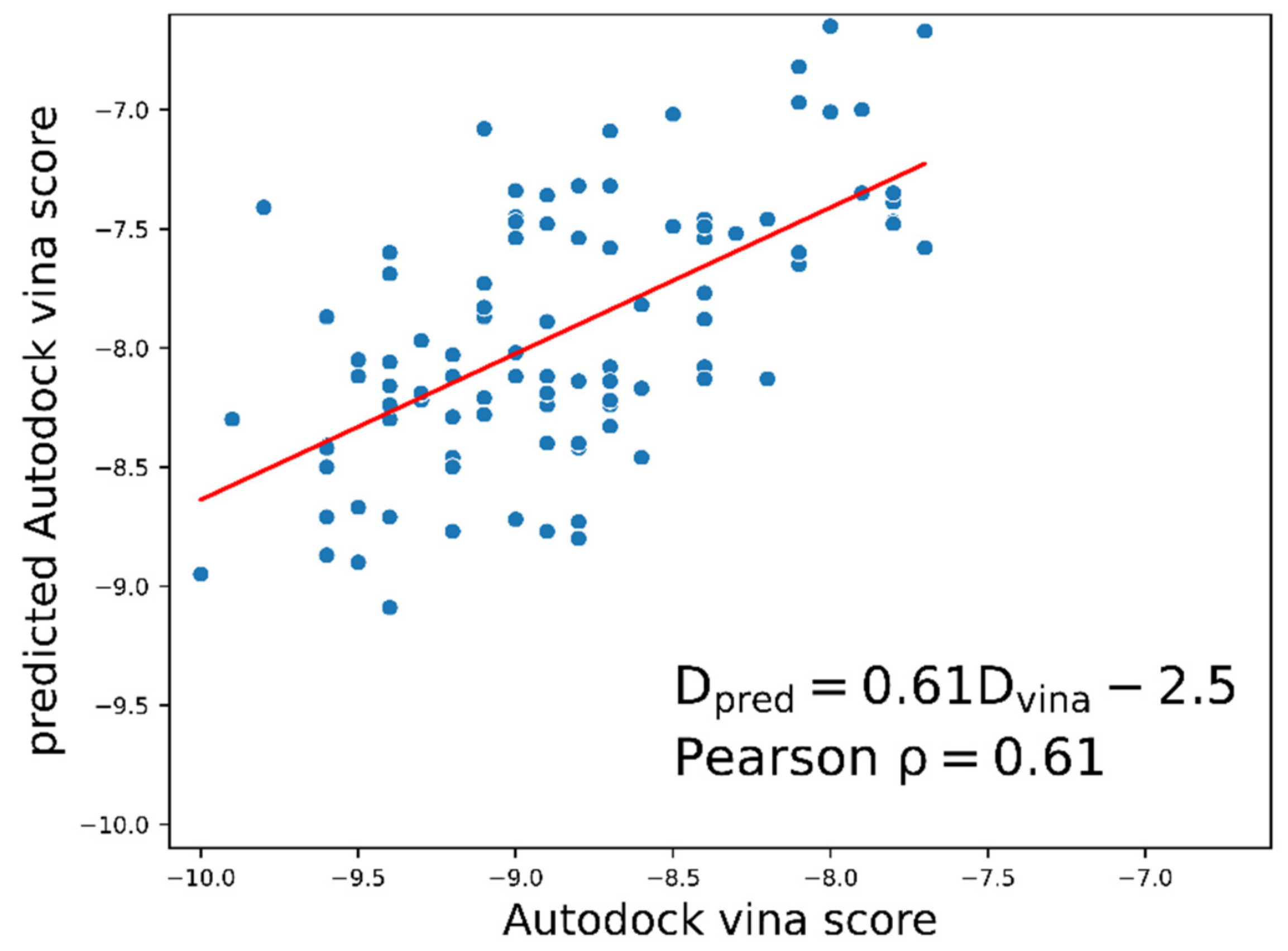
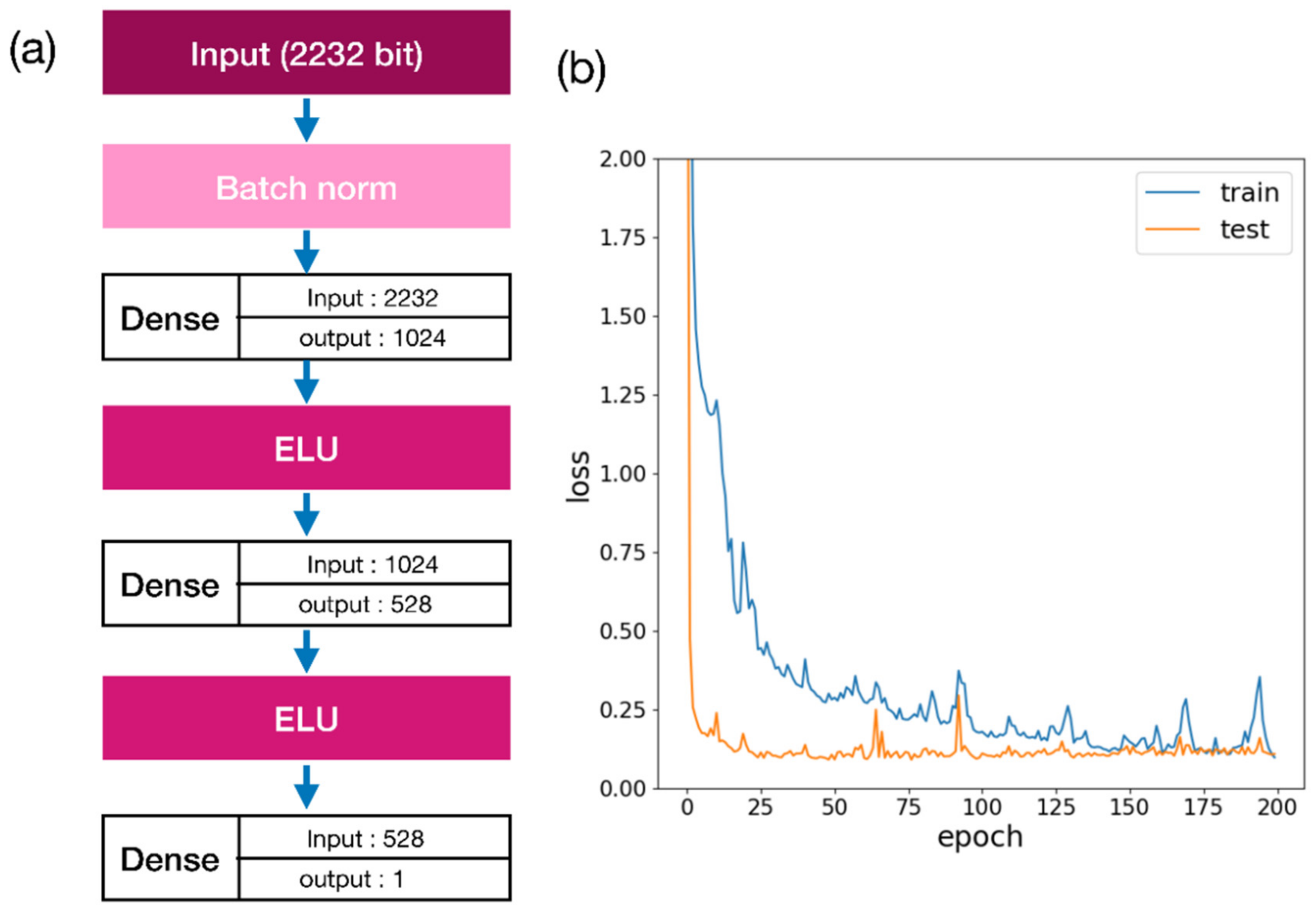
2.1. Docking Energy Prediction Model Training Result
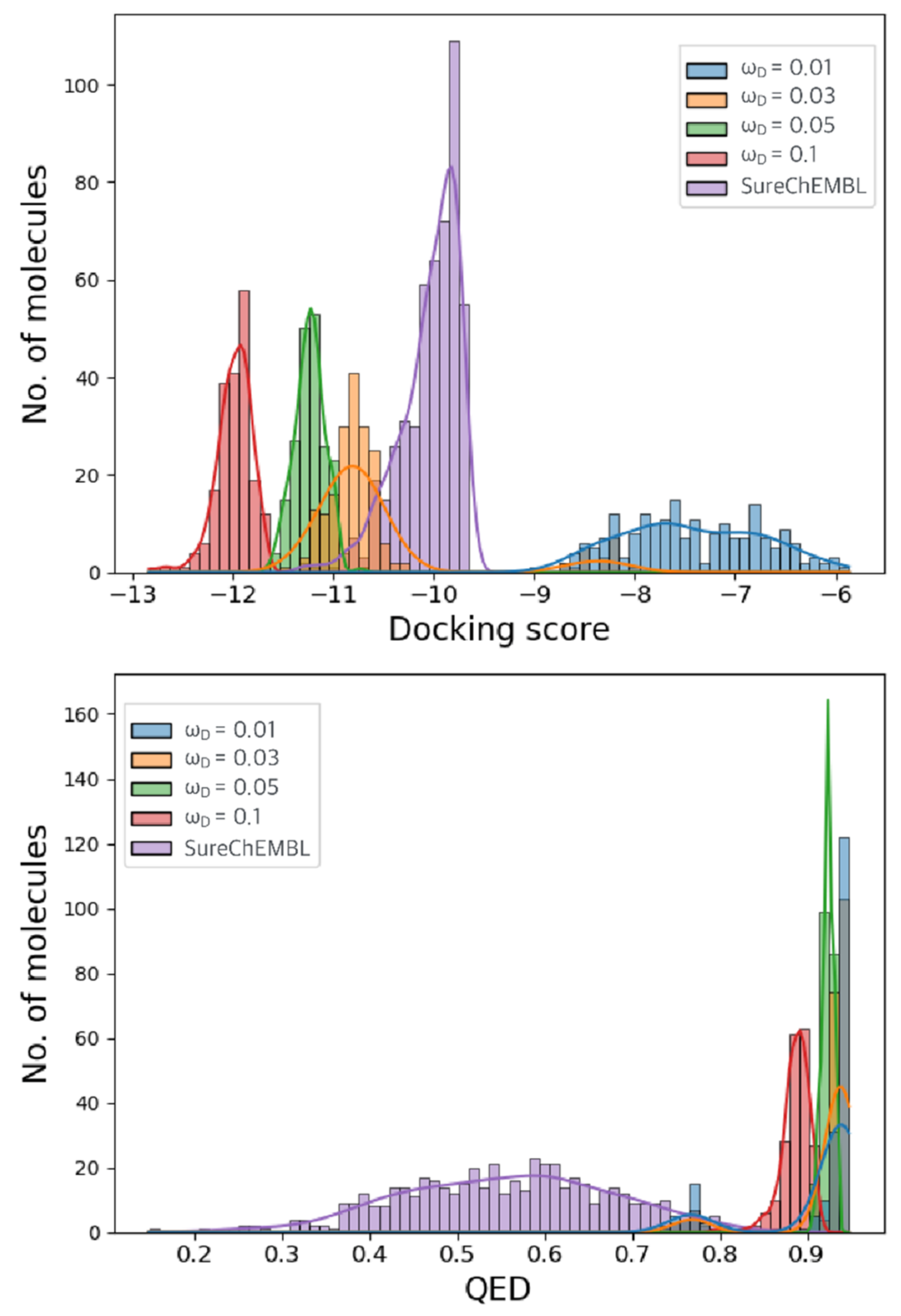
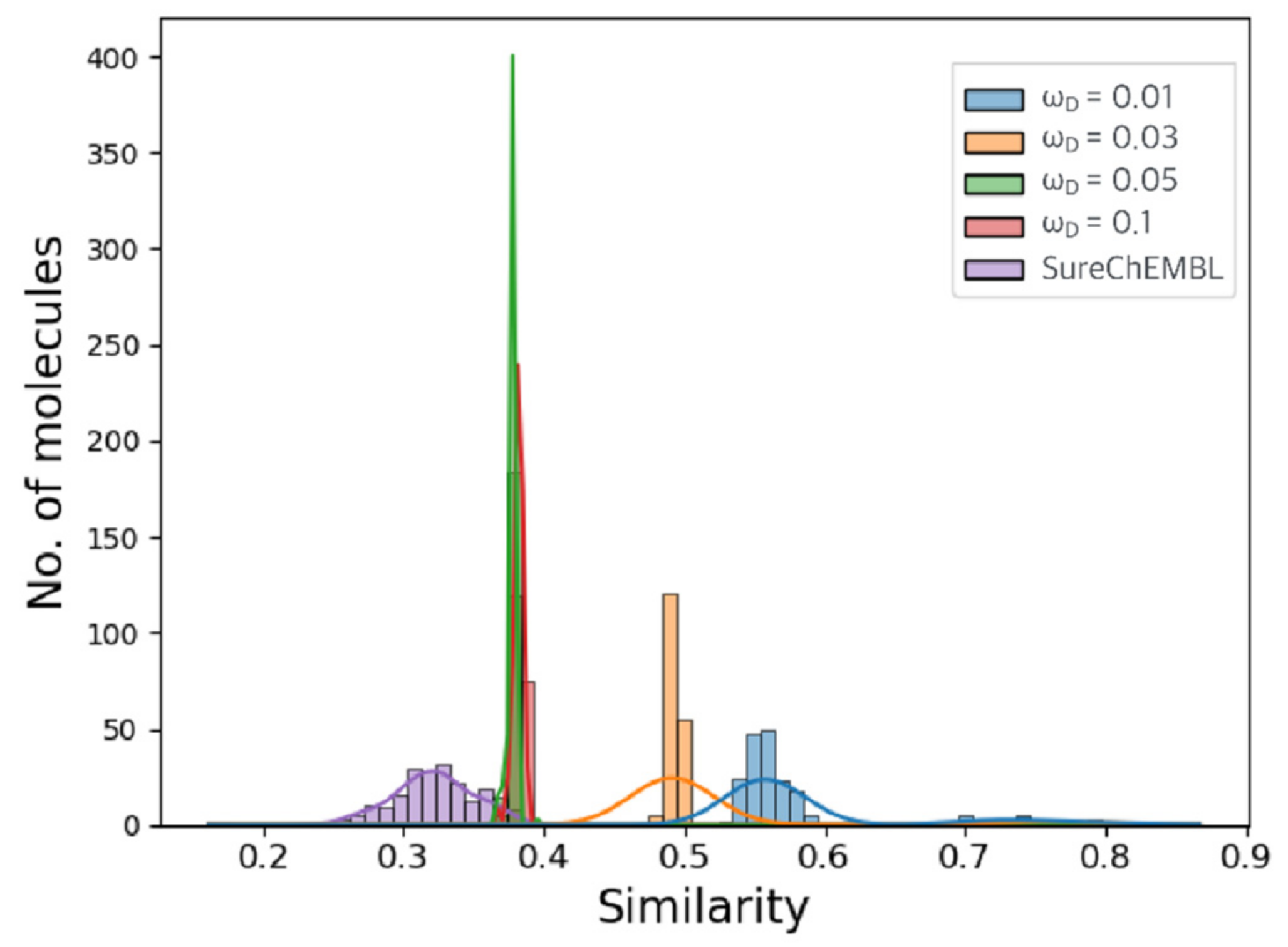
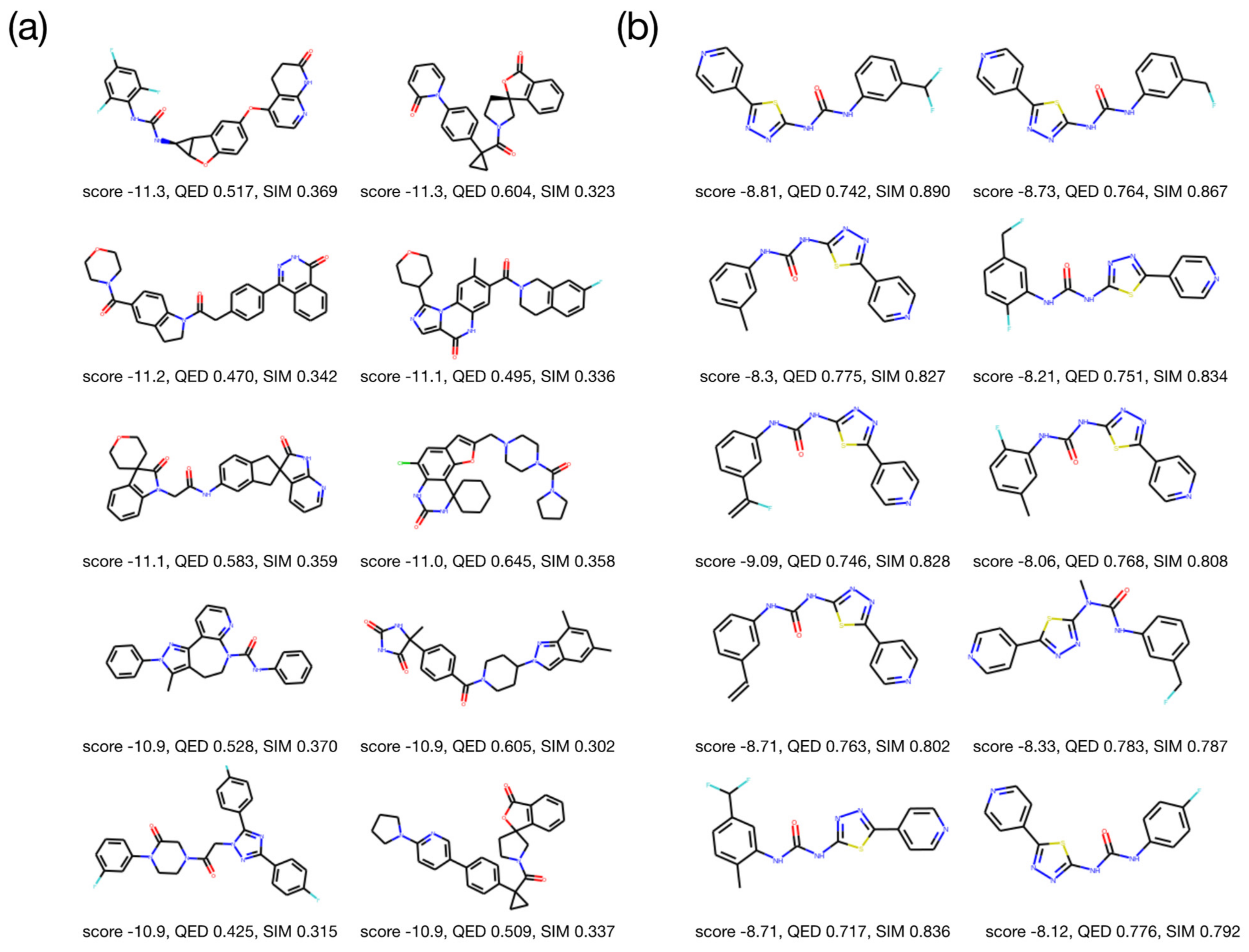
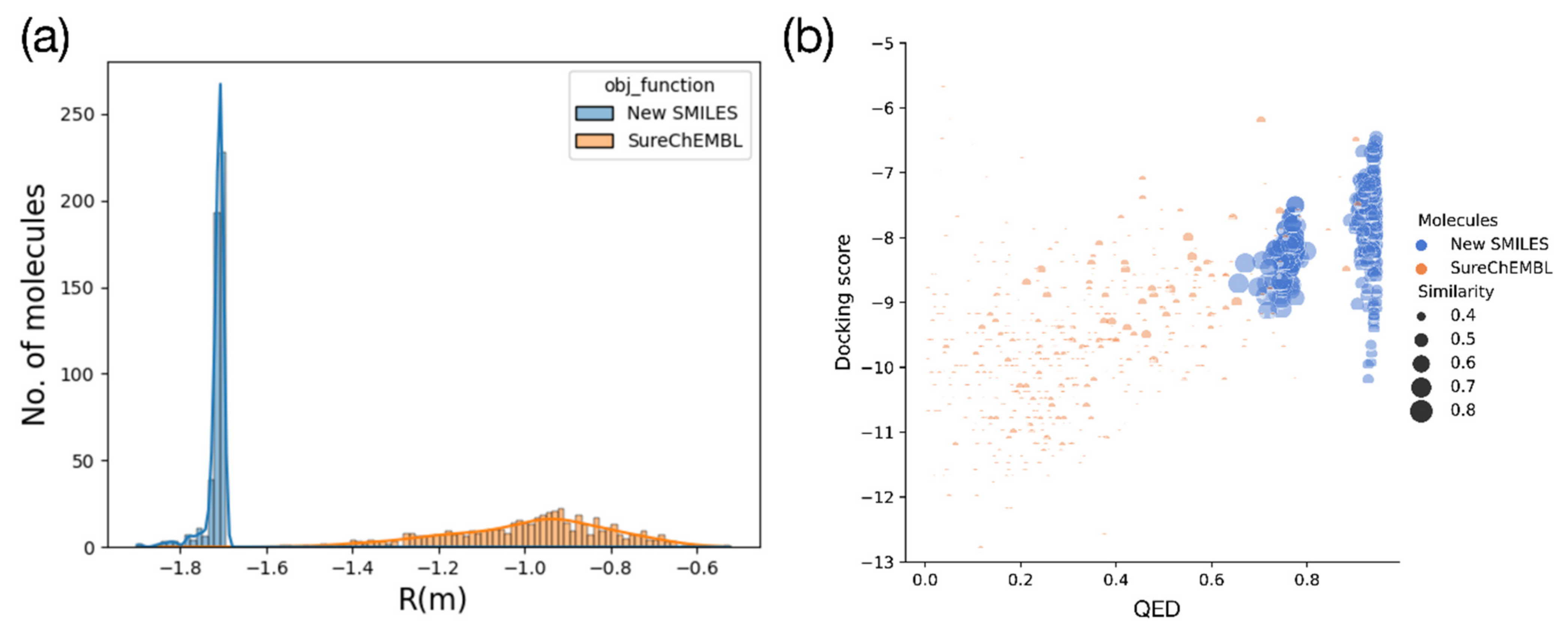
2.2. Optimization Results for Generated Molecules
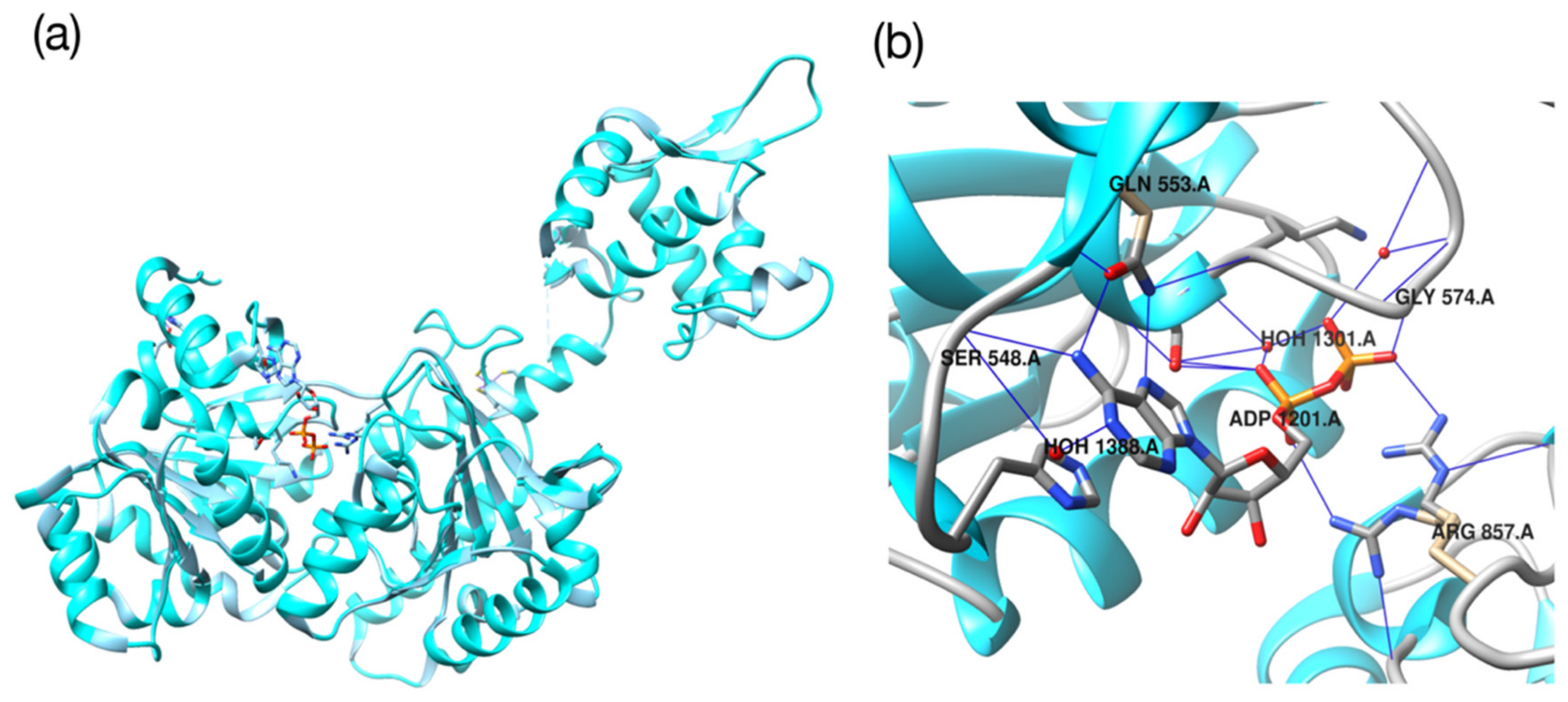
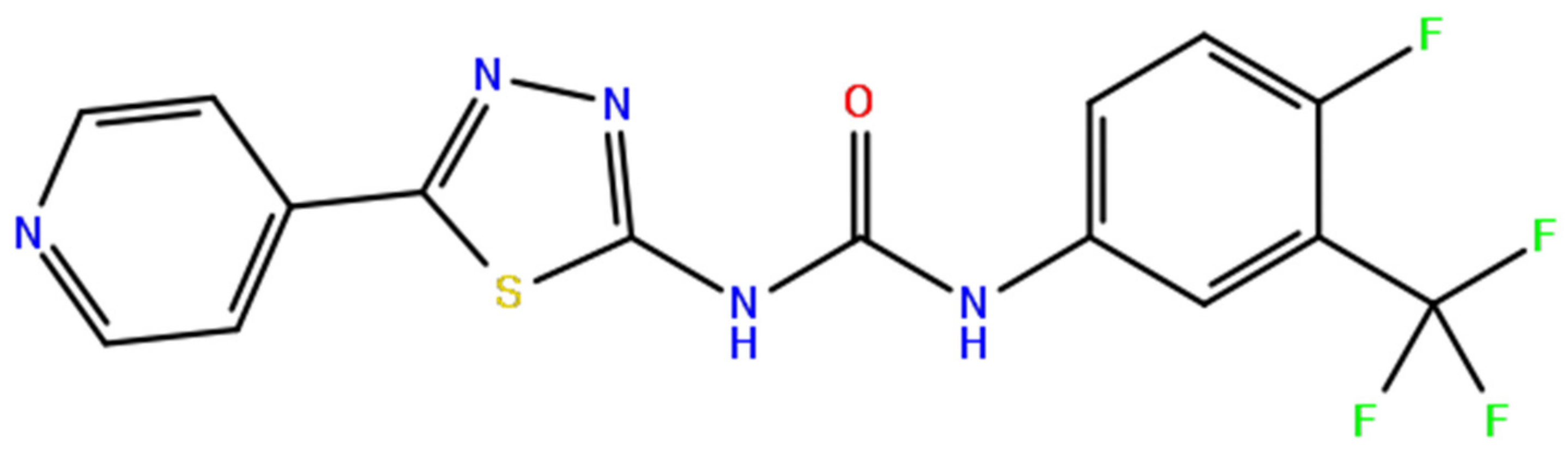
2.3. Molecular Validation Designed with V-Dock
2.4. Possible Limitations of the V-Dock Approach
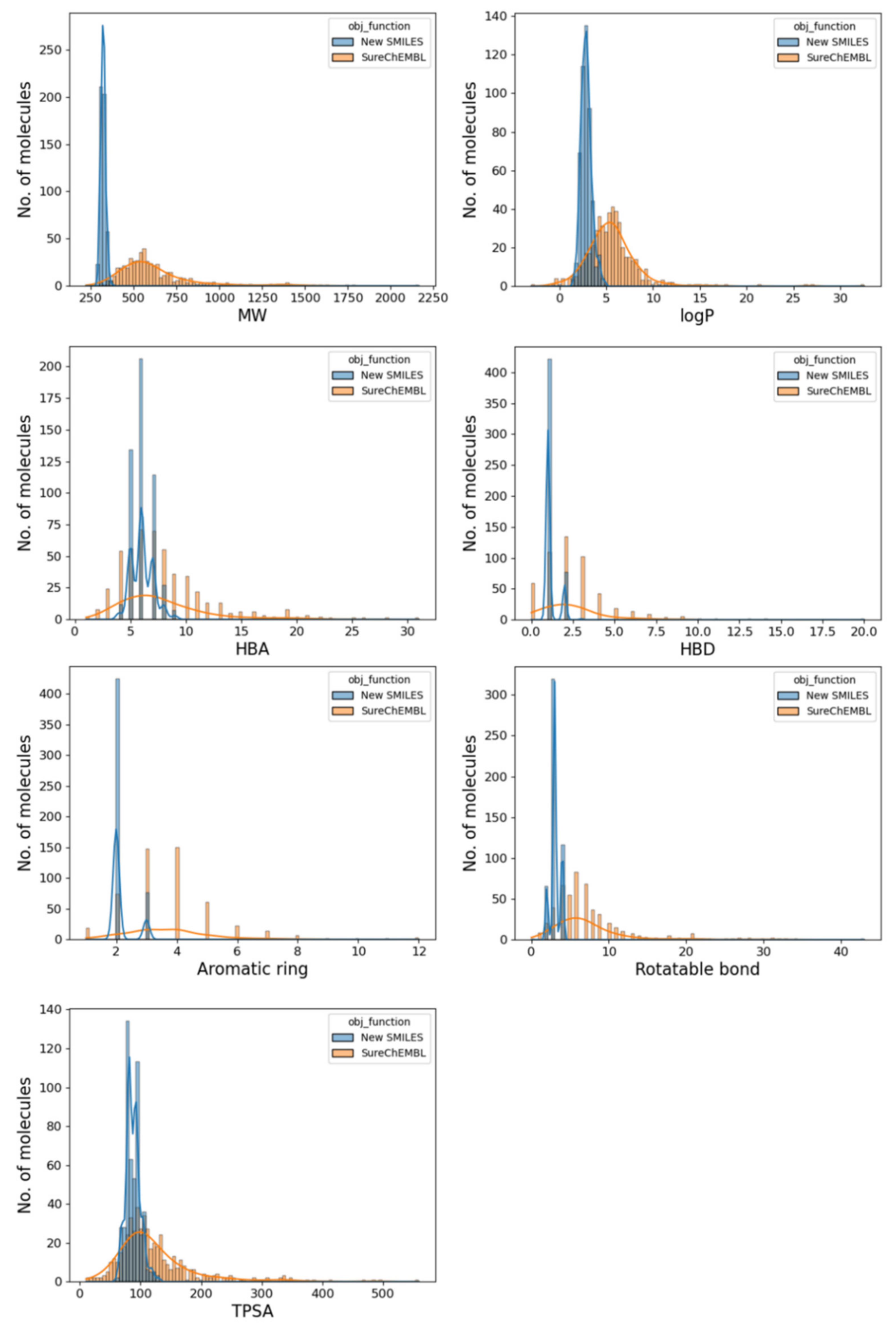
2.5. Drug-likeness of the Generated Molecule
3. Materials and Methods
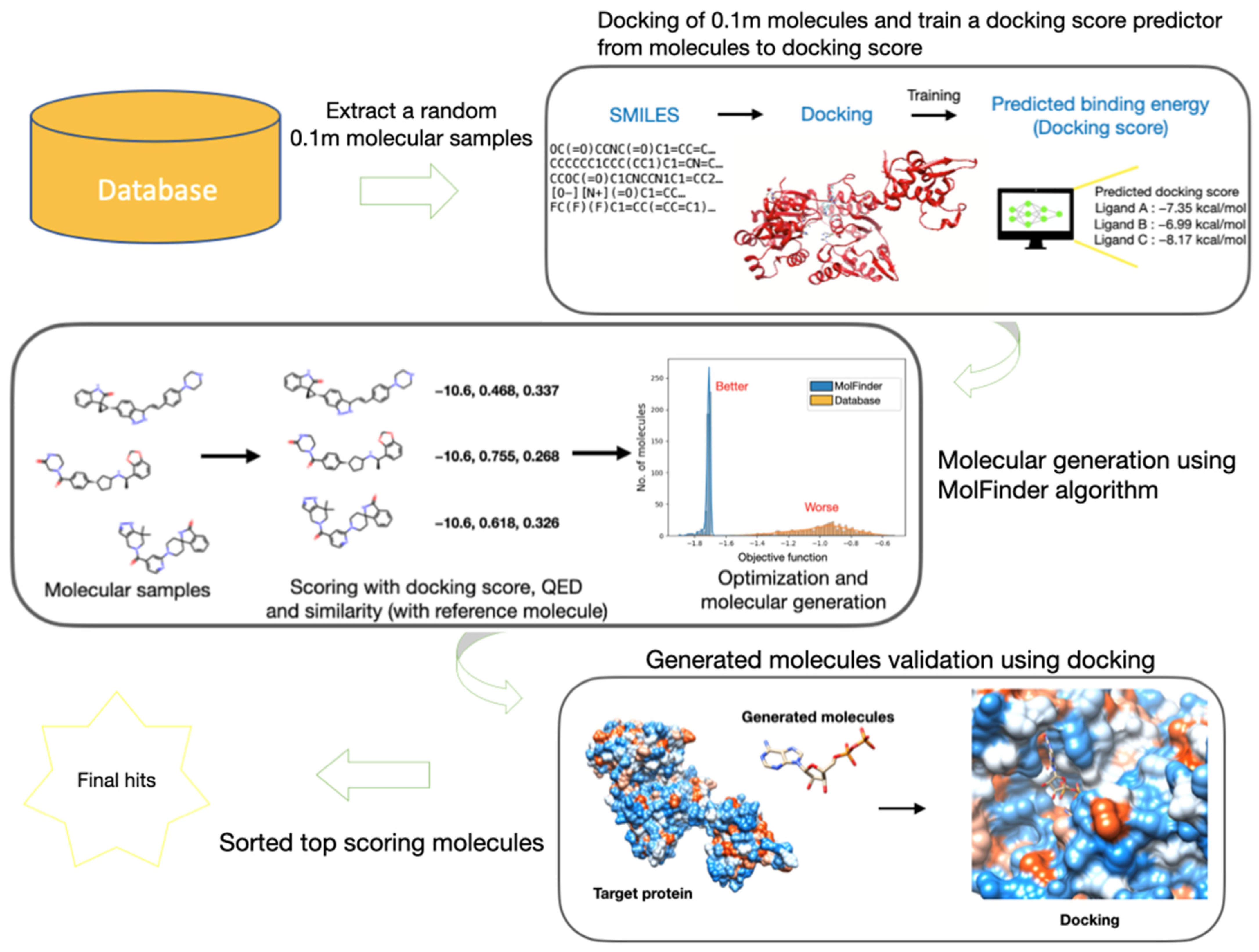
3.1. Overall Workflow of V-Dock
3.2. Molecular Database
3.3. Protein-Ligand Docking Energy Calculations
3.4. Docking Score Prediction Model
3.5. Objective Function
3.6. Molecular Generation Using MolFinder
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, M.-Q.; Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Lee, J. MolFinder: An evolutionary algorithm for the global optimization of molecular properties and the extensive exploration of chemical space using SMILES. J. Chemin. 2021, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Gottipati, S.K.; Sattarov, B.; Niu, S.; Pathak, Y.; Wei, H.; Liu, S.; Blackburn, S.; Thomas, K.; Coley, C.; Tang, J.; et al. Learning to navigate the synthetically accessible chemical space using reinforcement learning. In Proceedings of the 37th International Conference on Machine Learning (ICML 2020), Vienna, Austria, 12–18 July 2020; pp. 3668–3679. [Google Scholar]
- Zhou, Z.; Kearnes, S.; Li, L.; Zare, R.N.; Riley, P. Optimization of Molecules via Deep Reinforcement Learning. Sci. Rep. 2019, 9, 10752. [Google Scholar] [CrossRef] [PubMed]
- Horwood, J.; Noutahi, E. Molecular Design in Synthetically Accessible Chemical Space via Deep Reinforcement Learning. ACS Omega 2020, 5, 32984–32994. [Google Scholar] [CrossRef]
- Yoshimori, A.; Kawasaki, E.; Kanai, C.; Tasaka, T. Strategies for Design of Molecular Structures with a Desired Pharmacophore Using Deep Reinforcement Learning. Chem. Pharm. Bull. 2020, 68, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [Green Version]
- Domenico, A.; Nicola, G.; Daniela, T.; Fulvio, C.; Nicola, A.; Orazio, N. De Novo Drug Design of Targeted Chemical Libraries Based on Artificial Intelligence and Pair-Based Multiobjective Optimization. J. Chem. Inf. Model. 2020, 60, 4582–4593. [Google Scholar] [CrossRef]
- White, C.C.; White, D.J. Markov decision processes. Eur. J. Oper. Res. 1989, 39, 1–16. [Google Scholar] [CrossRef]
- Lim, J.; Hwang, S.-Y.; Moon, S.; Kim, S.; Kim, W.Y. Scaffold-based molecular design with a graph generative model. Chem. Sci. 2020, 11, 1153–1164. [Google Scholar] [CrossRef] [Green Version]
- Tran-Nguyen, V.-K.; Rognan, D. Benchmarking Data Sets from PubChem BioAssay Data: Current Scenario and Room for Improvement. Int. J. Mol. Sci. 2020, 21, 4380. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.-T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Berenger, F.; Kumar, A.; Zhang, K.Y.J.; Yamanishi, Y. Lean-Docking: Exploiting Ligands’ Predicted Docking Scores to Accelerate Molecular Docking. J. Chem. Inf. Model. 2021, 61, 2341–2352. [Google Scholar] [CrossRef]
- Svensson, F.; Norinder, U.; Bender, A. Improving Screening Efficiency through Iterative Screening Using Docking and Conformal Prediction. J. Chem. Inf. Model. 2017, 57, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, K.; Komine, S.; Suzuki, S.D.; Ohue, M.; Ishida, T.; Akiyama, Y. Spresso: An ultrafast compound pre-screening method based on compound decomposition. Bioinformatics 2017, 33, 3836–3843. [Google Scholar] [CrossRef] [Green Version]
- Cherkasov, A.; Ban, F.; Li, Y.; Fallahi, M.; Hammond, G.L. Progressive Docking: A Hybrid QSAR/Docking Approach for Accelerating In Silico High Throughput Screening. J. Med. Chem. 2006, 49, 7466–7478. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular docking with deep learning. J. Cheminformatics 2021, 13, 1–20. [Google Scholar] [CrossRef]
- Jeon, W.; Kim, D. Autonomous molecule generation using reinforcement learning and docking to develop potential novel inhibitors. Sci. Rep. 2020, 10, 22104. [Google Scholar] [CrossRef]
- Krishnan, S.R.; Bung, N.; Bulusu, G.; Roy, A. Accelerating De Novo Drug Design against Novel Proteins Using Deep Learning. J. Chem. Inf. Model. 2021, 61, 621–630. [Google Scholar] [CrossRef]
- Guo, J.; Janet, J.P.; Bauer, M.R.; Nittinger, E.; Giblin, K.A.; Papadopoulos, K.; Voronov, A.; Patronov, A.; Engkvist, O.; Margreitter, C. DockStream: A Docking Wrapper to Enhance De Novo Molecular Design. Theor. Comput. Chem. 2021. [Google Scholar] [CrossRef]
- Boitreaud, J.; Mallet, V.; Oliver, C.; Waldispühl, J. OptiMol: Optimization of Binding Affinities in Chemical Space for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5658–5666. [Google Scholar] [CrossRef]
- Brookes, D.; Park, H.; Listgarten, J. Conditioning by adaptive sampling for robust design. In Proceedings of the 36th International Conference on Machine Learning, Long Beach, CA, USA, 9–15 June 2019; Volume 97, pp. 773–782. [Google Scholar]
- Ma, B.; Terayama, K.; Matsumoto, S.; Isaka, Y.; Sasakura, Y.; Iwata, H.; Araki, M.; Okuno, Y. Structure-Based de Novo Molecular Generator Combined with Artificial Intelligence and Docking Simulations. J. Chem. Inf. Model. 2021, 61, 3304–3313. [Google Scholar] [CrossRef]
- Alhossary, A.; Handoko, S.D.; Mu, Y.; Kwoh, C.-K. Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics 2015, 31, 2214–2216. [Google Scholar] [CrossRef] [Green Version]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Chemin. 2009, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Mojumdar, A. Mutations in conserved functional domains of human RecQ helicases are associated with diseases and cancer: A review. Biophys. Chem. 2020, 265, 106433. [Google Scholar] [CrossRef]
- Kategaya, L.; Perumal, S.K.; Hager, J.H.; Belmont, L.D. Werner Syndrome Helicase Is Required for the Survival of Cancer Cells with Microsatellite Instability. iScience 2019, 13, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.K.; Adam, J.; Wimmerová, M.; Koča, J. In Silico Mutagenesis and Docking Study of Ralstonia solanacearum RSL Lectin: Performance of Docking Software To Predict Saccharide Binding. J. Chem. Inf. Model. 2012, 52, 1250–1261. [Google Scholar] [CrossRef]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef] [PubMed]
- Papadatos, G.; Davies, M.; Dedman, N.; Chambers, J.; Gaulton, A.; Siddle, J.; Koks, R.; Irvine, S.A.; Pettersson, J.; Goncharoff, N.; et al. SureChEMBL: A large-scale, chemically annotated patent document database. Nucleic Acids Res. 2016, 44, D1220–D1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulton, A.; Bellis, L.; Bento, A.P.S.F.F.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Crabbe, L.; Verdun, R.E.; Haggblom, C.I.; Karlseder, J. Defective Telomere Lagging Strand Synthesis in Cells Lacking WRN Helicase Activity. Science 2004, 306, 1951–1953. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.A.; Gavard, A.E.; Lieb, S.; Ravichandran, M.C.; Hauer, K.; Werni, P.; Geist, L.; Böttcher, J.; Engen, J.R.; Rumpel, K.; et al. Structure of the helicase core of Werner helicase, a key target in microsatellite instability cancers. Life Sci. Alliance 2021, 4, e202000795. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox-1758-2946-3-33.pdf. J. Cheminformatics 2011, 3, 1–14. [Google Scholar]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminformatics 2009, 1, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Landrum, G. RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 26 October 2021).
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 2019, 32, 8026–8037. [Google Scholar]
- Clevert, D.A.; Unterthiner, T.; Hochreiter, S. Fast and accurate deep network learning by exponential linear units (ELUS). In Proceedings of the 4th International Conference on Learning Representations, ICLR 2016—Conference Track Proceedings, San Juan, Puerto Rico, 2–4 May 2016; pp. 1–14. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. In Proceedings of the 3rd International Conference on Learning Representations, ICLR 2015—Conference Track Proceedings, San Diego, CA, USA, 7–9 May 2015; pp. 1–15. [Google Scholar]
- Rosenthal, A.S.; Dexheimer, T.S.; Gileadi, O.; Nguyen, G.H.; Chu, W.K.; Hickson, I.D.; Jadhav, A.; Simeonov, A.; Maloney, D.J. Synthesis and SAR studies of 5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine derivatives as potent inhibitors of Bloom helicase. Bioorganic Med. Chem. Lett. 2013, 23, 5660–5666. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, A.S.; Dexheimer, T.S.; Nguyen, G.; Gileadi, O.; Vindigni, A.; Simeonov, A.; Jadhav, A.; Hickson, I.; Maloney, D.J. Discovery of ML216, a Small Molecule Inhibitor of Bloom (BLM) Helicase. Probe Rep. 2013, 2013, 1–26. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature Name | Describe |
|---|---|
| HBD | Number of hydrogen bond donors |
| HBA | Number of hydrogen bond acceptors |
| Rotatable bond | Number of rotatable bonds |
| Ring | Number of rings |
| Radical | Number of radicals |
| Heteroatoms | Number of hetero atoms |
| Heterocycles | Number of heterocycles |
| LipinskiHBA | Number of Lipinski H-bond acceptors |
| LipinskiHBD | Number of Lipinski H-bond donors |
| AromaticCarbocycles | Number of Aromatic carbocycles |
| AromaticHeterocycles | Number of Aromatic heterocycles |
| AmideBonds | Number of Amide bond |
| AliphaticCarbocycles | Number of Aliphatic carbocycles |
| AliphaticHeterocycles | Number of Aliphatic heterocycles |
| FractionCSP3 | Fraction of C atoms that are SP3 hybridized |
| LabuteASA | Labute Accessible Surface Area |
| TPSA | Topological polar surface area |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.; Lee, J. V-Dock: Fast Generation of Novel Drug-like Molecules Using Machine-Learning-Based Docking Score and Molecular Optimization. Int. J. Mol. Sci. 2021, 22, 11635. https://doi.org/10.3390/ijms222111635
Choi J, Lee J. V-Dock: Fast Generation of Novel Drug-like Molecules Using Machine-Learning-Based Docking Score and Molecular Optimization. International Journal of Molecular Sciences. 2021; 22(21):11635. https://doi.org/10.3390/ijms222111635
Chicago/Turabian StyleChoi, Jieun, and Juyong Lee. 2021. "V-Dock: Fast Generation of Novel Drug-like Molecules Using Machine-Learning-Based Docking Score and Molecular Optimization" International Journal of Molecular Sciences 22, no. 21: 11635. https://doi.org/10.3390/ijms222111635
APA StyleChoi, J., & Lee, J. (2021). V-Dock: Fast Generation of Novel Drug-like Molecules Using Machine-Learning-Based Docking Score and Molecular Optimization. International Journal of Molecular Sciences, 22(21), 11635. https://doi.org/10.3390/ijms222111635