
Transient Fluorescence Labeling: Low Affinity—High Benefits


Abstract
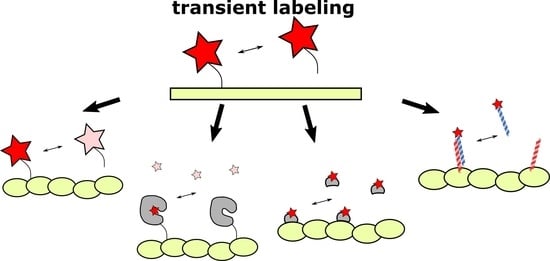
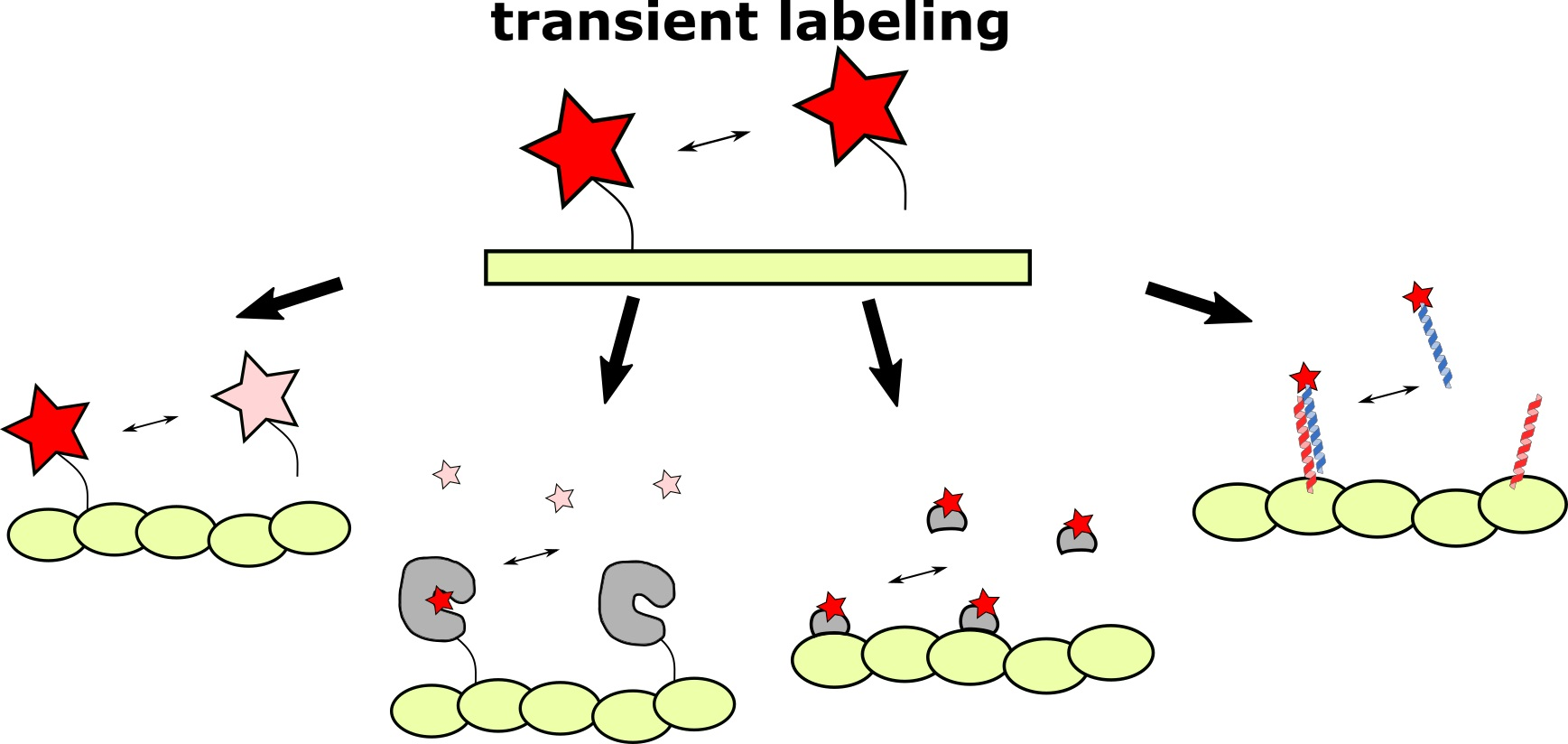
:
1. Introduction
2. PAINTing the Cell
3. Fluorogen-Activating Proteins (Protein-PAINT)
4. Cytoskeleton Labeling
5. Imaging by Peptide–Peptide Interactions
6. Exchange-STED
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Blc | bacterial lipocalin |
| BODIPY | 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene |
| CTPE | chemogenetic tag with probe exchange |
| DFHBI | difluoro-4-hydroxybenzylidene imidazolinone |
| DFHBI-1T | 3,5-difluoro-4-hydroxybenzylidene-2,2,2-trifluoroethyl imidazolinone |
| DiB | dye in Blc |
| dSTORM | direct STORM |
| FAP | fluorogen-activating protein |
| FPs | fluorescent proteins |
| GFP | green fluorescent protein |
| HBR-DOM | 4-hydroxy-3,5-dimethoxybenzylidene rhodanine |
| HMBR | 4-hydroxy-3-methylbenzylidene rhodanine |
| IRIS | image reconstruction by integrating exchangeable single-molecule localization |
| KECs | K/E-coils |
| MAPs | microtubule-associated proteins |
| MG-ester | malachite green ester |
| PAINT | point accumulation for imaging in nanoscale topography |
| PALM | photoactivated localization microscopy |
| PSF | point spread function |
| PYP | photoactive yellow protein |
| qPAINT | quantitative PAINT |
| RhoBAST | rhodamine-binding aptamer for super-resolution imaging techniques |
| scFv | single-chain antibodies |
| SiR | silicon-rhodamine |
| SMLM | single-molecule localization microscopy |
| SOFI | super-resolution optical fluctuation imaging |
| SRRF | super-resolution radial fluctuations |
| STED | stimulated emission depletion |
| STORM | stochastic optical reconstruction microscopy |
| TIRF | total internal reflection fluorescence |
| tPAINT | tension PAINT |
| TTDOM | time distribution optical microscopy |
| uPAINT | universal PAINT |
| Y-FAST | yellow fluorescence-activating and absorption-shifting tag |
References
- Sahl, S.J.; Hell, S.W.; Jakobs, S. Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Biol. 2017, 18, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Mishin, A.S.; Belousov, V.V.; Solntsev, K.M.; Lukyanov, K.A. Novel uses of fluorescent proteins. Curr. Opin. Chem. Biol. 2015, 27, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hoelzel, C.A.; Zhang, X. Visualizing and manipulating biological processes by using HaloTag and SNAP-Tag technologies. ChemBioChem 2020, 21, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ma, H.; Liu, Y. Stochastic Optical Reconstruction Microscopy (STORM). Curr. Protoc. Cytom. 2017, 81, 12.46.1–12.46.27. [Google Scholar] [CrossRef] [PubMed]
- Olivier, N.; Keller, D.; Rajan, V.S.; Gönczy, P.; Manley, S. Simple buffers for 3D STORM microscopy. Biomed. Opt. Express 2013, 4, 885–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, M. Localization microscopy coming of age: From concepts to biological impact. J. Cell Sci. 2013, 126, 3505–3513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiuchi, T.; Higuchi, M.; Takamura, A.; Maruoka, M.; Watanabe, N. Multitarget super-resolution microscopy with high-density labeling by exchangeable probes. Nat. Methods 2015, 12, 743–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavrikov, A.S.; Baranov, M.S.; Mishin, A.S. Live-cell nanoscopy with spontaneous blinking of conventional green fluorescent proteins. Biochem. Biophys. Res. Commun. 2020, 522, 852–854. [Google Scholar] [CrossRef]
- Paez-Segala, M.G.; Sun, M.G.; Shtengel, G.; Viswanathan, S.; Baird, M.A.; Macklin, J.J.; Patel, R.; Allen, J.R.; Howe, E.S.; Piszczek, G.; et al. Fixation-resistant photoactivatable fluorescent proteins for CLEM. Nat. Methods 2015, 12, 215–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W.; Mannherz, H.G.; Suck, D.; Pai, E.F.; Holmes, K.C. Atomic structure of the actin: DNase I complex. Nature 1990, 347, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Orm, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal structure of the aequorea victoria green fluorescent protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanchanawong, P.; Waterman, C.M. Localization-based super-resolution imaging of cellular structures. Methods Mol. Biol. 2013, 1046, 59–84. [Google Scholar] [PubMed] [Green Version]
- Riedl, J.; Crevenna, A.H.; Kessenbrock, K.; Yu, J.H.; Neukirchen, D.; Bista, M.; Bradke, F.; Jenne, D.; Holak, T.A.; Werb, Z.; et al. Lifeact: A versatile marker to visualize F-Actin. Nat. Methods 2008, 5, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, L.; Rothmann, C.; Lavie, R.; Malik, Z. Green fluorescent protein photobleaching: A model for protein damage by endogenous and exogenous singlet oxygen. Biol. Chem. 2000, 381, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Pakhomov, A.A.; Martynov, V.I.; Orsa, A.N.; Bondarenko, A.A.; Chertkova, R.V.; Lukyanov, K.A.; Petrenko, A.G.; Deyev, I.E. Fluorescent Protein Dendra2 as a Ratiometric Genetically Encoded pH-Sensor. Biochem. Biophys. Res. Commun. 2017, 493, 1518–1521. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Schwartz, S.L.; Maji, S.; Huang, F.; Szent-Gyorgyi, C.; Lidke, D.S.; Lidke, K.A.; Bruchez, M.P. Localization microscopy using noncovalent Fluorogen activation by genetically encoded Fluorogen-activating proteins. ChemPhysChem 2014, 15, 687–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, I.D.; Kang, H.C.; Haugland, R.P. Fluorescent membrane probes incorporating Dipyrrometheneboron Difluoride Fluorophores. Anal. Biochem. 1991, 198, 228–237. [Google Scholar] [CrossRef]
- Lukinavičius, G.; Reymond, L.; D’Este, E.; Masharina, A.; Göttfert, F.; Ta, H.; Güther, A.; Fournier, M.; Rizzo, S.; Waldmann, H.; et al. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 2014, 11, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Dubois, J.; Le Goff, M.T.; Guéritte-Voegelein, F.; Guénard, D.; Tollon, Y.; Wright, M. Fluorescent and biotinylated analogues of docetaxel: Synthesis and biological evaluation. Bioorg. Med. Chem. 1995, 3, 1357–1368. [Google Scholar] [CrossRef]
- Perfilov, M.M.; Gurskaya, N.G.; Serebrovskaya, E.O.; Melnikov, P.A.; Kharitonov, S.L.; Lewis, T.R.; Arshavsky, V.Y.; Baklaushev, V.P.; Mishin, A.S.; Lukyanov, K.A. Highly photostable fluorescent labeling of proteins in live cells using exchangeable coiled coils heterodimerization. Cell. Mol. Life Sci. 2020, 77, 4429–4440. [Google Scholar] [CrossRef] [PubMed]
- Belin, B.J.; Goins, L.M.; Dyche Mullins, R. Comparative analysis of tools for live cell imaging of Actin network architecture. BioArchitecture 2014, 4, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Mei, E.; Gao, F.; Hochstrasser, R.M. Controlled bimolecular collisions allow sub-diffraction limited microscopy of lipid vesicles. Phys. Chem. Chem. Phys. 2006, 8, 2077–2082. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Bardo, A.M.; Martinez, C.; Higgins, D.A. Characterization of molecular scale environments in polymer films by single molecule spectroscopy. J. Phys. Chem. B 2000, 104, 212–219. [Google Scholar] [CrossRef]
- Mei, E.; Hochstrasser, R.M. High-resolution optical imaging from trajectory time distributions. J. Phys. Chem. B 2006, 110, 25101–25107. [Google Scholar] [CrossRef] [PubMed]
- Sharonov, A.; Hochstrasser, R.M. Wide-field Subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl. Acad. Sci. USA 2006, 103, 18911–18916. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by Stochastic Optical Reconstruction Microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Hess, S.T.; Girirajan, T.P.K.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [Green Version]
- Jungmann, R.; Steinhauer, C.; Scheible, M.; Kuzyk, A.; Tinnefeld, P.; Simmel, F.C. Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Lett. 2010, 10, 4756–4761. [Google Scholar] [CrossRef]
- Giannone, G.; Hosy, E.; Levet, F.; Constals, A.; Schulze, K.; Sobolevsky, A.I.; Rosconi, M.P.; Gouaux, E.; Tampé, R.; Choquet, D.; et al. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys. J. 2010, 99, 1303–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, N.; Mitchison, T.J. Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science 2002, 295, 1083–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungmann, R.; Avendaño, M.S.; Dai, M.; Woehrstein, J.B.; Agasti, S.S.; Feiger, Z.; Rodal, A.; Yin, P. Quantitative super-resolution imaging with qPAINT. Nat. Methods 2016, 13, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, R.; Ke, Y.; Jungmann, R.; Schlichthaerle, T.; Woehrstein, J.B.; Yin, P. Polyhedra self-assembled from DNA tripods and characterized with 3D DNA-PAINT. Science 2014, 344, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Strauss, S.; Nickels, P.C.; Strauss, M.T.; Jimenez Sabinina, V.; Ellenberg, J.; Carter, J.D.; Gupta, S.; Janjic, N.; Jungmann, R. Modified aptamers enable quantitative sub-10-Nm cellular DNA-PAINT imaging. Nat. Methods 2018, 15, 685–688. [Google Scholar] [CrossRef]
- Jungmann, R.; Avendaño, M.S.; Woehrstein, J.B.; Dai, M.; Shih, W.M.; Yin, P. Multiplexed 3D Cellular super-resolution imaging with DNA-PAINT and exchange-PAINT. Nat. Methods 2014, 11, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.; Jungmann, R.; Yin, P. optical imaging of individual biomolecules in densely packed clusters. Nat. Nanotechnol. 2016, 11, 798–807. [Google Scholar] [CrossRef] [Green Version]
- Brockman, J.M.; Su, H.; Blanchard, A.T.; Duan, Y.; Meyer, T.; Quach, M.E.; Glazier, R.; Bazrafshan, A.; Bender, R.L.; Kellner, A.V.; et al. Live-cell super-resolved PAINT imaging of piconewton cellular traction forces. Nat. Methods 2020, 17, 1018–1024. [Google Scholar] [CrossRef]
- Dertinger, T.; Colyer, R.; Iyer, G.; Weiss, S.; Enderlein, J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc. Natl. Acad. Sci. USA 2009, 106, 22287–22292. [Google Scholar] [CrossRef] [Green Version]
- Endesfelder, U.; van de Linde, S.; Wolter, S.; Sauer, M.; Heilemann, M. Subdiffraction-resolution fluorescence microscopy of myosin-actin motility. ChemPhysChem 2010, 11, 836–840. [Google Scholar] [CrossRef]
- Geissbuehler, S.; Dellagiacoma, C.; Lasser, T. Comparison between SOFI and STORM. Biomed. Opt. Express 2011, 2, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klementieva, N.V.; Zagaynova, E.V.; Lukyanov, K.A.; Mishin, A.S. The principles of super-resolution fluorescence microscopy (Review). Sovrem. Tehnol. Med. 2016, 8, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Glogger, M.; Spahn, C.; Enderlein, J.; Heilemann, M. Multi-color, Bleaching-resistant super-resolution optical fluctuation imaging with oligonucleotide-based exchangeable fluorophores. Angew. Chem. Weinheim Bergstr. Ger. 2021, 133, 6380–6383. [Google Scholar] [CrossRef]
- Sharonov, A.; Hochstrasser, R.M. Single-molecule imaging of the association of the cell-penetrating peptide Pep-1 to model membranes. Biochemistry 2007, 46, 7963–7972. [Google Scholar] [CrossRef]
- Paige, J.S.; Wu, K.Y.; Jaffrey, S.R. RNA mimics of green fluorescent protein. Science 2011, 333, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Braselmann, E.; Rathbun, C.; Richards, E.M.; Palmer, A.E. Illuminating RNA biology: Tools for imaging RNA in live mammalian cells. Cell Chem. Biol. 2020, 27, 891–903. [Google Scholar] [CrossRef]
- Sunbul, M.; Lackner, J.; Martin, A.; Englert, D.; Hacene, B.; Grün, F.; Nienhaus, K.; Nienhaus, G.U.; Jäschke, A. Super-resolution RNA imaging using a rhodamine-binding aptamer with fast exchange kinetics. Nat. Biotechnol. 2021. [Google Scholar] [CrossRef]
- Kung, C.E.; Reed, J.K. Fluorescent molecular rotors: A new class of probes for tubulin structure and assembly. Biochemistry 1989, 28, 6678–6686. [Google Scholar] [CrossRef]
- Iwaki, T.; Torigoe, C.; Noji, M.; Nakanishi, M. Antibodies for fluorescent molecular rotors. Biochemistry 1993, 32, 7589–7592. [Google Scholar] [CrossRef]
- Szent-Gyorgyi, C.; Schmidt, B.F.; Creeger, Y.; Fisher, G.W.; Zakel, K.L.; Adler, S.; Fitzpatrick, J.A.J.; Woolford, C.A.; Yan, Q.; Vasilev, K.V.; et al. Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol. 2008, 26, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Ozhalici-Unal, H.; Pow, C.L.; Marks, S.A.; Jesper, L.D.; Silva, G.L.; Shank, N.I.; Jones, E.W.; Burnette, J.M., 3rd; Berget, P.B.; Armitage, B.A. A rainbow of fluoromodules: A promiscuous scFv protein binds to and activates a diverse set of fluorogenic cyanine dyes. J. Am. Chem. Soc. 2008, 130, 12620–12621. [Google Scholar]
- Zanotti, K.J.; Silva, G.L.; Creeger, Y.; Robertson, K.L.; Waggoner, A.S.; Berget, P.B.; Armitage, B.A. Blue fluorescent dye-protein complexes based on fluorogenic cyanine dyes and single chain antibody fragments. Org. Biomol. Chem. 2011, 9, 1012–1020. [Google Scholar] [CrossRef]
- He, J.; Wang, Y.; Missinato, M.A.; Onuoha, E.; Perkins, L.A.; Watkins, S.C.; St Croix, C.M.; Tsang, M.; Bruchez, M.P. A genetically targetable near-infrared photosensitizer. Nat. Methods 2016, 13, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Perkins, L.A.; Yan, Q.; Schmidt, B.F.; Kolodieznyi, D.; Saurabh, S.; Larsen, M.B.; Watkins, S.C.; Kremer, L.; Bruchez, M.P. Genetically targeted ratiometric and activated ph indicator complexes (TRApHIC) for receptor trafficking. Biochemistry 2018, 57, 861–871. [Google Scholar] [CrossRef]
- Wang, Y.; Ballou, B.; Schmidt, B.F.; Andreko, S.; St Croix, C.M.; Watkins, S.C.; Bruchez, M.P. Affibody-Targeted fluorogen activating protein for in vivo tumor imaging. Chem. Commun. 2017, 53, 2001–2004. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Telmer, C.A.; Schmidt, B.F.; Franke, J.D.; Ort, S.; Arndt-Jovin, D.J.; Bruchez, M.P. Fluorogen activating protein-affibody probes: Modular, no-wash measurement of epidermal growth factor receptors. Bioconjug. Chem. 2015, 26, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Lyakhov, I.; Zielinski, R.; Kuban, M.; Kramer-Marek, G.; Fisher, R.; Chertov, O.; Bindu, L.; Capala, J. HER2- and EGFR-Specific Affiprobes: Novel recombinant optical probes for cell imaging. ChemBioChem 2010, 11, 345–350. [Google Scholar] [CrossRef]
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A bilirubin-inducible fluorescent protein from eel muscle. Cell 2013, 153, 1602–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, S.; Toda, Y. A novel fluorescent protein purified from eel muscle. Fish. Sci. 2009, 75, 1461–1469. [Google Scholar] [CrossRef]
- Kwon, J.; Park, J.-S.; Kang, M.; Choi, S.; Park, J.; Kim, G.T.; Lee, C.; Cha, S.; Rhee, H.-W.; Shim, S.-H. Bright ligand-activatable fluorescent protein for high-quality multicolor live-cell super-resolution microscopy. Nat. Commun. 2020, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Plamont, M.-A.; Billon-Denis, E.; Maurin, S.; Gauron, C.; Pimenta, F.M.; Specht, C.G.; Shi, J.; Quérard, J.; Pan, B.; Rossignol, J.; et al. Small fluorescence-activating and absorption-shifting tag for tunable protein imaging in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 497–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebo, A.G.; Pimenta, F.M.; Zhang, Y.; Gautier, A. Improved chemical-genetic fluorescent markers for live cell microscopy. Biochemistry 2018, 57, 5648–5653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Plamont, M.-A.; Sladitschek, H.L.; Rodrigues, V.; Aujard, I.; Neveu, P.; Le Saux, T.; Jullien, L.; Gautier, A. Dynamic multicolor protein labeling in living cells. Chem. Sci. 2017, 8, 5598–5605. [Google Scholar] [CrossRef] [Green Version]
- Povarova, N.V.; Zaitseva, S.O.; Baleeva, N.S.; Smirnov, A.Y.; Myasnyanko, I.N.; Zagudaylova, M.B.; Bozhanova, N.G.; Gorbachev, D.A.; Malyshevskaya, K.K.; Gavrikov, A.S.; et al. Red-shifted substrates for FAST Fluorogen-activating protein based on the GFP-like chromophores. Chemistry 2019, 25, 9592–9596. [Google Scholar] [CrossRef]
- Myasnyanko, I.N.; Gavrikov, A.S.; Zaitseva, S.O.; Smirnov, A.Y.; Zaitseva, E.R.; Sokolov, A.I.; Malyshevskaya, K.K.; Baleeva, N.S.; Mishin, A.S.; Baranov, M.S. Color tuning of fluorogens for FAST Fluorogen-activating protein. Chemistry 2021, 27, 3986–3990. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tebo, A.G.; Thauvin, M.; Plamont, M.-A.; Volovitch, M.; Morin, X.; Vriz, S.; Gautier, A. A far-red emitting fluorescent chemogenetic reporter for in vivo molecular imaging. Angew. Chem. Int. Ed. Engl. 2020, 59, 17917–17923. [Google Scholar] [CrossRef] [PubMed]
- Tebo, A.G.; Gautier, A. A split fluorescent reporter with rapid and reversible complementation. Nat. Commun. 2019, 10, 2822. [Google Scholar] [CrossRef] [Green Version]
- Tebo, A.G.; Moeyaert, B.; Thauvin, M.; Carlon-Andres, I.; Böken, D.; Volovitch, M.; Padilla-Parra, S.; Dedecker, P.; Vriz, S.; Gautier, A. Orthogonal fluorescent chemogenetic reporters for multicolor imaging. Nat. Chem. Biol. 2021, 17, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Benaissa, H.; Ounoughi, K.; Aujard, I.; Fischer, E.; Goïame, R.; Nguyen, J.; Tebo, A.G.; Li, C.; Le Saux, T.; Danglot, L.; et al. Engineering of a fluorescent chemogenetic reporter with tunable color for advanced live-cell imaging. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mineev, K.S.; Goncharuk, S.A.; Goncharuk, M.V.; Povarova, N.V.; Sokolov, A.I.; Baleeva, N.S.; Smirnov, A.Y.; Myasnyanko, I.N.; Ruchkin, D.A.; Bukhdruker, S.; et al. NanoFAST: Structure-Based design of a small fluorogen-activating protein with only 98 amino acids. Chem. Sci. 2021, 12, 6719–6725. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Gautier, A.; Puchner, E.M. Single-Molecule localization microscopy with the fluorescence-activating and absorption-shifting tag (FAST) System. ACS Chem. Biol. 2019, 14, 1115–1120. [Google Scholar] [CrossRef]
- Venkatachalapathy, M.; Belapurkar, V.; Jose, M.; Gautier, A.; Nair, D. Live cell super resolution imaging by radial fluctuations using fluorogen binding tags. Nanoscale 2019, 11, 3626–3632. [Google Scholar] [CrossRef] [PubMed]
- Bozhanova, N.G.; Baranov, M.S.; Klementieva, N.V.; Sarkisyan, K.S.; Gavrikov, A.S.; Yampolsky, I.V.; Zagaynova, E.V.; Lukyanov, S.A.; Lukyanov, K.A.; Mishin, A.S. Protein labeling for live cell fluorescence microscopy with a highly photostable renewable signal. Chem. Sci. 2017, 8, 7138–7142. [Google Scholar] [CrossRef] [Green Version]
- Bozhanova, N.G.; Baranov, M.S.; Baleeva, N.S.; Gavrikov, A.S.; Mishin, A.S. Red-shifted aminated derivatives of GFP Chromophore for live-cell protein labeling with lipocalins. Int. J. Mol. Sci. 2018, 19, 3778. [Google Scholar] [CrossRef] [Green Version]
- Bozhanova, N.G.; Gavrikov, A.S.; Mishin, A.S.; Meiler, J. DiB-Splits: Nature-guided design of a novel fluorescent labeling split system. Sci. Rep. 2020, 10, 11049. [Google Scholar] [CrossRef]
- Muslinkina, L.; Gavrikov, A.S.; Bozhanova, N.G.; Mishin, A.S.; Baranov, M.S.; Meiler, J.; Pletneva, N.V.; Pletnev, V.Z.; Pletnev, S. Structure-based rational design of two enhanced bacterial lipocalin tags for protein-PAINT Super-Resolution Microscopy. ACS Chem. Biol. 2020, 15, 2456–2465. [Google Scholar] [CrossRef]
- Dou, J.; Vorobieva, A.A.; Sheffler, W.; Doyle, L.A.; Park, H.; Bick, M.J.; Mao, B.; Foight, G.W.; Lee, M.Y.; Gagnon, L.A.; et al. De novo design of a fluorescence-activating β-barrel. Nature 2018, 561, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Klima, J.C.; Doyle, L.A.; Lee, J.D.; Rappleye, M.; Gagnon, L.A.; Lee, M.Y.; Barros, E.P.; Vorobieva, A.A.; Dou, J.; Bremner, S.; et al. Incorporation of sensing modalities into de novo designed fluorescence-activating proteins. Nat. Commun. 2021, 12, 856. [Google Scholar] [CrossRef]
- Lyer, A.; Baranov, M.; Foster, A.J.; Chordia, S.; Roelfes, G.; Vlijm, R.; den Bogaart Geert, V.; Poolman, B. Chemogenetic Tags with probe exchange for live-cell fluorescence microscopy. ACS Chem. Biol. 2021, 16, 891–904. [Google Scholar]
- Fitzpatrick, J.A.J.; Yan, Q.; Sieber, J.J.; Dyba, M.; Schwarz, U.; Szent-Gyorgyi, C.; Woolford, C.A.; Berget, P.B.; Waggoner, A.S.; Bruchez, M.P. STED nanoscopy in living cells using fluorogen activating proteins. Bioconjug. Chem. 2009, 20, 1843–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazloom-Farsibaf, H.; Farzam, F.; Fazel, M.; Wester, M.J.; Meddens, M.B.M.; Lidke, K.A. Comparing lifeact and phalloidin for super-resolution imaging of actin in fixed cells. PLoS ONE 2021, 16, e0246138. [Google Scholar] [CrossRef] [PubMed]
- Pospich, S.; Merino, F.; Raunser, S. Structural effects and functional implications of phalloidin and jasplakinolide binding to actin filaments. Structure 2020, 28, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Visegrády, B.; Lorinczy, D.; Hild, G.; Somogyi, B.; Nyitrai, M. The effect of phalloidin and jasplakinolide on the flexibility and thermal stability of actin filaments. FEBS Lett. 2004, 565, 163–166. [Google Scholar] [CrossRef]
- Ashdown, G.W.; Burn, G.L.; Williamson, D.J.; Pandžić, E.; Peters, R.; Holden, M.; Ewers, H.; Shao, L.; Wiseman, P.W.; Owen, D.M. Live-cell super-resolution reveals F-actin and plasma membrane dynamics at the T Cell Synapse. Biophys. J. 2017, 112, 1703–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagaynova, E.V.; Furman, O.E.; Perfilov, M.M.; Klementieva, N.V.; Lukyanov, K.A.; Bozhanova, N.G.; Mishin, A.S. Dendra2-Tagged Lifeact and MAP4 as exchangeable probes for single-molecule fluorescence imaging of cytoskeleton in live cells. In Proceedings of the Biophotonics: Photonic Solutions for Better Health Care VI; International Society for Optics and Photonics: Bellingham, WA, USA, 2018; Volume 10685, p. 106850S. [Google Scholar]
- Sanders, T.A.; Llagostera, E.; Barna, M. Specialized Filopodia Direct Long-Range Transport of SHH during Vertebrate Tissue Patterning. Nature 2013, 497, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Munsie, L.N.; Caron, N.; Desmond, C.R.; Truant, R. Lifeact cannot visualize some forms of stress-induced twisted F-Actin. Nat. Methods 2009, 6, 317. [Google Scholar] [CrossRef]
- Burkel, B.M.; von Dassow, G.; Bement, W.M. Versatile Fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton 2007, 64, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Rybakova, I.N.; Ervasti, J.M. Identification of Spectrin-like Repeats Required for High Affinity Utrophin-Actin Interaction. J. Biol. Chem. 2005, 280, 23018–23023. [Google Scholar] [CrossRef] [Green Version]
- Belin, B.J.; Cimini, B.A.; Blackburn, E.H.; Dyche Mullins, R. Visualization of Actin Filaments and Monomers in Somatic Cell Nuclei. Molecular Biol. Cell 2013, 24, 982–994. [Google Scholar] [CrossRef]
- Schell, M.J.; Erneux, C.; Irvine, R.F. Inositol 1,4,5-Trisphosphate 3-Kinase A Associates with F-Actin and Dendritic Spines via Its N Terminus. J. Biol. Chem. 2001, 276, 37537–37546. [Google Scholar] [CrossRef] [PubMed]
- Tokuraku, K.; Matsushima, K.; Matui, T.; Nakagawa, H.; Katsuki, M.; Majima, R.; Kotani, S. The Number of Repeat Sequences in Microtubule-Associated Protein 4 Affects the Microtubule Surface Properties. J. Biol. Chem. 2003, 278, 29609–29618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monroy, B.Y.; Sawyer, D.L.; Ackermann, B.E.; Borden, M.M.; Tan, T.C.; Ori-McKenney, K.M. Competition between microtubule-associated proteins directs motor transport. Nat. Commun. 2018, 9, 1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhmanova, A. The microtubule plus-end-tracking protein CLIP-170 Associates with the spermatid manchette and is essential for spermatogenesis. Genes Dev. 2005, 19, 2501–2515. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [Green Version]
- Hooikaas, P.J.; Martin, M.; Mühlethaler, T.; Kuijntjes, G.-J.; Peeters, C.A.E.; Katrukha, E.A.; Ferrari, L.; Stucchi, R.; Verhagen, D.G.F.; van Riel, W.E.; et al. MAP7 Family Proteins Regulate Kinesin-1 Recruitment and Activation. J. Cell Biol. 2019, 218, 1298–1318. [Google Scholar] [CrossRef] [Green Version]
- Hijikata, T.; Nakamura, A.; Isokawa, K.; Imamura, M.; Yuasa, K.; Ishikawa, R.; Kohama, K.; Takeda, S.; Yorifuji, H. Plectin 1 links intermediate filaments to costameric sarcolemma through β-synemin, α-dystrobrevin and actin. J. Cell Sci. 2008, 121, 2062–2074. [Google Scholar] [CrossRef] [Green Version]
- Manley, S.; Gillette, J.M.; Lippincott-Schwartz, J. Single-particle tracking photoactivated localization microscopy for mapping single-molecule dynamics. Methods Enzymol. 2010, 475, 109–120. [Google Scholar]
- Tas, R.P.; Chazeau, A.; Cloin, B.M.C.; Lambers, M.L.A.; Hoogenraad, C.C.; Kapitein, L.C. Differentiation between Oppositely Oriented Microtubules Controls Polarized Neuronal Transport. Neuron 2017, 96, 1264–1271. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.; Houston, M.E., Jr.; Grothe, S.; Kay, C.M.; O’Connor-McCourt, M.; Irvin, R.T.; Hodges, R.S. Kinetic Study on the Formation of a de Novo Designed Heterodimeric Coiled-Coil: Use of Surface Plasmon Resonance to Monitor the Association and Dissociation of Polypeptide Chains. Biochemistry 1996, 35, 12175–12185. [Google Scholar] [CrossRef]
- Litowski, J.R.; Hodges, R.S. Designing heterodimeric two-stranded alpha-helical coiled-coils. Effects of Hydrophobicity and Alpha-Helical propensity on protein folding, stability, and specificity. J. Biol. Chem. 2002, 277, 37272–37279. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.; Bautista, D.L.; Litowski, J.; Irvin, R.T.; Hodges, R.S. Use of a Heterodimeric Coiled-Coil System for Biosensor Application and Affinity Purification. J. Chromatogr. B Biomed. Sci. Appl. 1998, 715, 307–329. [Google Scholar] [CrossRef]
- Yano, Y.; Yano, A.; Oishi, S.; Sugimoto, Y.; Tsujimoto, G.; Fujii, N.; Matsuzaki, K. Coiled-Coil Tag−Probe System for Quick Labeling of Membrane Receptors in Living Cells. ACS Chem. Biol. 2008, 3, 341–345. [Google Scholar] [CrossRef]
- Reinke, A.W.; Grant, R.A.; Keating, A.E. A Synthetic Coiled-Coil Interactome Provides Heterospecific Modules for Molecular Engineering. J. Am. Chem. Soc. 2010, 132, 6025–6031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, K.E.; Bashor, C.J.; Lim, W.A.; Keating, A.E. SYNZIP Protein Interaction Toolbox: In Vitro and in Vivo Specifications of Heterospecific Coiled-Coil Interaction Domains. ACS Synth. Biol. 2012, 1, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Zane, H.K.; Doh, J.K.; Enns, C.A.; Beatty, K.E. Versatile Interacting Peptide (VIP) Tags for Labeling Proteins with Bright Chemical Reporters. ChemBioChem 2017, 18, 470–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eklund, A.S.; Ganji, M.; Gavins, G.; Seitz, O.; Jungmann, R. Peptide-PAINT Super-Resolution imaging using transient coiled coil interactions. Nano Lett. 2020, 20, 6732–6737. [Google Scholar] [CrossRef]
- Oi, C.; Gidden, Z.; Holyoake, L.; Kantelberg, O.; Mochrie, S.; Horrocks, M.H.; Regan, L. LIVE-PAINT Allows Super-Resolution microscopy inside living cells using reversible peptide-protein interactions. Commun. Biol. 2020, 3, 458. [Google Scholar] [CrossRef]
- Pratt, S.E.; Speltz, E.B.; Mochrie, S.G.J.; Regan, L. Designed proteins as novel imaging reagents in living Escherichia coli. ChemBioChem 2016, 17, 1652–1657. [Google Scholar] [CrossRef]
- Lebar, T.; Lainšček, D.; Merljak, E.; Aupič, J.; Jerala, R. A tunable orthogonal coiled-coil interaction toolbox for engineering mammalian cells. Nat. Chem. Biol. 2020, 16, 513–519. [Google Scholar] [CrossRef]
- Gradišar, H.; Jerala, R. De novo design of orthogonal peptide pairs forming parallel coiled-coil heterodimers. J. Pept. Sci. 2011, 17, 100–106. [Google Scholar] [CrossRef]
- Ljubetič, A.; Lapenta, F.; Gradišar, H.; Drobnak, I.; Aupič, J.; Strmšek, Ž.; Lainšček, D.; Hafner-Bratkovič, I.; Majerle, A.; Krivec, N.; et al. Design of coiled-coil protein-origami cages that self-assemble in vitro and in vivo. Nat. Biotechnol. 2017, 35, 1094–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utterström, J.; Naeimipour, S.; Selegård, R.; Aili, D. Coiled Coil-Based Therapeutics and Drug Delivery Systems. Adv. Drug Deliv. Rev. 2021, 170, 26–43. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the Diffraction Resolution Limit by Stimulated Emission: Stimulated-Emission-Depletion Fluorescence Microscopy. Opt. Lett. 1994, 19, 780. [Google Scholar] [CrossRef]
- Harke, B.; Keller, J.; Ullal, C.K.; Westphal, V.; Schönle, A.; Hell, S.W. Resolution Scaling in STED Microscopy. Opt. Express 2008, 16, 4154–4162. [Google Scholar] [CrossRef]
- Spahn, C.; Grimm, J.B.; Lavis, L.D.; Lampe, M.; Heilemann, M. Whole-Cell, 3D and Multi-Color STED Imaging with Exchangeable Fluorophores. Nano Lett. 2019, 19, 500–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spahn, C.; Hurter, F.; Glaesmann, M.; Karathanasis, C.; Lampe, M.; Heilemann, M. Protein-Specific, Multicolor and 3D STED Imaging in Cells with DNA-Labeled Antibodies. Angew. Chem. Int. Ed. Engl. 2019, 58, 18835–18838. [Google Scholar] [CrossRef] [PubMed]
- Urban, N.T.; Willig, K.I.; Hell, S.W.; Nägerl, U.V. STED Nanoscopy of Actin Dynamics in Synapses Deep inside Living Brain Slices. Biophys. J. 2011, 101, 1277–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukinavičius, G.; Blaukopf, C.; Pershagen, E.; Schena, A.; Reymond, L.; Derivery, E.; Gonzalez-Gaitan, M.; D’Este, E.; Hell, S.W.; Gerlich, D.W.; et al. SiR–Hoechst Is a Far-Red DNA Stain for Live-Cell Nanoscopy. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schueder, F.; Strauss, M.T.; Hoerl, D.; Schnitzbauer, J.; Schlichthaerle, T.; Strauss, S.; Yin, P.; Harz, H.; Leonhardt, H.; Jungmann, R. Universal super-resolution multiplexing by DNA Exchange. Angew. Chem. Int. Ed. 2017, 56, 4052–4055. [Google Scholar] [CrossRef] [Green Version]
- Beater, S.; Holzmeister, P.; Lalkens, B.; Tinnefeld, P. Simple and Aberration-Free 4color-STED--Multiplexing by Transient Binding. Opt. Express 2015, 23, 8630–8638. [Google Scholar] [CrossRef] [PubMed]
- Spahn, C.K.; Glaesmann, M.; Grimm, J.B.; Ayala, A.X.; Lavis, L.D.; Heilemann, M. A Toolbox for Multiplexed Super-Resolution Imaging of the E. Coli Nucleoid and Membrane Using Novel PAINT Labels. Sci. Rep. 2018, 8, 14768. [Google Scholar] [CrossRef] [PubMed]
- Legant, W.R.; Shao, L.; Grimm, J.B.; Brown, T.A.; Milkie, D.E.; Avants, B.B.; Lavis, L.D.; Betzig, E. High-Density Three-Dimensional Localization Microscopy across Large Volumes. Nat. Methods 2016, 13, 359–365. [Google Scholar] [CrossRef] [Green Version]





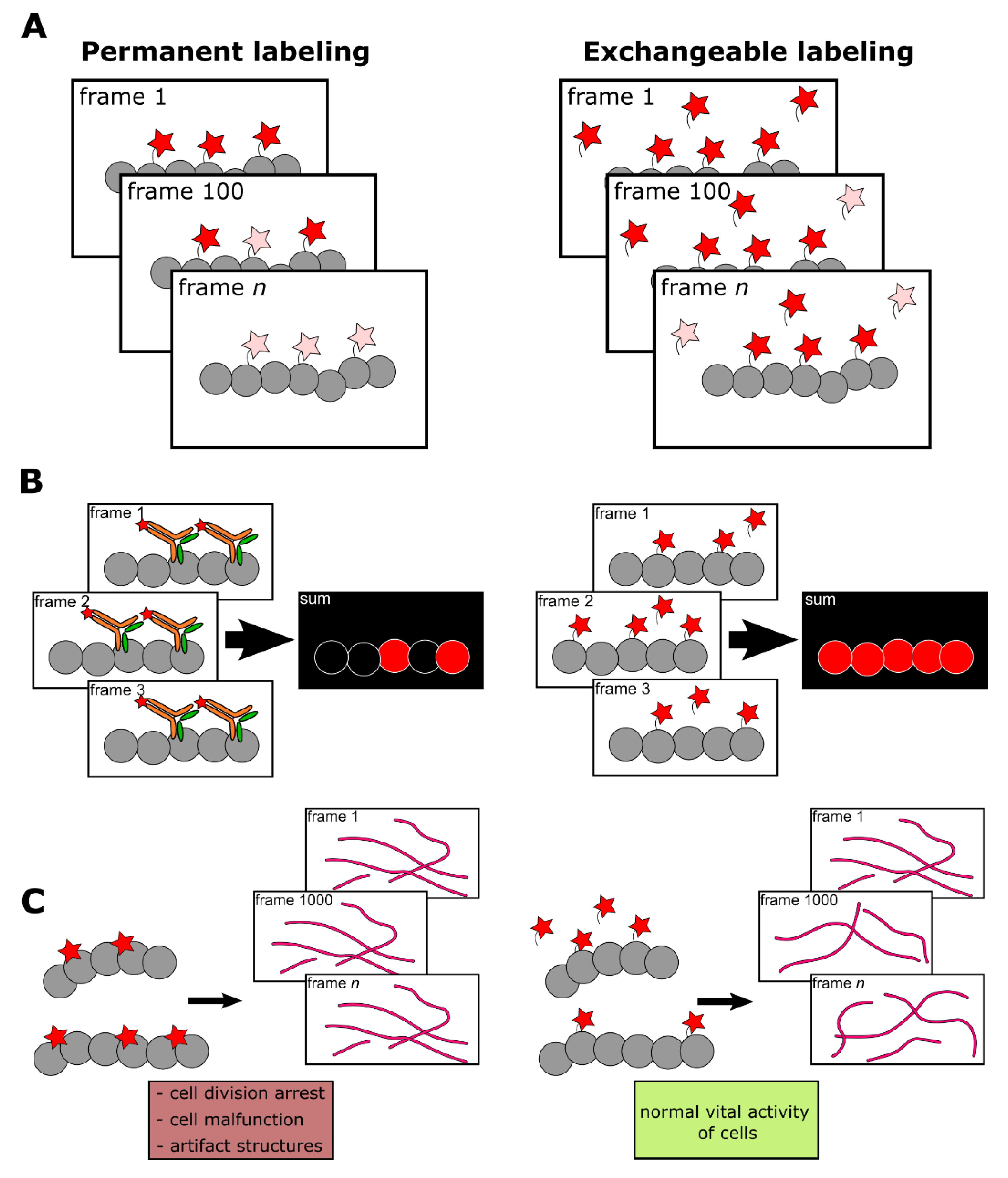
{kind=link}
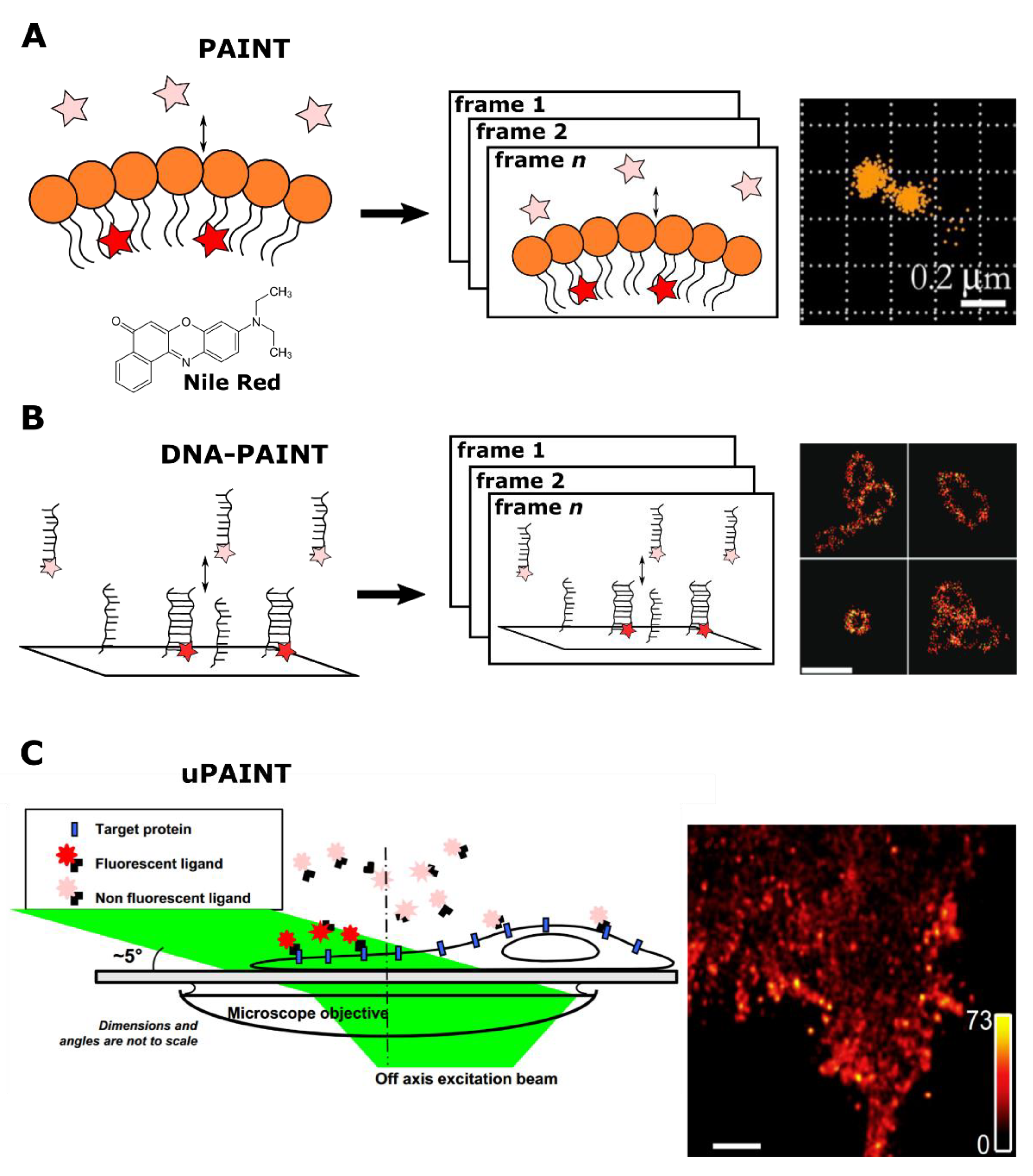
{kind=link}
{kind=link}
{kind=link}
{kind=link}
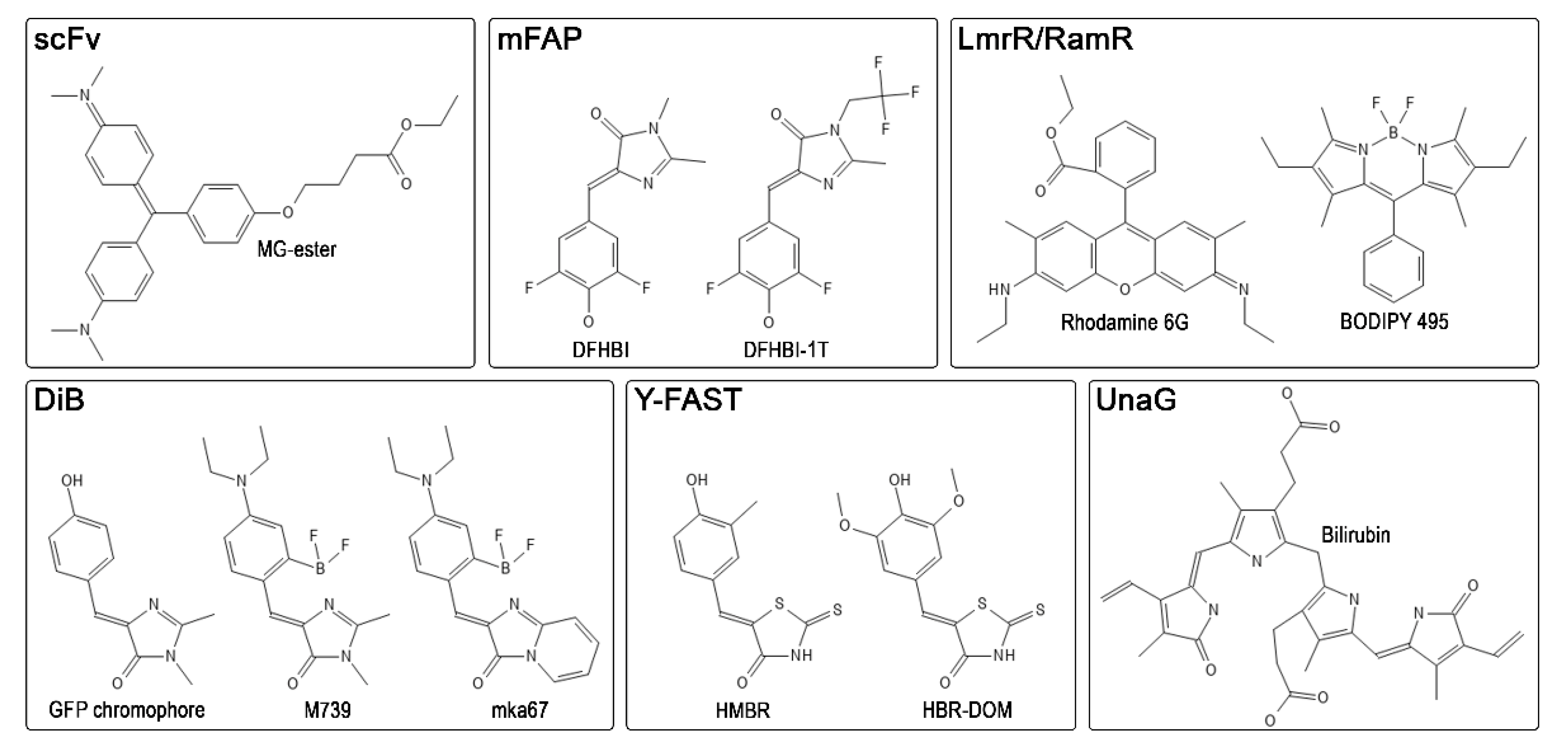
| System | Protein Size, kDa | KD, μM | Color | Super-Resolution Implementation | Reference |
|---|---|---|---|---|---|
| scFv | 11.2–26.5 | 0.0012–0.712 | blue—far red | SMLM, STED | [17,51,53,81] |
| UnaG | 15.6 | 0.000098 | green | SMLM | [59,61] |
| Y-FAST | 14.0 | 0.14–16.0 | blue—far red | SRRF | [62,65,73] |
| DiB | 18.1 | 0.1–9.0 | green—red | SMLM, STED | [74,75,77] |
| mFAP | 14.0 | 0.045–11.0 | green | not tested | [78,79] |
| LmrR/RamR | 15.0–23.0 | 0.2–10.0 | green—red | not tested | [80] |
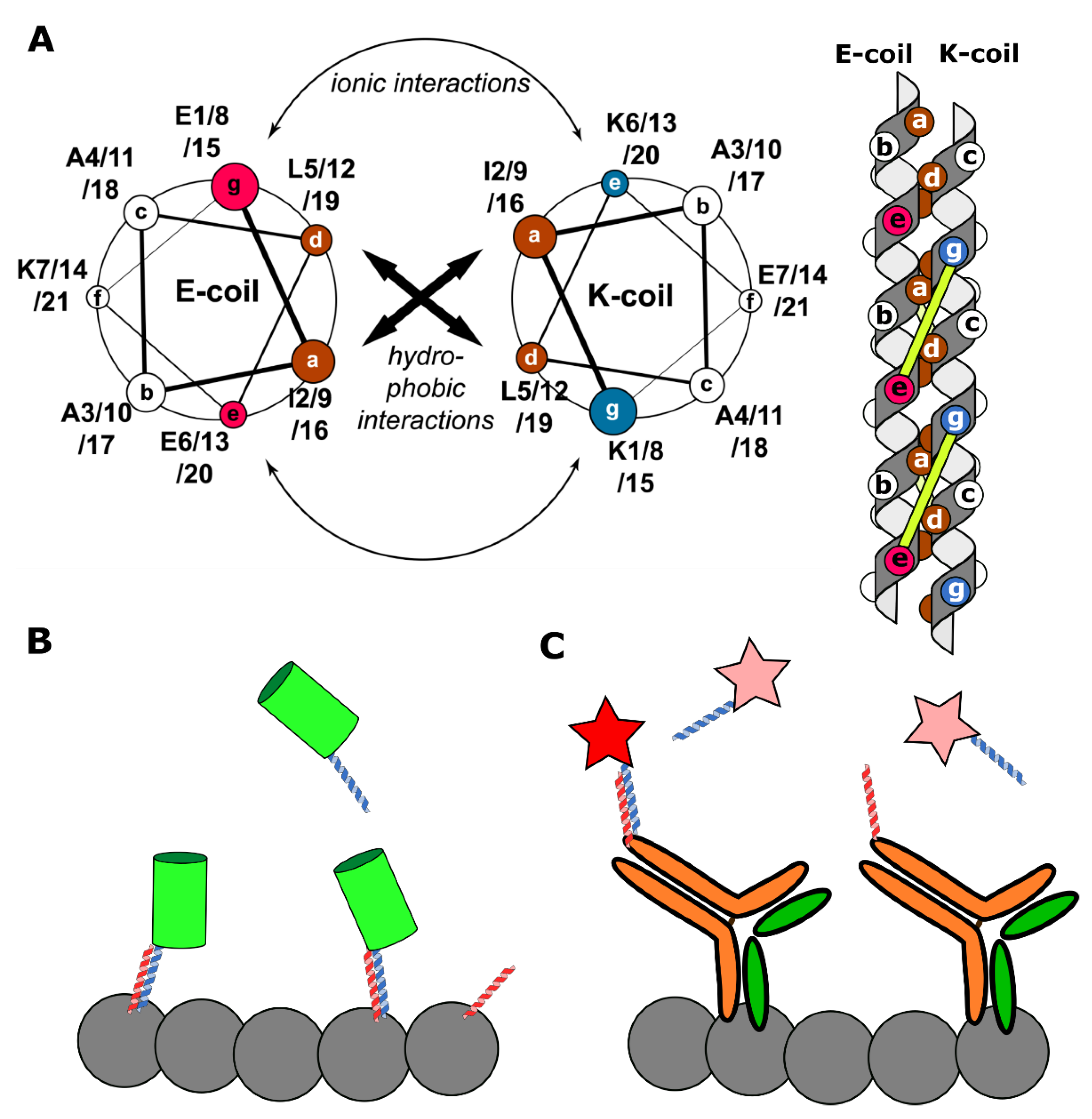
| Method | Target | Super-Resolution Implementation | Fixed/Live-Cell Imaging | Genetically Encoded? | Reference |
|---|---|---|---|---|---|
| PAINT | Membranes | SMLM, STED | Both | No | [26,117] |
| DNA-PAINT | DNA-origami, proteins | SMLM, STED, SOFI | Fixed | No | [30,34,44,118] |
| uPAINT | Proteins | SMLM | Live-cell | No | [31] |
| RNA-aptamers | RNA | SMLM | Both | Partially 1 | [48] |
| FAPs | Proteins | SMLM, STED, SRRF | Both | Partially 1 | [17,61,73,77] |
| IRIS 2 | Proteins | SMLM, STED | Both | Both 3 | [8,86,117] |
| KECs 4 | Proteins | SMLM | Both | Yes | [21,109] |
| Peptide-PAINT | DNA-origami, proteins | SMLM | Fixed | No | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perfilov, M.M.; Gavrikov, A.S.; Lukyanov, K.A.; Mishin, A.S. Transient Fluorescence Labeling: Low Affinity—High Benefits. Int. J. Mol. Sci. 2021, 22, 11799. https://doi.org/10.3390/ijms222111799
Perfilov MM, Gavrikov AS, Lukyanov KA, Mishin AS. Transient Fluorescence Labeling: Low Affinity—High Benefits. International Journal of Molecular Sciences. 2021; 22(21):11799. https://doi.org/10.3390/ijms222111799
Chicago/Turabian StylePerfilov, Maxim M., Alexey S. Gavrikov, Konstantin A. Lukyanov, and Alexander S. Mishin. 2021. "Transient Fluorescence Labeling: Low Affinity—High Benefits" International Journal of Molecular Sciences 22, no. 21: 11799. https://doi.org/10.3390/ijms222111799
APA StylePerfilov, M. M., Gavrikov, A. S., Lukyanov, K. A., & Mishin, A. S. (2021). Transient Fluorescence Labeling: Low Affinity—High Benefits. International Journal of Molecular Sciences, 22(21), 11799. https://doi.org/10.3390/ijms222111799