
Mitochondrial Effects in the Liver of C57BL/6 Mice by Low Dose, High Energy, High Charge Irradiation


Abstract
:1. Introduction
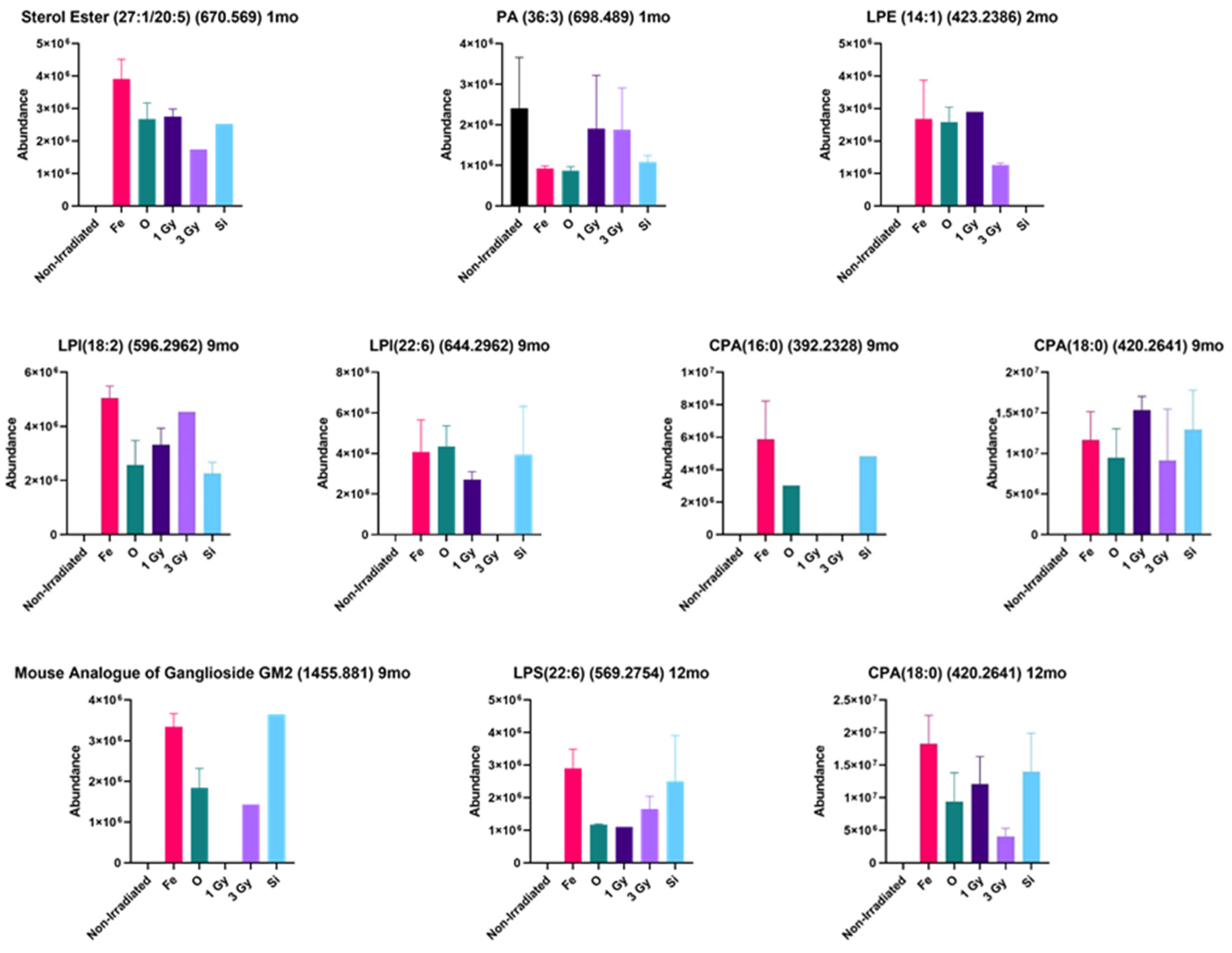
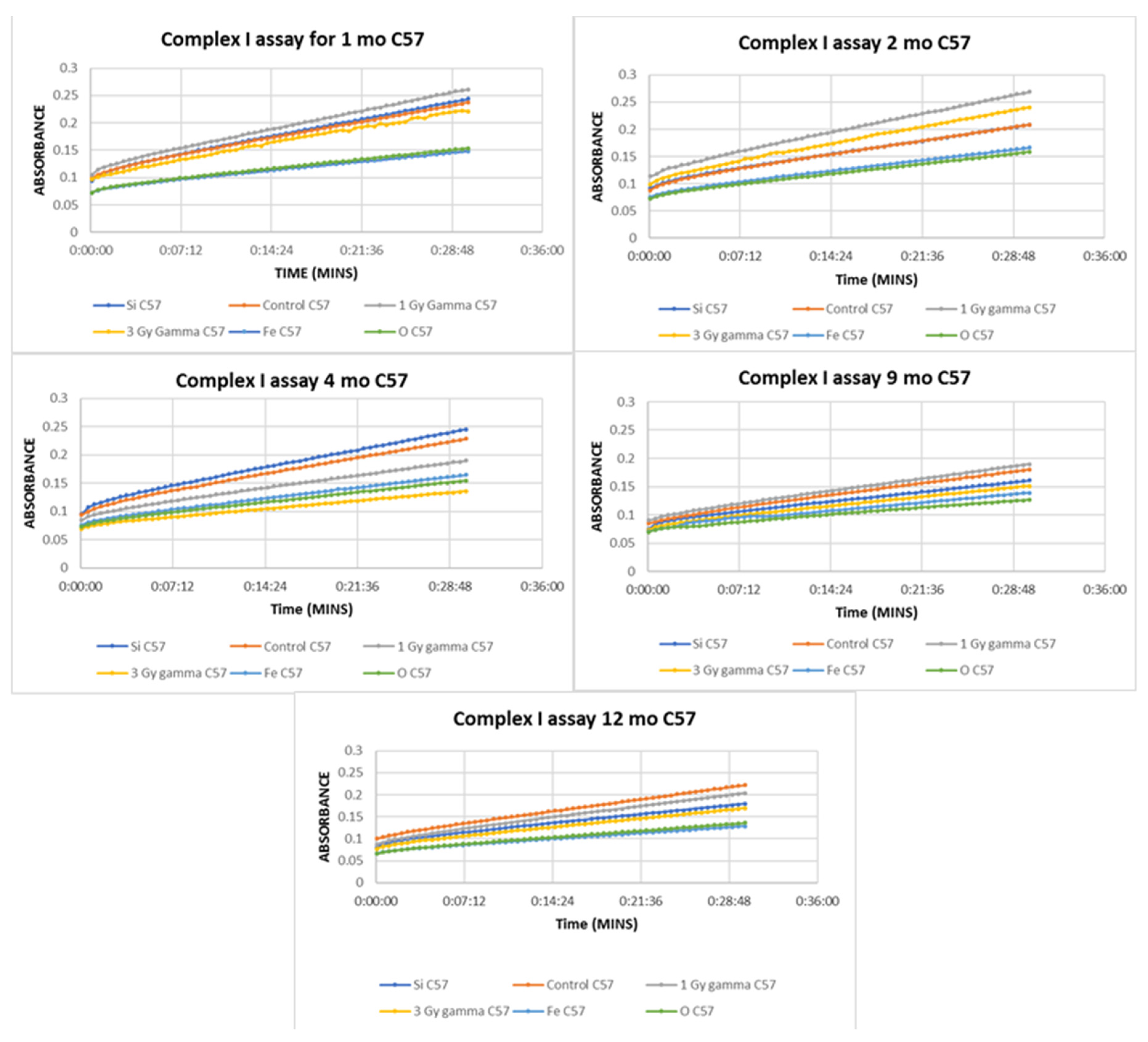
2. Results
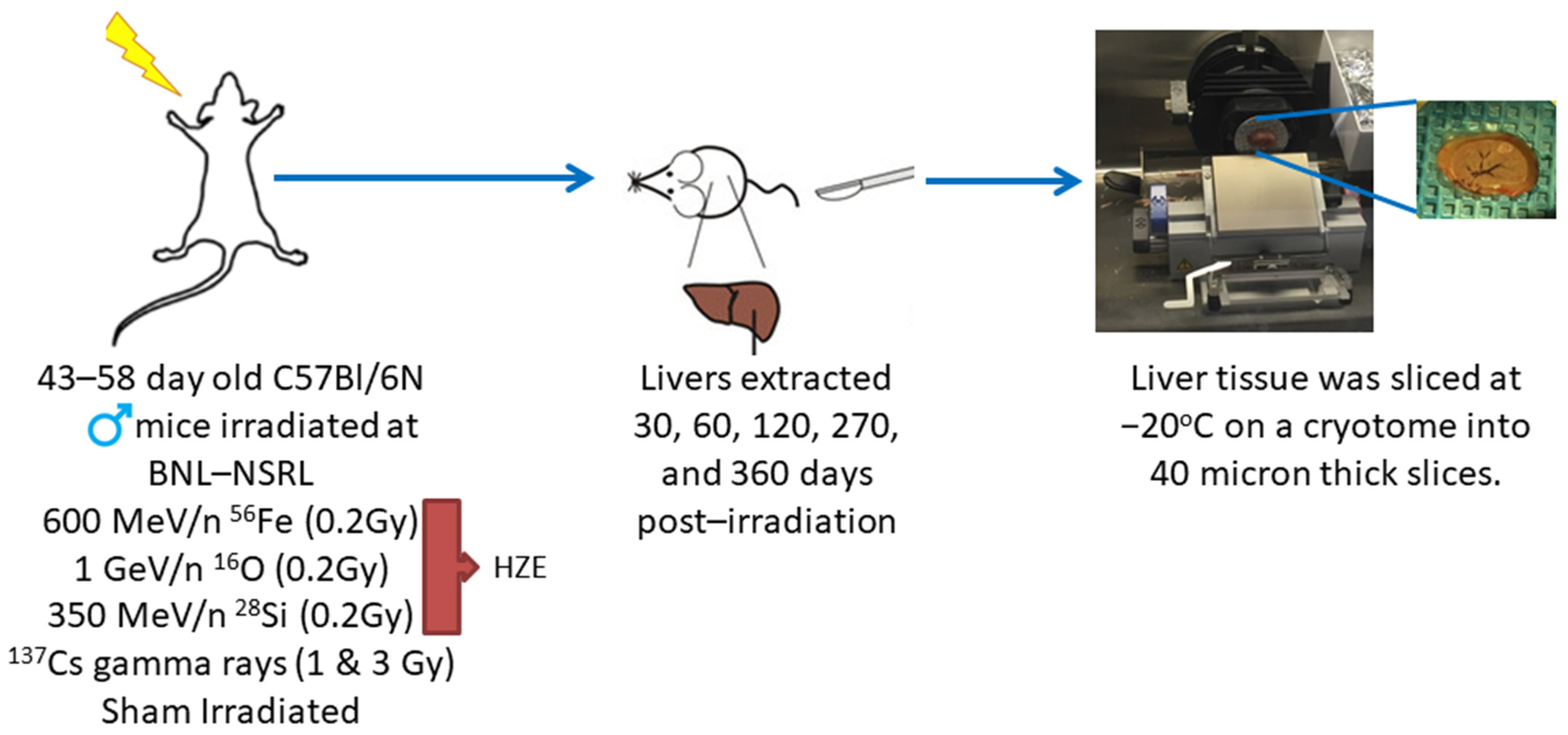
3. Materials and Methods
- Number of peptides per protein, 15;
- Number of transitions per peptide, 5;
- Peptide confidence threshold %, 95;
- False discovery rate threshold %, 1.0.
- XIC extraction window (min), 8.0;
- XIC width (ppm), 30.
- Forward: 5-TGC TAG CCG CAG GCA TTA C-3;
- Reverse: 5-GGG TGC CCA AAG AAT CAG AAC-3.
- Forward: 5-CTTCCCCACTGGCCTCAA G-3;
- Reverse: 5-CCA AAA CCC AGT GAT CCA GC-3 [20].
4. Discussion
4.1. ROS Scavengers
4.2. Mitochondrial Centric
4.3. Anti-Inflammatory
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species, (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Hashemi, E.; Akhavan, O.; Shamsara, M.; Rahighi, R.; Esfandiard, A.; Tayefeha, A.R. Cyto and genotoxicities of graphene oxide and reduced graphene oxide sheets on spermatozoa. Rsc. Adv. 2014, 52, 27213–27223. [Google Scholar] [CrossRef]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Bio. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nugent, S.; Mothersill, C.E.; Seymour, C.; McClean, B.; Lyng, F.M.; Murphy, J.E. International Journal of Radiation Biology Altered mitochondrial function and genome frequency post exposure to γ-radiation and bystander factors. Int. J. Radiat. Biol. 2010, 86, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, K.; Suman, S.; Kallakury, B.V.S.; Fornace, A.J. Exposure to Heavy Ion Radiation Induces Persistent Oxidative Stress in Mouse Intestine. PLoS ONE 2012, 7, e42224. [Google Scholar] [CrossRef] [PubMed]
- Datta, K.; Suman, S.; Kallakury, B.V.S.; Fornace, A.J. Heavy Ion Radiation Exposure Triggered Higher Intestinal Tumor Frequency and Greater β-Catenin Activation than γ Radiation in APCMin/+ Mice. PLoS ONE 2013, 8, e59295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by mitochondrial reactive oxygen species in physiology and pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [Green Version]
- De Barcelos, I.P.; Haas, R.H. CoQ10 and aging. Biology 2019, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatti, J.S.; Kumar, S.; Vijayan, M.; Bhatti, G.K.; Reddy, P.H. Therapeutic Strategies for Mitochondrial Dysfunction and Oxidative Stress in Age-Related Metabolic Disorders. Prog. Mol. Biol. Transl. Sci. 2017, 146, 13–46. [Google Scholar]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weil, M.M.; Bedford, J.S.; Bielefeldt-Ohmann, H.; Ray, F.A.; Genik, P.C.; Ehrhart, E.J.; Fallgren, C.M.; Hailu, F.; Battaglia, C.L.; Charles, B.; et al. Incidence of acute myeloid leukemia and hepatocellular carcinoma in mice irradiated with 1 GeV/nucleon 56Fe ions. Radiat. Res. 2009, 172, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Weil, M.M.; Ray, F.A.; Genik, P.C.; Yu, Y.; McCarthy, M.; Fallgren, C.M.; Ullrich, R.L. Effects of 28Si Ions, 56Fe Ions, and Protons on the Induction of Murine Acute Myeloid Leukemia and Hepatocellular Carcinoma. PLoS ONE 2014, 9, e104819, eCollection 2014. [Google Scholar] [CrossRef] [PubMed]
- Lichti, C.F.; Liu, H.; Shavkunov, A.S.; Mostovenko, E.; Sulman, E.P.; Ezhilarasan, R.; Wang, Q.; Kroes, R.A.; Moskal, J.C.; Fenyö, D.; et al. Integrated Chromosome 19 Transcriptomic and Proteomic Data Sets Derived from Glioma Cancer Stem-Cell Lines. J. Proteome Res. 2014, 13, 191–199. [Google Scholar] [CrossRef]
- Mastro, R.; Hall, M. Protein Delipidation and Precipitation by Tri-n-butylphosphate, Acetone, and Methanol Treatment for Isoelectric Focusing and Two-Dimensional Gel Electrophoresis. Anal. Biochem. 1999, 273, 313–315. [Google Scholar] [CrossRef]
- Almeida, R.; Pauling, J.K.; Sokol, E.; Hannibal-Bach, H.K.; Ejsing, C.S. Comprehensive Lipidome Analysis by Shotgun Lipidomics on a Hybrid Quadrupole-Orbitrap-Linear Ion Trap Mass Spectrometer. J. Am. Soc. Mass. Spectrom. 2015, 26, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Rupasinghe, T.W.T. Lipidomics: Extraction Protocols for Biological Matrices. In Methods in Molecular Biology; Clifton, N.J., Ed.; Humana Press: Totowa, NJ, USA, 2013; Volume 1055, pp. 71–80. [Google Scholar]
- He, H.; Conrad, C.A.; Nilsson, C.L.; Ji, Y.; Schaub, T.M.; Marshall, A.G.; Emmett, M.R. Method for Lipidomic Analysis: p53 Expression Modulation of Sulfatide, Ganglioside, and Phospholipid Composition of U87 MG Glioblastoma Cells. Anal. Chem. 2007, 79, 8423–8430. [Google Scholar] [CrossRef] [Green Version]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2006, 35, D527–D532. [Google Scholar] [CrossRef] [Green Version]
- Amthor, H.; Macharia, R.; Navarrete, R.; Schuelke, M.; Brown, S.; Otto, A.; Voit, T.; Muntoni, F.; Vrbóva, G.; Partridge, T.; et al. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc. Natl. Acad. Sci. USA 2007, 104, 1835–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.B.; Zhang, H.W.; Fu, R.Q.; Hu, X.Y.; Liu, L.; Li, Y.N.; Liu, Y.X.; Liu, X.; Hu, J.J.; Deng, Q.; et al. Mitochondrial fission and mitophagy depend on cofilin-mediated actin depolymerization activity at the mitochondrial fission site. Oncogene 2018, 37, 1485–1502. [Google Scholar] [CrossRef]
- Nia, A.M.; Chen, T.; Barnette, B.L.; Khanipov, K.; Ullrich, R.L.; Bhavnani, B.; Emmett, M.R. Efficient identification of multiple pathways: RNA-Seq analysis of livers from 56Fe ion irradiated mice. BMC Bioinform. 2020, 21, 118. [Google Scholar] [CrossRef]
- Verdin, E.; Hirschey, M.D.; Finley, L.W.S.; Haigis, M.C. Sirtuin regulation of mitochondria—Energy production. Trends in Biochem. Sci. 2011, 35, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H. Critical Review ER Stress Response, Peroxisome Proliferation, Mitochondrial Unfolded Protein Response and Golgi Stress Response. IUBMB Life 2009, 61, 871–879. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci 2019, 9, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainbolt, T.K.; Atanassova, N.; Genereux, J.C.; Wiseman, R.L. Stress-Regulated Translational Attenuation Adapts Mitochondrial Protein Import through Tim17A Degradation. Cell Metab. 2013, 18, 908–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melone, M.A.B.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The carnitine system and cancer metabolic plasticity. Cell Death Dis. 2018, 9, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franko, A.; Hrabe de Angelis, M.; Wiesner, R.J. Mitochondrial Function, Dysfunction and Adaptation in the Liver during the Development of Diabetes. In Mitochondria in Liver Disease; Han, D., Kaplowitz, N., Eds.; CRC Press: Abingdon, UK, 2015; pp. 383–411. [Google Scholar]
- Han, C.Y. Update on FXR biology: Promising therapeutic target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelcer, N.; Tontonoz, P. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Inv. 2006, 116, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Tian, G.; Sawashita, J.; Kubo, H.; Nishio, S.Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; et al. Ubiquinol-10 supplementation activates mitochondria functions to decelerate senescence in senescence-accelerated mice. Antioxid. Redox Signal. 2014, 20, 2606–2620. [Google Scholar] [CrossRef]
- Poulose, S.M.; Bielinski, D.F.; Carrihill-Knoll, K.; Rabin, B.M.; Shukitt-Hale, B. Exposure to 16 O-Particle Radiation Causes Aging-Like Decrements in Rats through Increased Oxidative Stress, Inflammation and Loss of Autophagy. Radiat. Res. 2011, 176, 761–769. [Google Scholar] [CrossRef]
- Haas, R.H. Mitochondrial dysfunction in aging and diseases of aging. Biology 2019, 8, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneiter, R. Intracellular sterol transport in eukaryotes, a connection to mitochondrial function? Biochimie 2007, 89, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Ohta, A.; Horiuchi, H.; Fukuda, R. Evaluation of sterol transport from the endoplasmic reticulum to mitochondria using mitochondrially targeted bacterial sterol acyltransferase in Saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 2015, 79, 1608–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Devaiah, S.P.; Zhang, W.; Welti, R. Signaling functions of phosphatidic acid. Prog. Lipid Res. 2006, 45, 250–278. [Google Scholar] [CrossRef] [PubMed]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2018, 28, 67–76. [Google Scholar] [CrossRef]
- Yang, C.Y.; Frohman, M.A. Mitochondria: Signaling with phosphatidic acid. Int. J. Biochem. Cell Biol. 2012, 44, 1346–1350. [Google Scholar] [CrossRef] [Green Version]
- Schenkel, L.; Bakovic, M. Formation and Regulation of Mitochondrial Membranes. Int. J. Cell Biol. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami-Murofushi, K.; Uchiyama, A.; Fujiwara, Y.; Kobayashi, T.; Kobayashi, S.; Mukai, M.; Murofushi, H.; Tigyi, G. Biological functions of a novel lipid mediator, cyclic phosphatidic acid. Biochim. Biophys. Acta 2002, 1582, 1–7. [Google Scholar] [CrossRef]
- Oishi-Tanaka, Y.; Glass, C.K. A new role for cyclic phosphatidic acid as a PPARγ antagonist. Cell Metab. 2010, 12, 207–208. [Google Scholar] [CrossRef] [Green Version]
- Arifin, S.A.; Falasca, M. Lysophosphatidylinositol signaling and metabolic diseases. Metabolites 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heringdorf, D.M. Lysophospholipids—Encyclopedia of Molecular Pharmacology; Offermanns, S., Rosenthal, W., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 710–716. [Google Scholar]
- Higashi, H.; Hirabayashi, Y.; Hirota, M.; Matsumoto, M.; Kato, S. Detection of ganglioside GM2 in sera and tumor tissues of hepatoma patients. Jpn. J. Cancer Res. 1987, 78, 1309–1313. [Google Scholar]
- Ohwada, K.; Takeda, H.; Yamazaki, M.; Isogai, H.; Nakano, M.; Shimomura, M.; Fukui, K.; Urano, S. Pyrroloquinoline Quinone (PQQ) Prevents Cognitive Deficit Caused by Oxidative Stress in Rats. J. Clin. Biochem. Nutr. 2008, 42, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, G.; Jasoliya, M.; Sahdeo, S.; Saccà, F.; Pane, C.; Filla, A.; Marsili, A.; Puorro, G.; Lanzillo, R.; Brescia Morra, V.; et al. Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum. Mol. Genet. 2017, 26, 2864–2873. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.Y.; Chen, J.; Lu, X.Y.; Zheng, M.Z.; Wang, L.L.; Shen, Y.L.; Chen, Y.Y. Dimethyl fumarate attenuates lipopolysaccharide-induced mitochondrial injury by activating Nrf2 pathway in cardiomyocytes. Life Sci. 2019, 235, 116863. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.Y.; Chen, J.; Lu, X.Y.; Zheng, M.Z.; Wang, L.L.; Shen, Y.L.; Chen, Y.Y. Elamipretide, (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J. Neuroinflammation 2019, 16, 1–19. [Google Scholar]
- Steele, V.E.; Holmes, C.A.; Hawk, E.T.; Kopelovich, L.; Lubet, R.A.; Crowell, J.A.; Sigman, C.C.; Kelloff, G.J. Lipoxygenase Inhibitors as Potential Cancer Chemopreventives. Cancer Epidemiol. Biomark. Prev. 1999, 8, 467–483. [Google Scholar]
- Yin, H.; Zhou, Y.; Zhu, M.; Hou, S.; Li, Z.; Zhong, H.; Lu, J.; Meng, T.; Wang, J.; Xia, L.; et al. Role of mitochondria in programmed cell death mediated by arachidonic acid-derived eicosanoids. Mitochondrion 2013, 13, 209–224. [Google Scholar] [CrossRef]
- Li, J.-G.; Barrero, C.; Merali, S.; Praticò, M. Five lipoxygenase hypomethylation mediates the homocysteine effect on Alzheimer’s phenotype. Sci. Rep. 2017, 7, 46002. [Google Scholar] [CrossRef] [Green Version]
- Laiakis, E.C.; Shuryak, I.; Deziel, A.; Wang, Y.W.; Barnette, B.L.; Yu, Y.; Ullrich, R.L.; Fornace, A.J.; Emmett, M.R. Effects of low dose space radiation exposures on the splenic metabolome. Int. J. Mol. Sci. 2021, 22, 3070. [Google Scholar] [CrossRef] [PubMed]
- da Silveira, W.A.; Fazelinia, H.; Rosenthal, S.B.; Laiakis, E.C.; Kim, M.S.; Meydan, C.; Kidane, Y.; Rathi, K.S.; Smith, S.M.; Stear, B.; et al. Comprehensive Multi-omics Analysis Reveals Mitochondrial Stress as a Central Biological Hub for Spaceflight Impact. Cell 2020, 183, 1185–1201. [Google Scholar] [CrossRef]
- Jonscher, K.R.; Alfonso-Garcia, A.; Suhalim, J.L.; Orlicky, D.J.; Potma, E.O.; Ferguson, V.L.; Bouxsein, M.L.; Bateman, T.A.; Stodieck, L.S.; Levi, M.; et al. Spaceflight activates lipotoxic pathways in mouse liver. PLoS ONE 2016, 11, 1–26. [Google Scholar]
- Blaber, E.A.; Pecaut, M.J.; Jonscher, K.R. Spaceflight activates autophagy programs and the proteasome in mouse liver. Int. J. Mol. Sci. 2017, 18, 2062. [Google Scholar] [CrossRef]
- Beheshti, A.; Chakravarty, K.; Fogle, H.; Fazelinia, H.; da Silveira, W.A.; Boyko, V.; Polo, S.-H.L.; Saravia-Butler, A.M.; Hardiman, G.; Taylor, D.; et al. Multi-omics analysis of multiple missions to space reveal a theme of lipid dysregulation in mouse liver. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.; McCubbin, F.M.; Kaur, J.; Smirnov, A.; Galdanes, K.; Schoonen, M.A.A.; Chen, L.C.; Tsirka, S.E.; Gorden, T. Acute Meteorite Dust Exposure and Pulmonary Inflammation—Implications for Human Space Exploration. 2017, NASA Tech Rep 20170004717.
- Schoonen, M.A.A.; Harrington, A.D.; Laffers, R.; Strongin, D.R. Role of hydrogen peroxide and hydroxyl radical in pyrite oxidation by molecular oxygen. Geochim. Cosmochim. Acta 2010, 74, 4971–4987. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
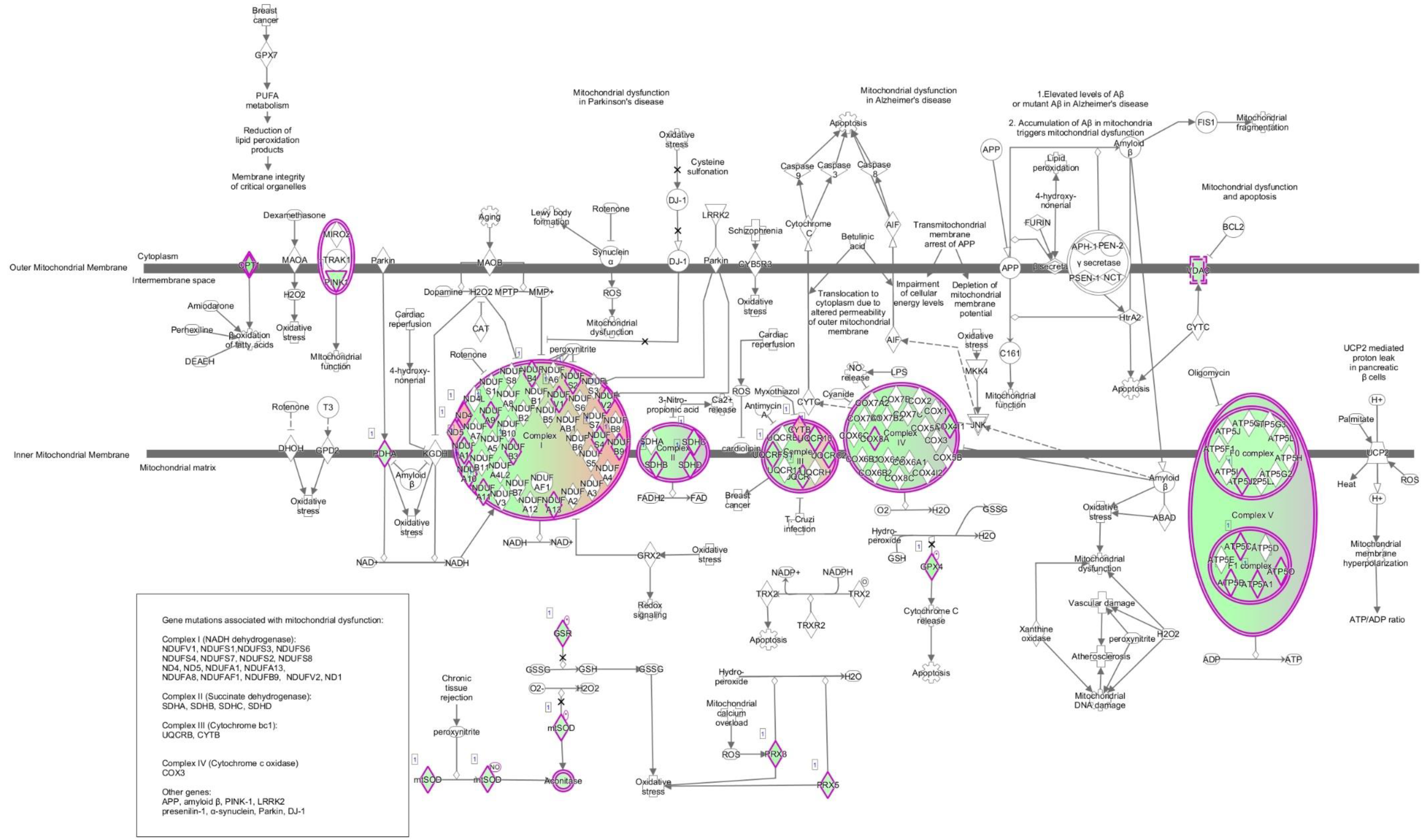
| Conditions with Mitochondria Dysfunction Pathway Effected | Number of Transcripts in the Pathway | Dysregulated Transcripts within Pathway | Number of Proteins in the Pathway | Dysregulated Proteins within Pathway |
|---|---|---|---|---|
| 1 mo 18O | 46 | aconitase 2, ATP synthase (H+ transporting mito F1 complex epsilon subunit, F1 subunit alpha beta & gamma, membrane subunit F, & peripheral stalk subunit OSCP), cytochrome c oxidase (subunit4I1, 6C, 7A2 like), carnitine palmitoyltransferase 1A, cytochrome c1, glutathione peroxidase 4, glutathione-disulfide reductase, cytochrome b, MTND2, NADH dehydrogenase subunits 4, 5 & 6 (complex I), NADH:ubiquinone oxidoreductase subunits A9, A10, A11, A13, B3, B4, B9, core subunit V1 & V2, pyruvate dehydrogenase E1 alpha 1 subunit, PTEN induced putative kinase 1, peroxiredoxin 3 & 5, succinate dehydrogenase complex iron sulfur subunit B, subunit C, & subunit D, superoxide dismutase 2, ubiquinol-cytochrome c reductase complex III subunit X, core protein 1, core protein 2 & Rieske iron-sulfur polypeptide 1, voltage dependent anion channel 1 & 2 | 8 | ATP synthase membrane subunit f, catalase, NADH dehydrogenase subunit 1 (complex I), NADH:ubiquinone oxidoreductase subunit V3, peroxiredoxin 3, synuclein alpha, SURF1 cytochrome c oxidase assembly factor, xanthine dehydrogenase |
| 1 mo 56Fe | 9 | ATP synthase F1 subunit gamma, carnitine palmitoyltransferase 1A, cytochrome b, NADH dehydrogenase subunit 1, 4, 5 & 6 (complex I), MTND2, NADH:ubiquinone oxidoreductase subunit B4 | 0 | |
| 1 mo 28Si | 3 | cytochrome b, MTND2, NADH dehydrogenase subunit 6 (complex I) | 6 | ATP synthase F1 subunit delta, catalase, mitogen-activated protein kinase 9, NADH dehydrogenase subunit 4(complex I), nicastrin, ras homolog family member T2 |
| 1 mo 3 Gy | 0 | 7 | ATP synthase membrane subunit f, catalase, NADH dehydrogenase subunit 1 (complex I), MTND2, NADH dehydrogenase subunit 4 (complex I), peroxiredoxin 3, synuclein alpha | |
| 2 mo 18O | 37 | ATP synthase (H+ transporting mito F1 complex epsilon subunit, F1 subunit alpha beta, gamma & delta, membrane subunit c locus 1 membrane subunit F & G, & peripheral stalk subunit OSCP & D), cytochrome c oxidase (subunit4I1, 6B1, 6C, 7B, & 8A), carnitine palmitoyltransferase 1A, cytochrome c1, glutathione peroxidase 4, glutathione-disulfide reductase, leucine rich repeat kinase 2, NADH dehydrogenase subunits 4 (complex I), NADH:ubiquinone oxidoreductase subunits A1, A2, A6, A8, A9, A10, A12, A13, B4, B7, B9, B11, S4, V3 core subunit S2 V1 & V2, oxoglutarate dehydrogenase, PTEN induced putative kinase 1, peroxiredoxin 3 & 5, parkin RBR E3 ubiquitin protein ligase, succinate dehydrogenase complex iron sulfur subunit B, subunit C, & subunit D, ubiquinol-cytochrome c reductase complex III subunit X, core protein 2 & Rieske iron-sulfur polypeptide 1, voltage dependent anion channel 1 | 10 | ATP synthase membrane subunit f, catalase, cytochrome c oxidase subunit 7A2 like, mitogen-activated protein kinase, kinase 4, mitogen-activated protein kinase 9, NADH:ubiquinone oxidoreductase subunit A11, NADH:ubiquinone oxidoreductase complex assembly factor 1, NADH:ubiquinone oxidoreductase subunit B6, peroxiredoxin 3, thioredoxin reductase 2 |
| 2 mo 56Fe | 19 | aph-1 homolog B gamma-secretase subunit, ATP synthase F1 subunit gamma, ATP synthase membrane subunit g, ATP synthase peripheral stalk subunit OSCP, cytochrome c oxidase copper chaperone COX17, cytochrome c oxidase subunit 4I1, cytochrome c oxidase subunit 6C, glutathione peroxidase 4, leucine rich repeat kinase 2, cytochrome b, NADH dehydrogenase subunit 4 (complex I), NADH:ubiquinone oxidoreductase subunits A1, A2, B2, B4, B9, & core subunit V2, synuclein alpha & ubiquinol-cytochrome c reductase binding protein | 0 | |
| 2 mo 3 Gy | 26 | ATP synthase (H+ transporting mito F1 complex epsilon subunit, F1 subunit gamma & delta, membrane subunit F & G, & peripheral stalk subunit OSCP), cytochrome c oxidase (subunit4I1, 6C, 7A2 like, & 8A), carnitine palmitoyltransferase 1A, cytochrome c1, glutathione peroxidase 4, glutathione-disulfide reductase, cytochrome b, NADH dehydrogenase subunits 1,4,5 &6 (complex I), MTND2, NADH:ubiquinone oxidoreductase subunits A2, A6, A9, A13, B2, B9, core subunit S2, S7 & V1, PTEN induced putative kinase 1, peroxiredoxin 5, parkin RBR E3 ubiquitin protein ligase, succinate dehydrogenase complex subunit C & D, ubiquinol-cytochrome c reductase complex III subunit X & XI & Rieske iron-sulfur polypeptide 1 | 0 | |
| 4 mo 18O | 11 | ATP synthase membrane subunit f, ATP synthase peripheral stalk subunit OSCP, cytochrome c oxidase subunit 5A & 6B1, NADH:ubiquinone oxidoreductase subunit A3, A7, A11, A12 & S6, thioredoxin 2, ubiquinol-cytochrome c reductase complex III subunit X | 0 | |
| 4 mo 3 Gy | 6 | cytochrome c oxidase subunit 5A & I, cytochrome b, MTND2, NADH dehydrogenase subunit 4 & 5 (complex I) | 0 | |
| 4 mo 1 Gy | 0 | 6 | caspase 8, NADH dehydrogenase subunit 4 (complex I), nicastrin, NADH:ubiquinone oxidoreductase subunit B6, NADH:ubiquinone oxidoreductase core subunit S3, thioredoxin reductase 2 | |
| 9 mo 18O | 12 | ATP synthase membrane subunit c locus 2, cytochrome c oxidase copper chaperone COX11, cytochrome c oxidase subunit 5A, glutathione peroxidase 4, cytochrome b, MTND2, NADH dehydrogenase subunit 4 & 5 (complex I), NADH:ubiquinone oxidoreductase subunit A8, A11, & S6, thioredoxin 2 | 0 | |
| 9 mo 3 Gy | 7 | cytochrome c oxidase copper chaperon COX11, cytochrome c oxidase subunit 1, MTND2, NADH dehydrogenase subunit 4 & 6 (complex 1), NADH:ubiquinone oxidoreductase subunit B4, ubiquinol-cytochrome c reductase binding protein | 0 | |
| 9 mo 28Si | 0 | 7 | caspase 3, cytochrome c oxidase subunit 7A2 like, mitogen-activated protein kinase 9, MTND2, NADH dehydrogenase subunit 4 (complex I), NADH:ubiquinone oxidoreductase core subunit S3, ras homolog family member T2 | |
| 12 mo 28Si | 0 | 6 | ATP synthase F1 subunit delta, catalase, cytochrome c oxidase subunit 6A1, MTND2, NADH:ubiquinone oxidoreductase complex assembly factor 2, ras homolog family member T2 | |
| 12 mo 1 Gy | 9 | cytochrome c oxidase subunit 7A2, 7A2 like, & 8A, fission mitochondrial 1, furin paired basic amino acid cleaving enzyme, NADH:ubiquinone oxidoreductase subunit B9, S6 & core subunit S2 | 11 | ATP synthase F1 subunit delta, catalase, cytochrome c oxidase assembly homolog COX15, cytochrome c oxidase subunit 7A2 like, mitogen-activated protein kinase 9, MTND2, NADH:ubiquinone oxidoreductase subunit A9, NADH:ubiquinone oxidoreductase complex assembly factor 1, NADH:ubiquinone oxidoreductase subunit B6, ras homolog family member T2, SURF1 cytochrome c oxidase assembly factor |
| Condition | Top 5 Transcript Based Canonical Pathways | Top Upstream Regulators Transcript Based | Other interesting Dysregulated Transcripts and Pathways | Top 5 Protein Based Canonical Pathway | Top Upstream Regulators Protein Based | Other Interesting Dysregulated Proteins and Pathways |
|---|---|---|---|---|---|---|
| 1 mo 18O | Mitochondrial dysfunction Oxidative phosphorylation Sirtuin Signaling pathway FXR/RXR activation Fatty Acid beta oxidation I | PPARA Pirin ixic acid POR TO-901317 Mono-(2-ethylhexyl) phthalate | TCA Cycle II, Gluconeogenesis I (aldolase, fructose-bisphosphate C; enolase 1; malate dehydrogenase 1 & 2, phosphoglycerate kinase 1 all down), ubiquinol-10 biosynthesis, mitochondrial L-carnitine shuttle pathway (acyl-coA synthetase long chain family member 1, carnitine palmitoyltransfease 1A & solute carrier family 27 member 5 all down), ceramide signaling | Sirtuin Signaling Pathway RhoA Signaling (cytoskeleton organization) Clathrin-mediated Endocytosis signaling LXR/RXR activation Actin Cytoskeleton Signaling | HNF4A-hepatocyte nuclear factor 4 alpha Insulin D-glucose Methylprednisolone ABCB6 | PI3K (down) mTOR (up) cyclin dependent kinase 2(up) cyclin dependent kinase inhibitor 1B (down) glycogen synthase kinase 3 beta (down) insulin signaling receptor (down) mannose-6-phosphate receptor (down) |
| 2 mo 18O | EIF2 signaling Oxidative phosphorylation Regulation of eIF4 and p70S6K signaling Mitochondrial dysfunction Sirtuin signaling pathway | MLXIPL RICTOR YAP1 MYC MYCN | mTOR signaling, Protein Ubiquitination Pathway, NRF2-mediated oxidative stress response, Unfolded protein response, TCA cycle II (fumarate hydratase, succinate dehydrogenase complex (iron sulfur subunit B, C & D all down) | Huntington’s Disease Signaling (HIP1 down) (HSP40 up) Iron homeostasis signaling pathway ILK signaling Tight Junction Signaling Glycogen Degradation III | HNF4A CST5 Maslinic acid miR-30c-5p (and other miRNAs w/seed GUAAACA) desmopressin | Also see sirtuin signaling and ceramide signaling (SMPD (sphingomyelin phosphodiesterase 4) up) |
| 4 mo 18O | EIF2 signaling Unfolded protein response Regulation of eIF4 and p70S6K signaling Protein Ubiquitination Pathway ER stress pathway-CALR (calreticulin) down, DDIT3 (DNA damage inducible transcript 3) down, EIF2AK3 down, HSP90B1 down, HSPA5 down | XBP1 ERN1 Tunicamycin RICTOR ATF6 | mTOR signaling, sirtuin signaling pathway | Acute phase response signaling Clathrin-mediated endocytosis signaling Phagosome maturation Role of JAK family kinases in IL-6 type cytokine signaling FXR/RXR activation | HNF4A CST5 TP53 Methylprednisolone D-glucose | Also see B cell receptor signaling, production of nitric oxide & ROS macrophages, cellular senescence (predicted inhibition), CDKN1B cyclin dependent kinase inhibitor (down), CDK2 cyclin dependent kinase 2-activation of s-phase progression, sumoylation pathway-SAE1(SUMO1 activating enzyme subunit 1), LXR/RXR activation, insulin receptor signaling |
| 9 mo 18O | EIF2 signaling Mitochondrial dysfunction Oxidative phosphorylation Regulation of eIF4 and p70S6K signaling Sirtuin signaling pathway | MLXIPL RICTOR MYC YAP1 CTNNB1 | mTOR signaling | Clathrin-mediated endocytosis signaling RhoA signaling Acute phase response signaling Huntington’s disease signaling Signaling by Rho family GTPases | NHF4A RHOJ XBP1 Bvr Sirolimus | Coenzyme A biosynthesis, Sirtuin signaling, heme oxygenase 1 & 2 (down) |
| 12 mo 18O | Mouse Embryonic Stem Cell Pluripotency-ID1 (inhibitor of DNA binding), ID2 down Trans, trans-farnesyl diphosphate biosynthesis-FDPS (farnesyl disphosphate synthase) up Pregnenolone biosynthesis-CYP26A1 up Histidine Degradation VI-same Superpathway of Geranylgeranyldiphosphate biosynthesis I (via Mevalonate)-FDPS up | ACVR2A LTBP4 SMAD1/5 Notch BMP10 | Ubiquinol-10 biosynthesis- CYP26A1 up, Role of lipids/lipid rafts in the pathogenesis of influenza-FDPS up | Acute phase response signaling Tight junction signaling IL-8 signaling LXR/RXR activation L-glutamine Biosynthesis II(tRNA-dependent) | HNF4A CST5 Tetrachlorodibenzodioxin GPD1 SLC25A13 | L-carnitine shuttle pathway (acyl-CoA synthetase long chain family member 3 (down)) & member 4 (up), phospholipase c signaling, type II diabetes signaling, role of NFAT in regulation of the immune response, CDK2 (down), Unfolded protein response |
| 1 mo 56Fe | PXR/RXR activation LPS/IL-1 mediated inhibition of RXR Function Sirtuin Signaling Pathway Nicotine Degradation II Circadian Rhythm Signaling-ARNTL (aryl hydrocarbon receptor nuclear translocator) up, BHLHE40 (basic helix-loop-helix family member e40) down; CLOCK (clock circadian regulator) up; CRY1 (cryptochrome circadian regulator 1) up; PER1 (period circadian regulator 1) down; PER2 down; PER3 down | RORC RORA PPARA Methylprednisolone NR1I2 | Ubiquinol-10 biosynthesis, acyl-CoA hydrolysis | Acute phase response signaling Glycogen degradation III Phagosome maturation Huntington’s Disease signaling Clathrin-mediated endocytosis signaling | TO-901317 Ciprofibrate Nitrofurantoin SCAP ACOX1 | Calcium transport I (ATPase sarcoplasmic/ER Ca2+ transporting 2 (down) & ATPase plasma membrane Ca2+ transporting 1 (up)), LXR/RXR (apolipoprotein A5-activates cholesterol efflux), Death receptor signaling (CASP7 inhibits DNA repair (down)) |
| 2 mo 56Fe | EIF2 Signaling Acute phase response signaling-exported Regulation of eIF4 and p70S6K signaling-exported list mTOR signaling-exported Production of nitric oxide and ROS in macrophages | Lipopolysaccharide MLXIPL IL1B YAP1 IFNG | Sirtuin signaling pathway, FXR/RXR, NRF2-mediated oxidative stress response | Acute phase response signaling Xenobiotic metabolism signaling NAD salvage pathway II UVB-induced MAPK signaling NF-kB activation by viruses | HNF4A Pregnenolone carbonitrile Pirinixic acid Estrone RAB1B | ceramide signaling (sphingosine kinase 2 (up)) |
| 4 mo 56Fe | Unfolded protein response Aldosterone signaling in Epithelial cells Endoplasmic reticulum stress pathway Protein ubiquitination pathway NRF2-mediated oxidative stress response | XBP1 Tunicamycin ERN1 ATF6 1,2-dithiol-3-thione | EIF2 signaling, Regulation of eIF4 and p70S6K signaling mTOR, Glucocorticoid receptor signaling, Sirtuin signaling pathway, insulin receptor signaling | Cell cycle regulation by BTG family proteins Inhibition of ARE-mediated mRNA degradation pathway 5-aminoimidazole ribonucleotide biosynthesis I Dopamine receptor signaling Breast cancer regulation by Stathmin1 | HNF4A Camptothecin (+-)-2-hydroxyoleic acid FAS HNRNPH1 | Cyclins and cell cycle regulation, regulation of eIF4 & p70S6K signaling, ceramide signaling (different protein that O) sphingosine kinase 2, role of CHK proteins in cell cycle checkpoint, mitochondrial L-carnitine shuttle pathway (acyl-CoA synthetase long chain family member 4 down & solute carrier family 27 member 4 (down), thrombin signaling, mTOR signaling |
| 9 mo 56Fe | Unfolded protein response-CEBPE down, all up ERN1, HSPA8, Hspa1b, HSPH1, MAP3K5 NRF2-mediated oxidative stress response-ACTG1, BACH1, DNAJA1, DNAJB1, FKBP5, GSTP1, MAP3K5 all up Aldosterone signaling in epithelial cells-DNAJA1, DNAJB1, HSP90AA1, HSPA8, HSPA4L, HSPH1 all up Protein Ubiquitination pathway same as above Agranulocyte-adhesion and diapedesis-ACTG1 up, CLDN3 down, CXCL2 (C-X-C chemokine ligand) up, CXCL13 up, IL1R1 (interleukin 1 receptor type 1) up | NR5A2 Lipopolysaccharide Cisplatin Thapsigargin NR1I3 | GADD45 signaling-CDKN1A (cyclin dependent kinase inhibitor 1A) up, GADD45G (growth arrest and DNA damage inducible gamma) up LXR/RXR signaling-C4A/C4B (complement C4B), CYP7A1, HMGCR (3-hydroxy-3-methylgutaryl-CoA reductase), IL1R1 (interleukin 1 receptor type 1) all up Asparagine biosynthesis I-ASNS(asparagine synthetase glutamine-hydrolyzing) up, FXR/RXR-C4A/C4B, CYP7A1, FOXA3, SLC10A2 all up, PXR/RXR ER stress pathway-ALAS1, CYP7A1, IGFBP1 all up, PI3K/AKT signaling-CDKN1A up, GDF15 (growth differentiation factor 15) down, HSP90AA1 up, MAP3K5 up, Sirtuin signaling pathway-BCL2L11, GADD45G, MT-ND2, MT-ND4, MT-ND6, PFKFB3 (6-phosphofructo-2 kinase/fructose 2,6-biphosphatase 3) all up Apoptosis signaling-BCL2L11 up, ENDOG down, MAP3K5 up | Purine nucleotides de novo biosynthesis II 5-aminoimidazole ribonucleotide biosynthesis I Regulation of cellular mechanics by calpain protease Acute phase responses signaling Sertoli cell-Sertoli cell junction signaling | TO-901317 Ciprofibrate Diethyl nitrosamine HNF4A ACOX1 | |
| 12 mo 56Fe | only 6 genes dysregulated all up | Gm23442 Gm25394 Gm25835 Gm26397 Snora78 Snord13 (small nucleolar RNA) | Acute phase response signaling Mechanisms of viral exit from host cells Wnt/beta catenin signaling Endothelin-1 signaling Endometrial cancer signaling | HNF4A Let-7a-5p (and other miRNAs w/seed GAGGUAG) 1,2-dithiol-3-thione Methylprednisolone miR-30c-5p (other miRNAs w/seed GUAAACA) | Sirtuin signaling pathway | |
| 1 mo 28Si | Mitochondrial Dysfunction Asparagine Biosynthesis I-ASNS (asparagine synthetase (glutamine-hydrolyzing) down Sirtuin Signaling Pathway-MT-CYB; MT-ND2; MT-ND6Alpha tocopherol degradation-CYP4A11 Oxidative phosphorylation | Actinonin MRPL12 DAP3 MT-TM SIRT3 | Ubiquinol-10 biosynthesis | Huntington’s Disease signaling Stearate Biosynthesis I (animals) FAT10 signaling pathway D-myo-inositol (1,4,5,6)-tetraphosphate biosynthesis D-myo-inositol (3,4,5,6)-tetrabiphosphate biosynthesis | HNF4A Glucagon Desmopressin CALCA BCAN | Acyl-CoA hydrolysis |
| 2 mo 28Si | FXR/RXR activation-FOXA2 up, HPX down, RARA down, SAA1 * down Role of Oct4 in Mammalian Embryonic stem cell pluripotency-FOXA2 up, RARA down Acute phase response signaling-HP, HPX (hemopexin), SAA1 * all down TR/RXR activation-HP (haptoglobin) and THRSP (thyroid hormone responsive) | Lipopolysaccharide TNF RORC IL1B NR1H4 | Acute phase response signaling Fcy receptor mediated phagocytosis in macrophages and monocytes Stearate biosynthesis I (animals) Caveolae-mediated endocytosis signaling Leukocyte extravasation signaling | HNF4A miR-1-3p(and other miRNAs w/seed GGAAUGU) ACOX1 ESR1 Fulvestrant | ||
| 4 mo 28Si | Unfolded protein response-DNAJB9, HSPA5, Hspa1b, SYVN1 (synoviolin 1) all down IL-7 signaling pathway-IGHG1, Ighg2b, Ighg2c all up Phagosome formation-IGHG1, Ighg2b, MARCO (macrophage receptor with collagenous structure) all up Autoimmune thyroid disease signaling-IGHG1, Ighg2b both up Hematopoiesis from pluripotent stem cells-same as above | CLOCK ATF6 Thapsigargin HTT tunicamycin | Phosphatidylethanolamine biosynthesis II (choline kinase alpha up), ER stress pathway (heat shock protein A(HSP70) member 5 down) | Huntington’s Disease Signaling 3-phoshoinsositide degradation D-myo-inositol-5-phosphate metabolism Superpathway of inositol phosphate compounds 3-phosphoinositide biosynthesis | HNF4A miR-30c-5p(and other miRNAs w/seed GUAAACA) PPARA KLF3 SREBF1 | |
| 9 mo 28Si | Acetone Degradation I (to Methylglyoxal)-CYP2A6 down, CYP2C8 down, CYP4A22 up Stearate biosynthesis I (animals)-ACOT1 down, CYP4A22 up, ELOVL6(ELOVL fatty acid elongase 6) down Nicotine Degradation III-CYP2A6, CYP2C8, UGT1A4 all down Melatonin Degradation I-same as above Nicotine Degradation II-same | STAT5B Pirinixic acid RORC L-triiodothyronine PPARA | Acyl-CoA hydrolysis, ubiquinol-10 biosynthesis | Remodeling of epithelial adherens junctions Caveolar-mediated endocytosis signaling Clathrin-mediated endocytosis signaling Paxillin signaling Integrin signaling | HNF4A ERBB3 IL4 RHOJ ciprofibrate | |
| 12 mo 28Si | only 1 gene upregulated | Gm22154 | Aldosterone signaling in epithelial cells Protein ubiquitination pathway Stearate Biosynthesis I (animals) Acute phase response signaling FAT10 signaling pathway | Pirinixic acid ELL2 HNF4A CYP2E1 ciprofibrate | sirtuin signaling pathway | |
| 1 mo 1 Gy | Stearate Biosynthesis I (animals)-ACOT1 (acyl-CoA thioesterase 1) down; ACOT4 down; CYP4A11 down; FASN (fatty acid synthase) up Superpathway of cholesterol biosynthesis-FDPS (farnesyl diphosphate synthase) up; MVD (mevalonate diphosphate decarboxylase) up; SQLE (squalene epoxidase) up Acyl-CoA hydrolysis-ACOT1; ACOT4 Superpathway of geranylgeranyl diphosphate biosynthesis I (via Mevalonate) TR/RXR activation-BCL3 (B cell CLL/lymphoma 3) up; FASN up; HP (haptoglobin) up | FGR19 PPARA SCAP CFTR SREBF1 | Palmitate biosynthesis I, ubiquinol-10 biosynthesis | Sirtuin signaling pathway Antigen presentation pathway Phagosome maturation Remodeling of epithelia adherens junctions Molybdenum cofactor biosynthesis | HNF4A POR SREBF1 SCAP ESR1 | PTEN signaling, sumoylation, death receptor signaling |
| 2 mo 1 Gy | EIF2 signaling mTOR signaling Regulation of eIF4 and p70S6K signaling Unfolded protein response Complement system | MLXIPL Lipopolysaccharide YAP1 IL6 CTNNB1 | Ubiquinol-10 biosynthesis, sphingosine-1-phosphate signaling | Stearate Biosynthesis I (animals) 3-phosphoinositide degradation Clathrin-mediated endocytosis signaling 3-phosphoinositide biosynthesis D-myo-inositol (1,4,5,6)-tetra bisphosphate biosynthesis | CFTR TO-901317 Pirinixic acid HNF4A PPARA | |
| 4 mo 1 Gy | Unfolded protein response ER stress pathway-CALR, DDIT3, DNAJC3, EIF2AK3, HSP90B1, HSPA5, XBP1 all down LXR/RXR activation FXR/RXR activation Acute phase response signaling | XBP1 Tunicamycin ERN1 RORC RORA | NRF2-mediated oxidative stress response, acyl-coA hydrolysis, ubiquinol-10 biosynthesis, palmitate biosynthesis I (fatty acid synthase up), fatty acid biosynthesis Initiation II(same from palmitate pathway) | NAD Salvage Pathway II Cholesterol Biosynthesis I Cholesterol Biosynthesis II (via 24,25-dihydrolanosterol) Cholesterol Biosynthesis III (via Desmosterol) TWEAK signaling | HNF4A Methylprednisolone miR-155-5p (miRNAs w/seed UAAUGCU) PML DMD | |
| 9 mo 1 Gy | Role of JAK2 in hormone-like cytokine signaling-IRS2 (insulin receptor substrate 2), SOCS2 (suppressor of cytokine signaling), SOCS3 all up Acute phase response signaling-IL1R1, SAA1 * (serum amyloid A1), SOCS2, SOCS3, all up TF (transferrin) down Sirtuin signaling pathway-ARNTL, CDH1, GADD45G, MT-ND4, MT-ND5 all up, MYCN (MYCN proto-oncogene, bHLH TF) down IGF-1 signaling-IGFBP1, IRS2, SOCS2, SOC3 all up IL-9 signaling-IRS2, SOCS2, SOCS3 all up | RORC Dexamethasone IL1B RORA tretinoin | PXR/RXR signaling-CYP2A6 down, CYP2B6 down, IGFBP1 up, LXR/RXR-IL1R1 up, SAA1 * up, TF down, FXR/RXR-SAA1 * up, SLC22A7 up, TF down Phosphatidylethanolamine biosynthesis II (choline kinase alpha up) | Glycine Betaine Degradation Clathrin-mediated endocytosis signaling Methionine salvage II (mammalian) L-serine degradation Caveolar-mediated endocytosis signaling | Diethylnitrosamine miR-155-3p (miRNAs w/seed UCCUACC) RORA TP53 NCOA6 | Sirtuin signaling pathway, superoxide radicals degradation |
| 12 mo 1 Gy | Mitochondrial dysfunction VDR/RXR activation-EP300, FOXO1, GADD45A, NCOR2, and PPARD all down, HR up B cell receptor signaling-9 molecules exported Estrogen receptor signaling-EP300, HNRNPD, NCOR2, RRAS, SPEN, TAF15, TAF4B all down Acute phase response signaling-IL1R1, IL6R, MAP3K5, RRAS, SAA1 *, SAA2-SAA4, SERPINA3, SOCS3 all down | ST1926 IL3 EPOR Lipopolysaccharide IL1B | Sirtuin signaling, mTOR | Stearate Biosynthesis I (animals) Acyl-CoA hydrolysis Acetone Degradation I (to methylglyoxal) Clathrin-mediated endocytosis signaling Sirtuin signaling pathway | NKX2-2-AS1 HNF4A Pirinixic acid Ciprofibrate di(2-ethylhexyl) phthalate | mitochondrial L-carnitine shuttle pathway |
| 1 mo 3 Gy | IL-17A signaling in fibroblasts-CEBPB (CCAAT enhancer binding protein beta) up; CEBPD (delta) up; JUN (Jun proto-oncogene) up; LCN2 (lipocalin 2) down Glucocorticoid receptor signaling-AR (androgen receptor) up; CDKN1A (cyclin dependent kinase inhibitor 1A) up; CEBPB up; DUSP1 (dual specificity phosphatase 1) up; FKBP5 (FK506 binding protein 5) up; JUN up; TSC22D3 (TSC22 domain family member 3) up Acute phase response signaling-APCS (amyloid P component, serum) down; CEBPB up; IL6R (interleukin 6 receptor) up; JUN up; SAA1 (serum amyloid A1) down JAK/Stat Signaling-CDKN1A (cyclin dependent kinase inhibitor 1A) up; CEBPB up; Jun up Hepatic Fibrosis/Hepatic Stellate Cell Activation-COL15A1 (collagen type XV alpha 1 chain) down; COL25A1 (collagen type XXV alpha chain) down; CTGF (connective tissue growth factor) up, IL6R up | TNF IL1B IFNG Glucocorticoid dexamethasone | Sumoylation pathway | Remodeling of epithelial adherns junctions Clathrin-mediated endocytosis signaling Systemic lupus erythematosus signaling Fcy receptor-mediated phagocytosis in macrophages and monocytes Caveolar-mediated endocytosis signaling | HNF4A SREBF1 TLE3 miR-30c-5p (and other miRNAs w/seed GUAAACA) COMMD1 | Insulin receptor signaling, sirtuin signaling pathway, mTOR signaling, PPAR signaling, PI3K/AKT signaling, ceramide signaling (sphingomyelin phosphodiesterase 4 (up)), PTEN Signaling |
| 2 mo 3 Gy | EIF2 signaling Regulation of eIF4 and p70S6K signaling mTOR signaling Unfolded protein response NRF2-mediated oxidative stress response | MLXIPL YAP1 Tunicamycin XBP1 MYCN | Sirtuin signaling pathway, ceramide signaling, ER stress pathway | Acute phase response signaling Clathrin-mediated endocytosis signaling tRNA charging RhoGDI signaling Death Receptor signaling | Monobutyl phthalate HNF4A CST5 OGA 2,4,5,2′,4′,5′-heachlorobiphenyl | |
| 4 mo 3 Gy | Oxidative phosphorylation Mito dysfunction Sirtuin signaling pathway-FOXO1, MT-CYB, MT-ND2, MT-ND4, MT-ND5, PPARGC1A all up, RRP9 down Unfolded protein response-HSPA5, Hspa1b, SYVN1 all down Granzyme B signaling-ENDOG (endonuclease G), LMNB2 (lamin B2) both down PXR/RXR activation-CYP2C8, FOXO1, PPARGC1A all up FXR/RXR activation-FOXO1 up, PPARGC1A up, SAA1 * down | IL1B THBS4 STAT3 ALKBH1 NSUN3 | JAK/Stat signaling IL-7 signaling pathway Reelin Signaling in Neurons Clathrin-mediated endocytosis signaling Systemic lupus erythematosus signaling | HNF4A Glucagon SREBF1 Insulin PPARGC1B | Sirtuin signaling, sphigosine-1-phosphate signaling, ubiquinol-10 biosynthesis (coenzyme Q3, methyltransferase up & cytochrome P450 family 46 subfamily A member 1 (down)) | |
| 9 mo 3 Gy | Mito dysfunction Oxidative phosphorylation IL-7 signaling pathway-IGHG1, Ighg2b, Ighg2c, MYC all up, JUN down Sirtuin signaling pathway-HIST1H4J & JUN down, MT-ND2, MT-ND4, MT-ND6, MYC, NDUFB4 all up LPS/IL-1 mediated inhibition of RXR function-ALAS1, CYP2A6, CYP7A1, GSTA5, Sult1d1 all up and JUN down | Lipopolysaccharide RORC NR1I2 RORA Cadmium chloride | Actin cytoskeleton signaling Acute phase response signaling Integrin signaling Signaling by Rho Family GTPases Remodeling of Epithelial Adherens Junctions | Desmopressin HNF4A Levodopa PPARA Gcg | Sirtuin signaling pathway, sphingosine-1-phosphate signaling | |
| 12 mo 3 Gy | Aryl hydrocarbon receptor signaling-DCT (dopachrome tautomerase) up, EP300, NCROR2, NFIA, NFIC, TP53 all down RAR activation-CYP26A1 up, ARID1A, EP300, NCOR2, ZBTB16 all down Iron homeostasis signaling pathway-ATP6V0C, EPAS1, IL6R all down, TFRC up Superpathway of cholesterol biosynthesis-DHCR7 down, FDPS up Hereditary breast cancer signaling-ARID1A, EP300, TP53 all down, UBC (ubiquitin) up | Diethylnitrosamine ACOX1 Hydrogen peroxide Pirinixic acid TAS-103 | PPARalpha/RXRalpha-BCL3, EP300, NCOR2 all down, Cyp2c54 up, sumoylation pathway | Tight Junction Signaling RhoGDI Signaling Huntington’s Disease Signaling Mechanisms of Viral Exit from Host cells LXR/RXR activation | HNF4A CST5 SREBF1 D-glucose IPMK |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barnette, B.L.; Yu, Y.; Ullrich, R.L.; Emmett, M.R. Mitochondrial Effects in the Liver of C57BL/6 Mice by Low Dose, High Energy, High Charge Irradiation. Int. J. Mol. Sci. 2021, 22, 11806. https://doi.org/10.3390/ijms222111806
Barnette BL, Yu Y, Ullrich RL, Emmett MR. Mitochondrial Effects in the Liver of C57BL/6 Mice by Low Dose, High Energy, High Charge Irradiation. International Journal of Molecular Sciences. 2021; 22(21):11806. https://doi.org/10.3390/ijms222111806
Chicago/Turabian StyleBarnette, Brooke L., Yongjia Yu, Robert L. Ullrich, and Mark R. Emmett. 2021. "Mitochondrial Effects in the Liver of C57BL/6 Mice by Low Dose, High Energy, High Charge Irradiation" International Journal of Molecular Sciences 22, no. 21: 11806. https://doi.org/10.3390/ijms222111806
APA StyleBarnette, B. L., Yu, Y., Ullrich, R. L., & Emmett, M. R. (2021). Mitochondrial Effects in the Liver of C57BL/6 Mice by Low Dose, High Energy, High Charge Irradiation. International Journal of Molecular Sciences, 22(21), 11806. https://doi.org/10.3390/ijms222111806