
Latently KSHV-Infected Cells Promote Further Establishment of Latency upon Superinfection with KSHV



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
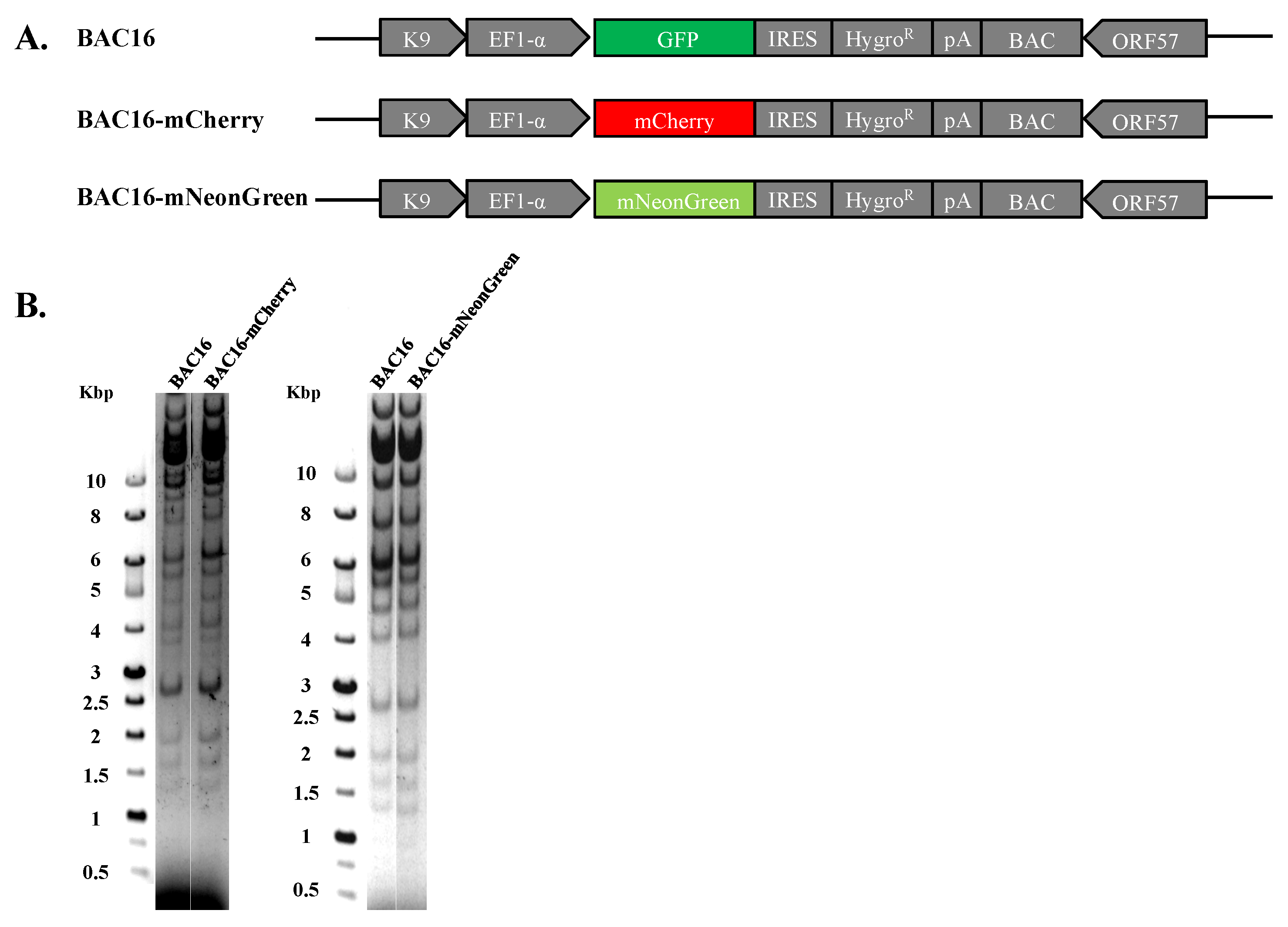
2.1. Construction of BAC16-mCherry and BAC16-mNeonGreen Recombinant Viruses
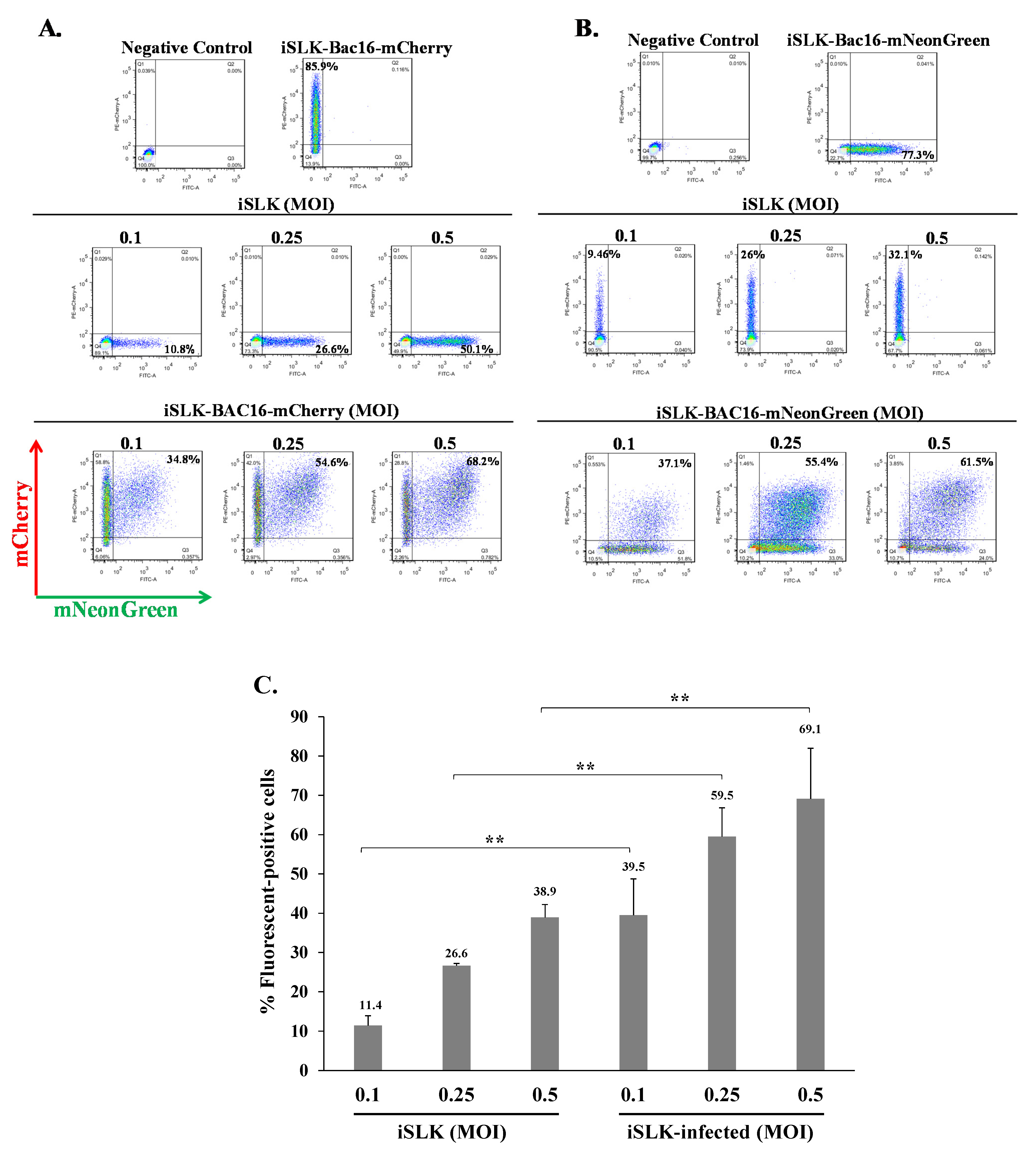
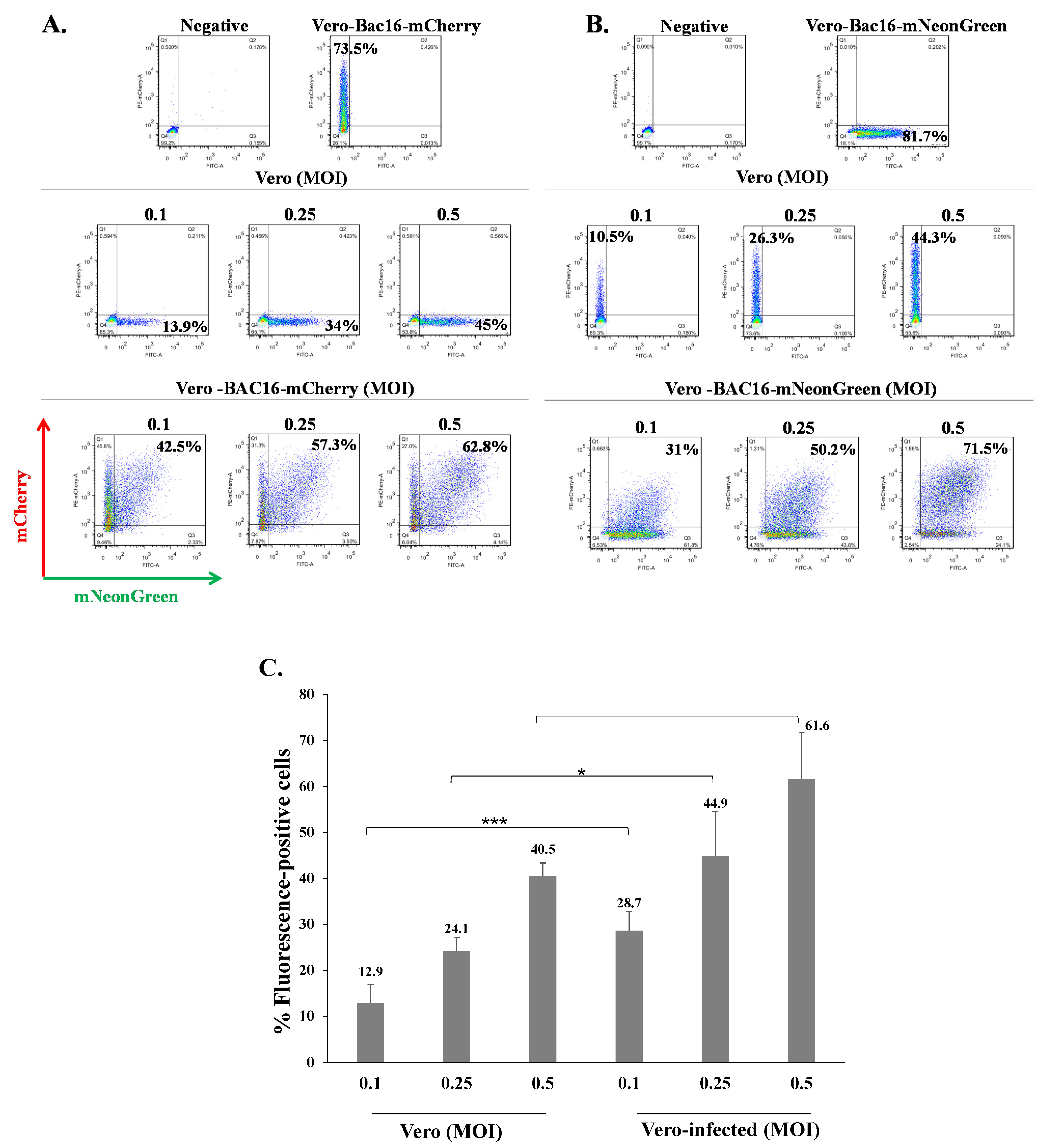
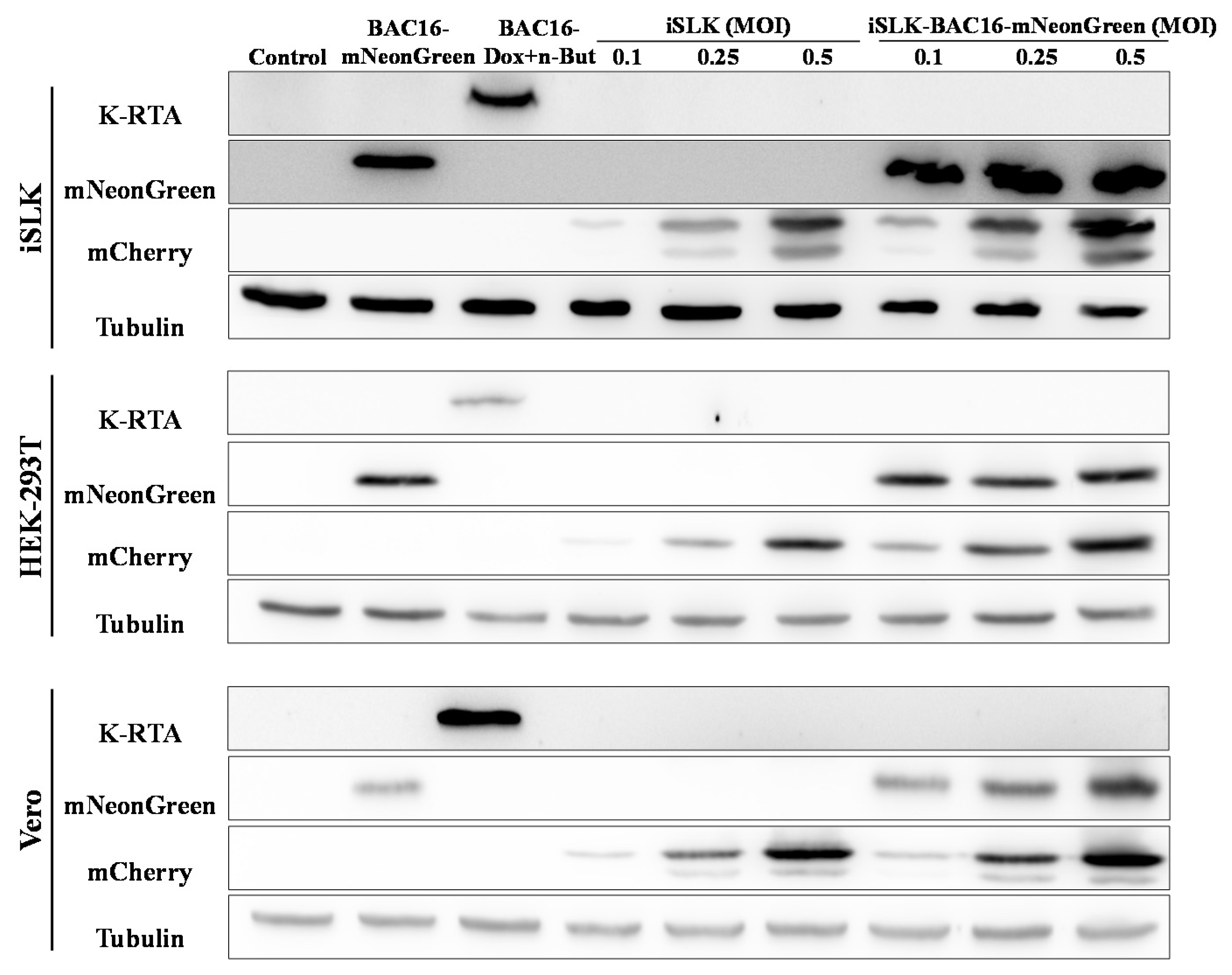
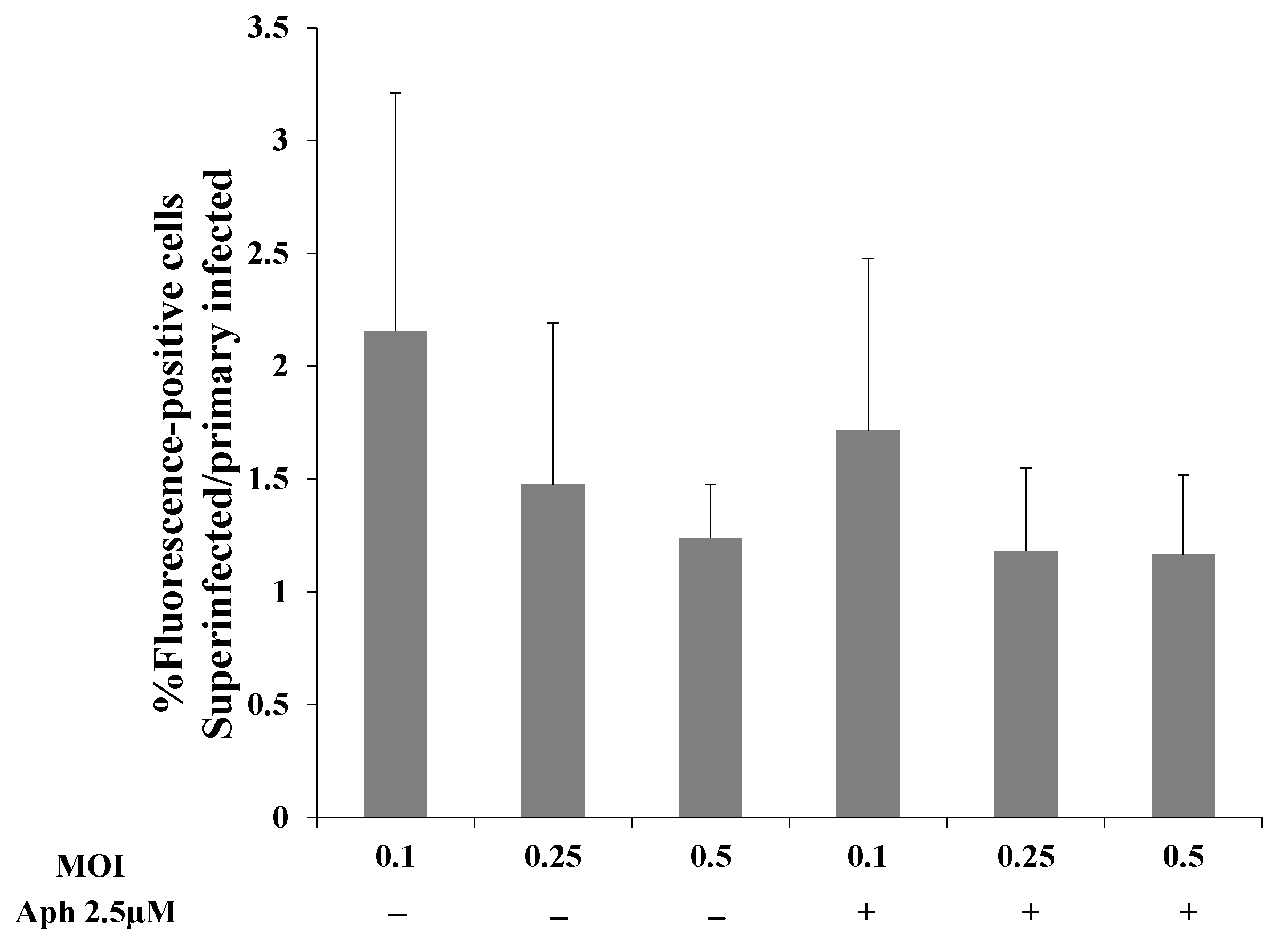
2.2. Latently KSHV-Infected Cells Permit Superinfection and Display Enhanced Expression of the Fluorescent Marker of the Incoming Virions
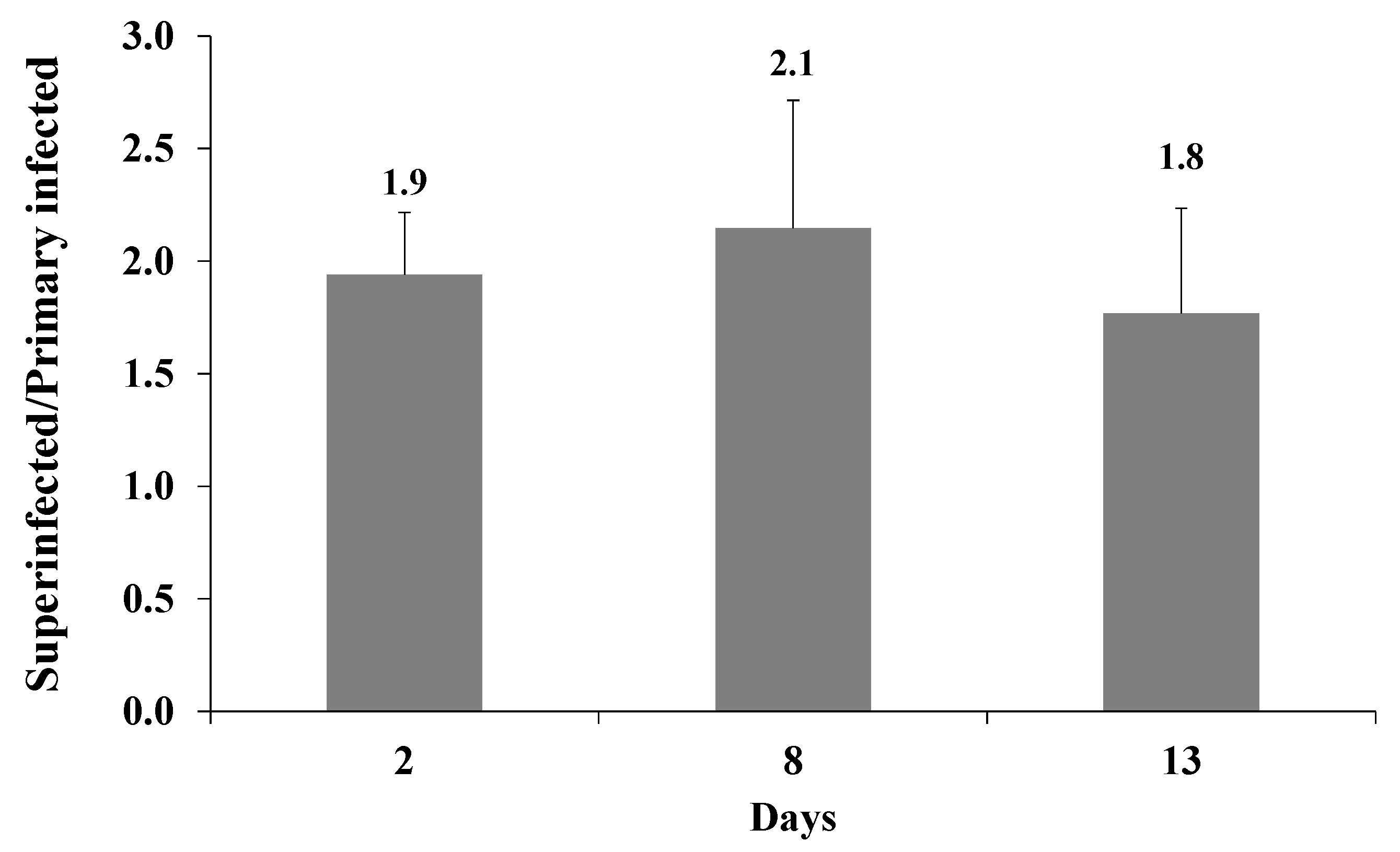
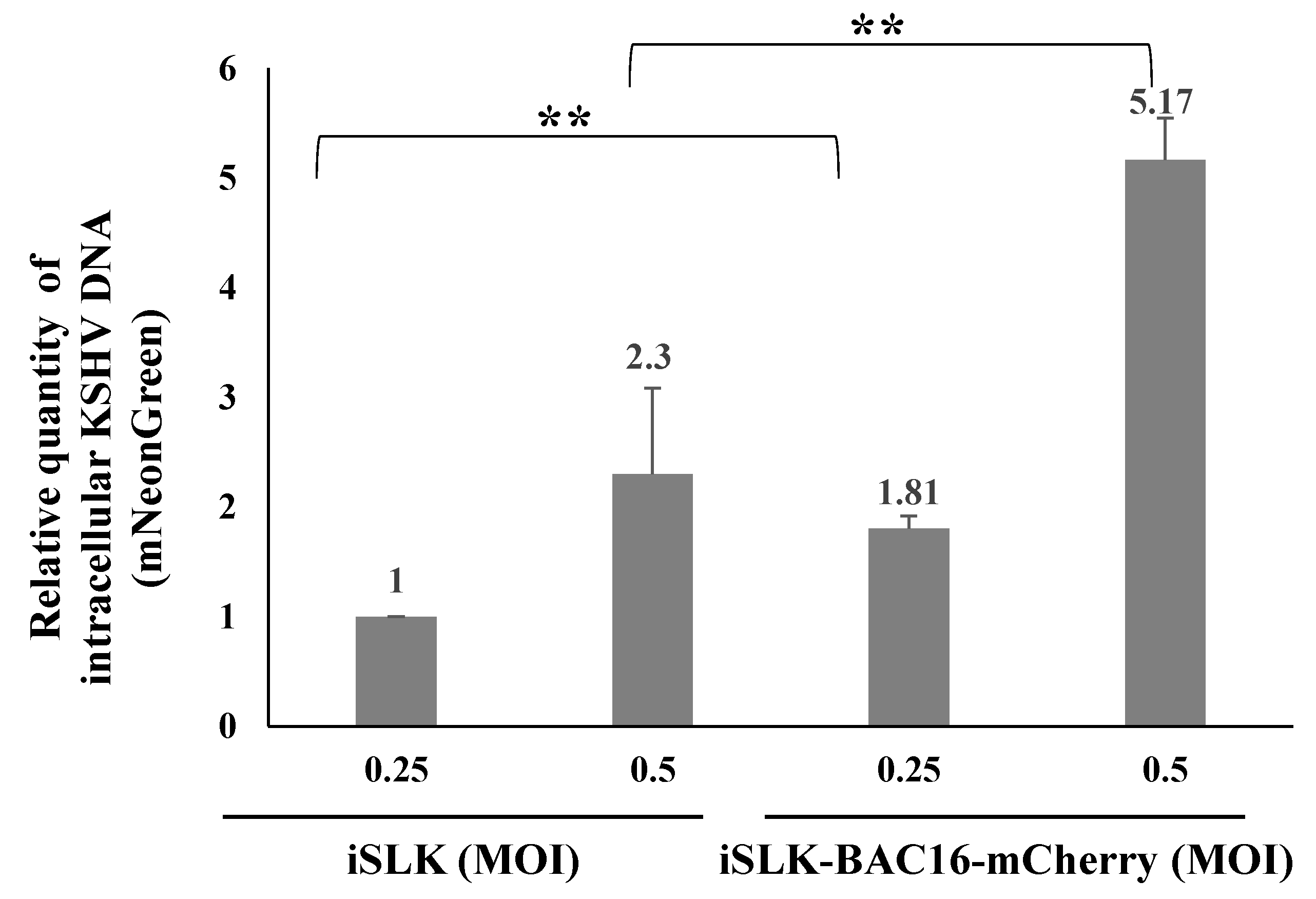
2.3. Latently KSHV-Infected Cells That Were Superinfected Contain Higher Levels of the Incoming Viral DNA Compared to Primary Infected Cells
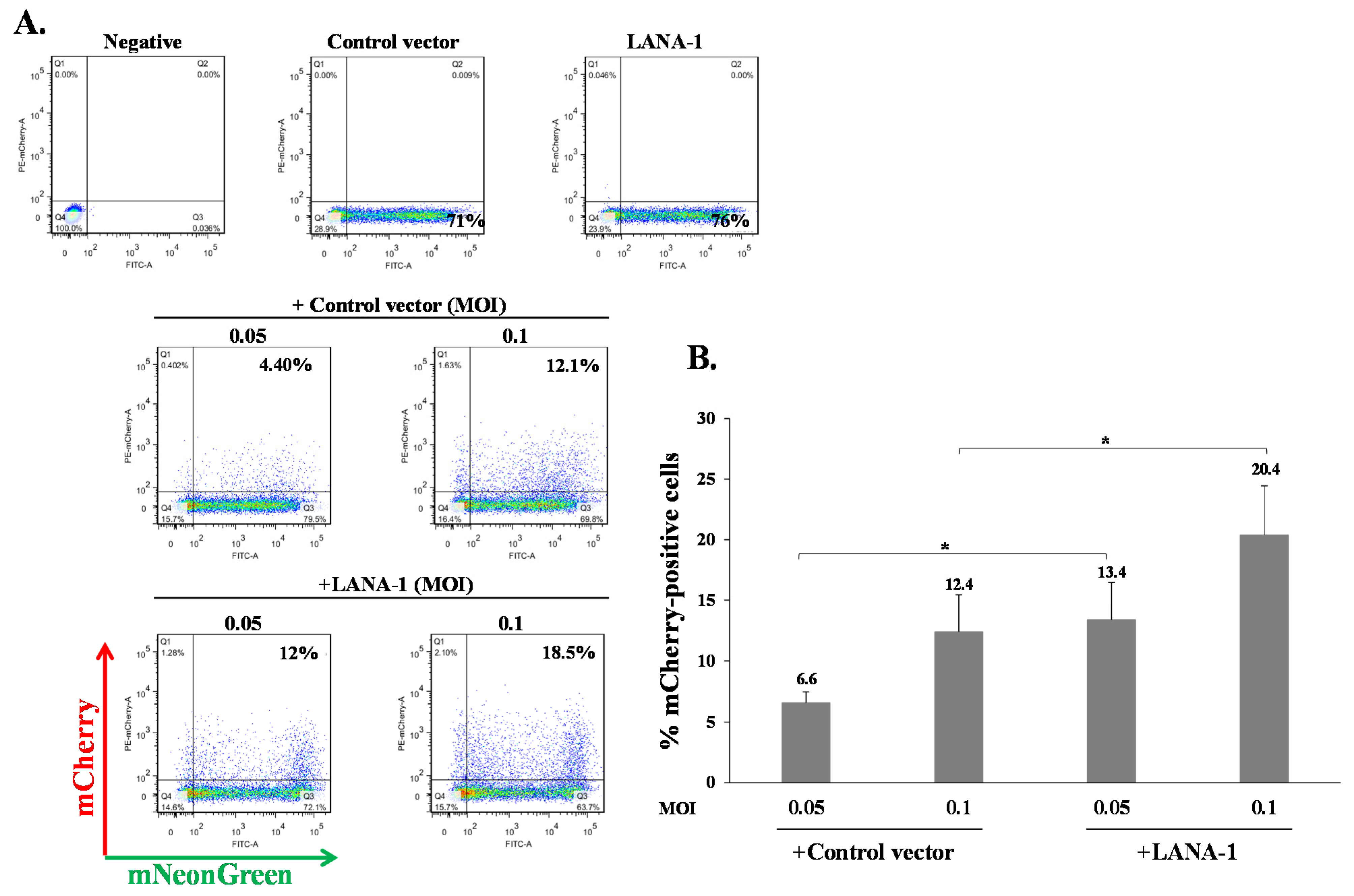
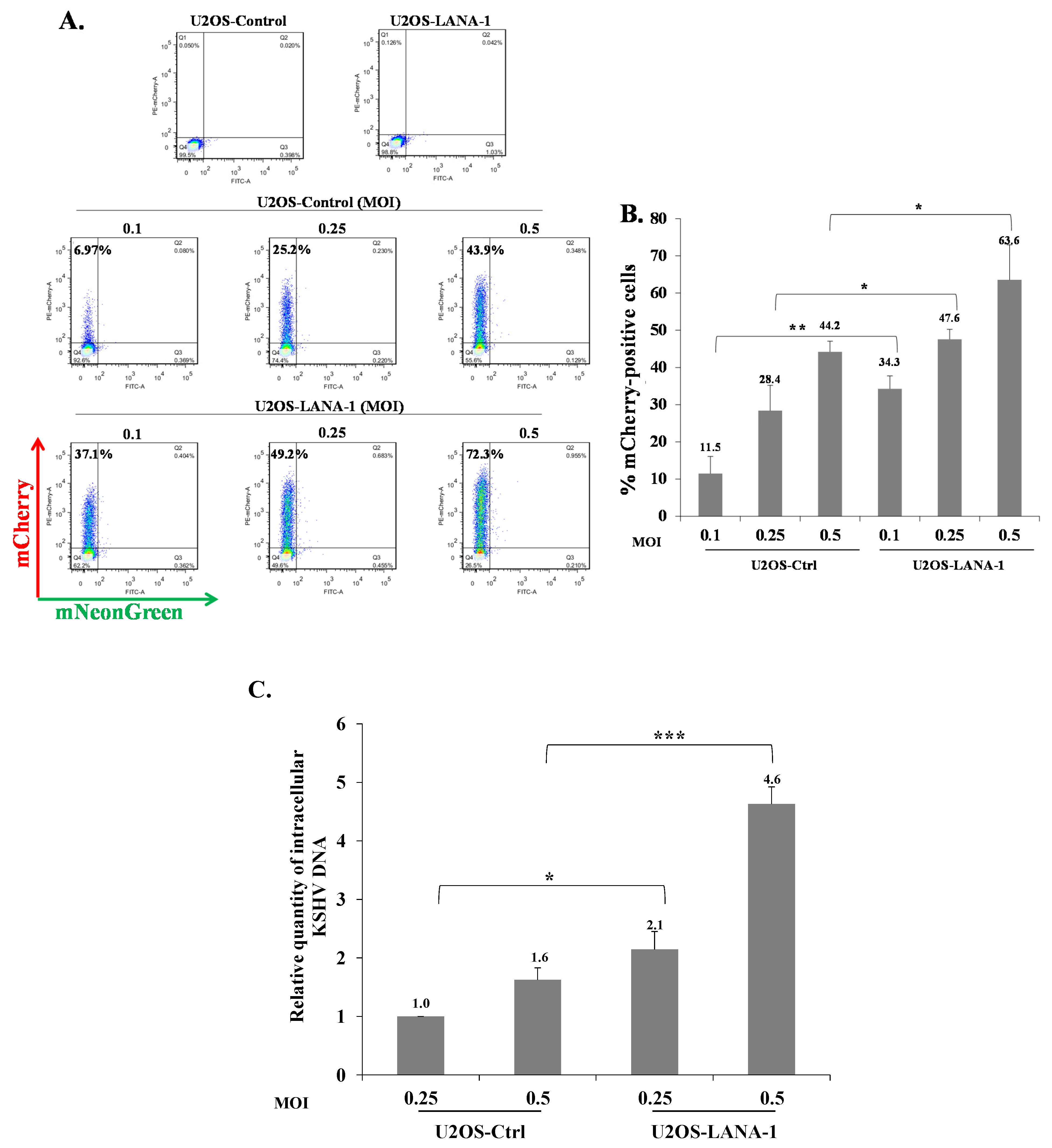
2.4. Increased Latency Establishment in Cells Expressing LANA-1
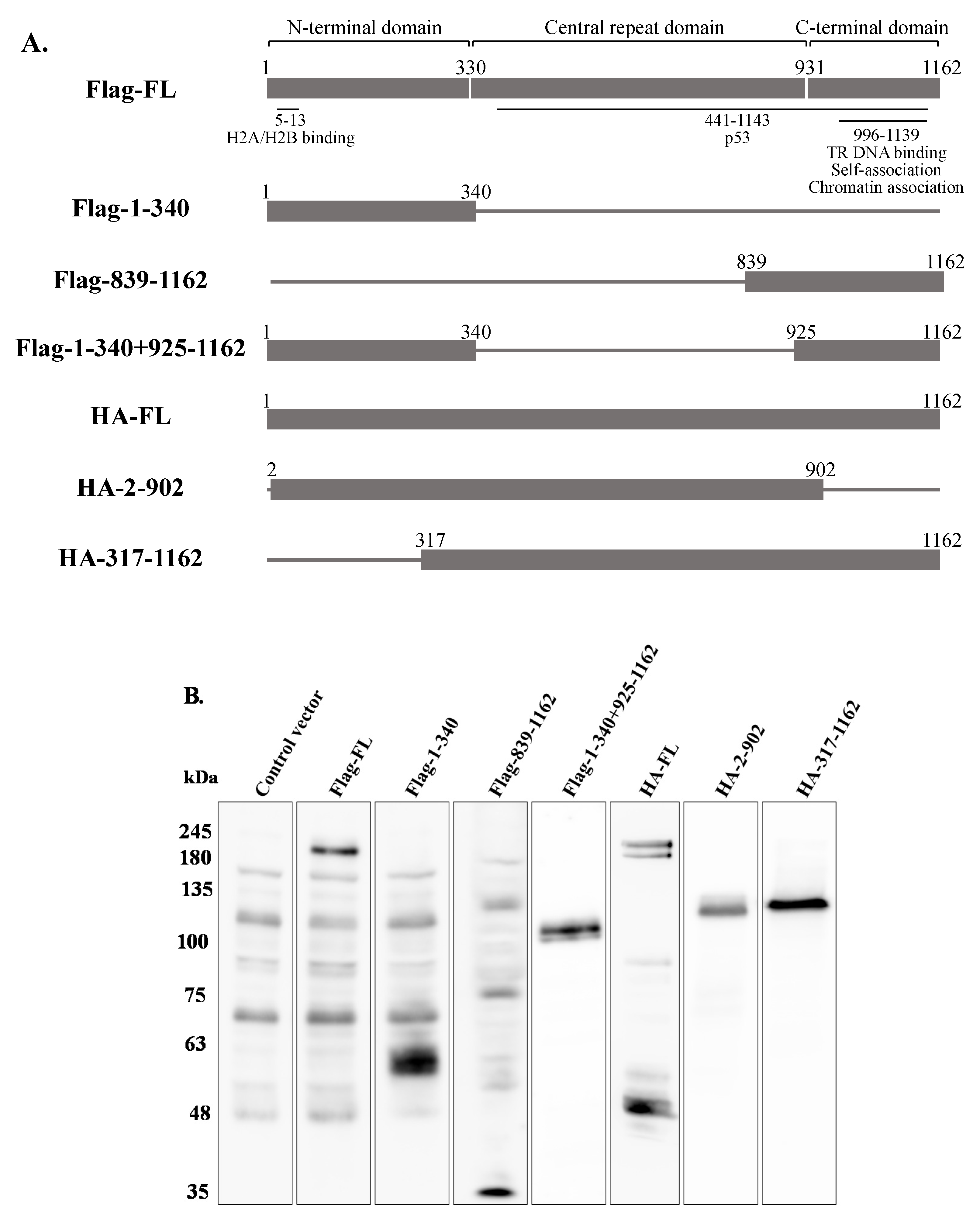
2.5. Full-Length LANA-1 Is Required to Promote Latency Establishment of KSHV
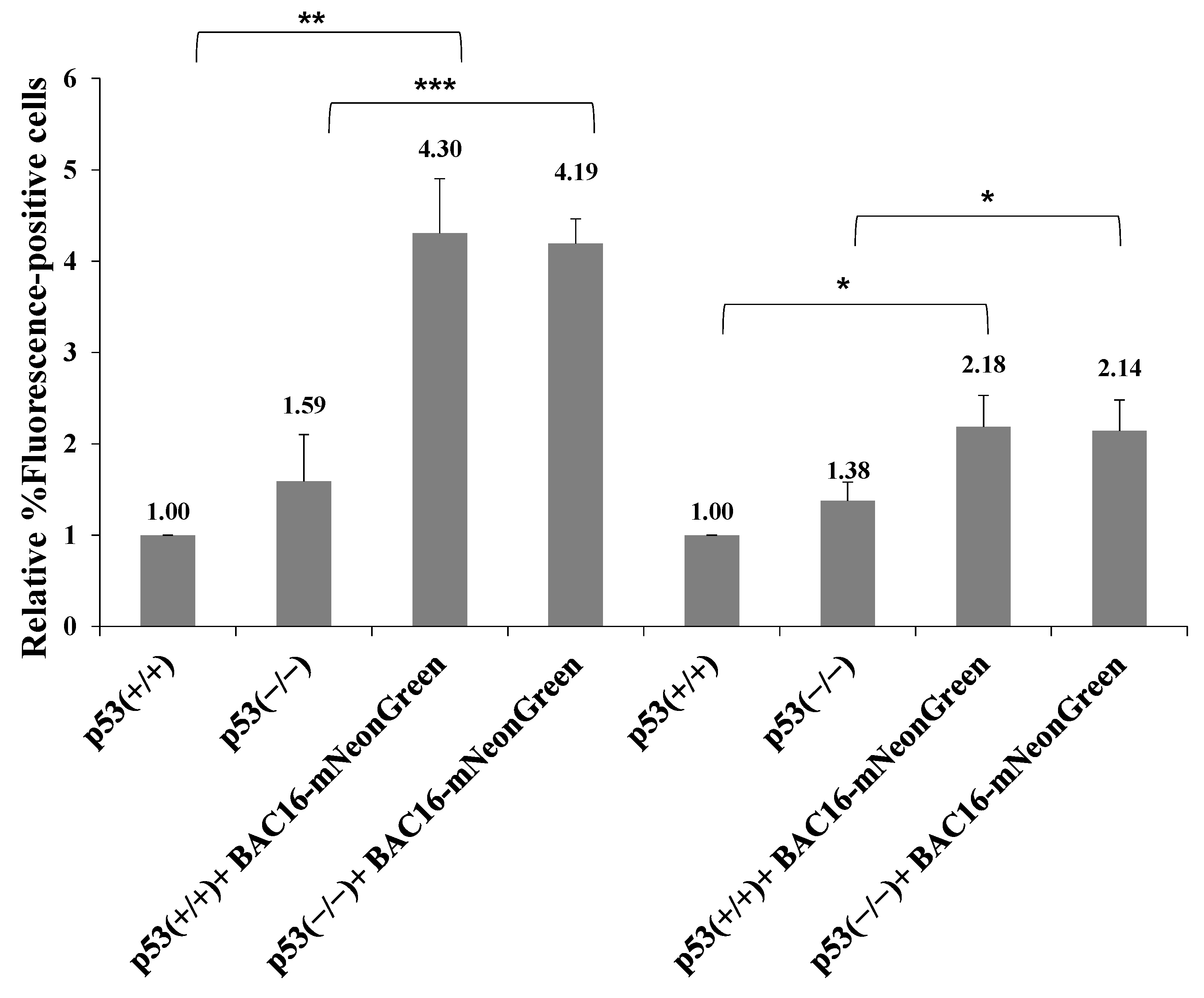
2.6. p53 Protein Does Not Affect Latency Establishment of KSHV during Superinfection
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Construction of BAC16-mNeonGreen and BAC16-mCherry Recombinant Viruses
4.3. Transfection of KSHV BAC16 DNA, Virus Reconstitution, Titration and Infection
4.4. Fluorescence-Activated Cell Sorting (FACS) Analysis
4.5. Quantitative TaqMan Real-Time Polymerase Chain Reaction
4.6. Antibodies and Western Blot Analysis
4.7. Plasmids and Transfections
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Polizzotto, M.N.; Uldrick, T.S.; Hu, D.; Yarchoan, R. Clinical Manifestations of Kaposi Sarcoma Herpesvirus Lytic Activation: Multicentric Castleman Disease (KSHV-MCD) and the KSHV Inflammatory Cytokine Syndrome. Front. Microbiol. 2012, 3, 73. [Google Scholar] [CrossRef] [Green Version]
- Kalt, I.; Masa, S.R.; Sarid, R. Linking the Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) to human malignancies. Methods Mol. Biol. 2009, 471, 387–407. [Google Scholar] [CrossRef]
- Katano, H. Pathological Features of Kaposi’s Sarcoma-Associated Herpesvirus Infection. Adv. Exp. Med. Biol. 2018, 1045, 357–376. [Google Scholar] [CrossRef]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, P.H.; Ziegelbauer, J.; Uldrick, T.S.; Yarchoan, R. Kaposi sarcoma herpesvirus-associated cancers and related diseases. Curr. Opin. HIV AIDS 2017, 12, 47–56. [Google Scholar] [CrossRef]
- Gramolelli, S.; Schulz, T.F. The role of Kaposi Sarcoma-associated Herpesvirus in the pathogenesis of Kaposi Sarcoma. J. Pathol. 2014, 235, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Ganem, D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Investig. 2004, 113, 124–136. [Google Scholar] [PubMed] [Green Version]
- Aneja, K.K.; Yuan, Y. Reactivation and Lytic Replication of Kaposi’s Sarcoma-Associated Herpesvirus: An Update. Front. Microbiol. 2017, 8, 613. [Google Scholar] [CrossRef]
- Uppal, T.; Jha, H.C.; Verma, S.C.; Robertson, E.S. Chromatinization of the KSHV Genome During the KSHV Life Cycle. Cancers 2015, 7, 112–142. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.F.; Sugden, A.U.; Fox, K.; Hayes, M.; Sugden, B. Kaposi’s sarcoma-associated herpesvirus stably clusters its genomes across generations to maintain itself extrachromosomally. J. Cell. Biol. 2017, 216, 2745–2758. [Google Scholar] [CrossRef]
- Haddad, C.O.; Kalt, I.; Shovman, Y.; Xia, L.; Schlesinger, Y.; Sarid, R.; Parnas, O. Targeting the Kaposi’s sarcoma-associated herpesvirus genome with the CRISPR-Cas9 platform in latently infected cells. Virol. J. 2021, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Lee, H.R.; Izumiya, Y.; Tepper, C.; Kung, H.J.; Jung, J.U. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog. 2013, 9, e1003813. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Wong, J.H.; Robertson, E.S. Herpesvirus Epigenetic Reprogramming and Oncogenesis. Annu. Rev. Virol. 2020, 7, 309–331. [Google Scholar] [CrossRef]
- Sun, R.; Tan, X.; Wang, X.; Wang, X.; Yang, L.; Robertson, E.S.; Lan, K. Epigenetic Landscape of Kaposi’s Sarcoma-Associated Herpesvirus Genome in Classic Kaposi’s Sarcoma Tissues. PLoS Pathog. 2017, 13, e1006167. [Google Scholar] [CrossRef] [Green Version]
- Uppal, T.; Banerjee, S.; Sun, Z.; Verma, S.C.; Robertson, E.S. KSHV LANA—The master regulator of KSHV latency. Viruses 2014, 6, 4961–4998. [Google Scholar] [CrossRef] [Green Version]
- De Leo, A.; Calderon, A.; Lieberman, P.M. Control of Viral Latency by Episome Maintenance Proteins. Trends Microbiol. 2020, 28, 150–162. [Google Scholar] [CrossRef]
- Folimonova, S.Y. Superinfection exclusion is an active virus-controlled function that requires a specific viral protein. J. Virol. 2012, 86, 5554–5561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumby, N.; Davidson, A.R.; Maxwell, K.L. The moron comes of age. Bacteriophage 2012, 2, 225–228. [Google Scholar] [CrossRef]
- Huang, I.C.; Li, W.; Sui, J.; Marasco, W.; Choe, H.; Farzan, M. Influenza A virus neuraminidase limits viral superinfection. J. Virol. 2008, 82, 4834–4843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doceul, V.; Hollinshead, M.; van der Linden, L.; Smith, G.L. Repulsion of superinfecting virions: A mechanism for rapid virus spread. Science 2010, 327, 873–876. [Google Scholar] [CrossRef] [Green Version]
- Schaller, T.; Appel, N.; Koutsoudakis, G.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J. Virol. 2007, 81, 4591–4603. [Google Scholar] [CrossRef] [Green Version]
- Tscherne, D.M.; Evans, M.J.; von Hahn, T.; Jones, C.T.; Stamataki, Z.; McKeating, J.A.; Lindenbach, B.D.; Rice, C.M. Superinfection exclusion in cells infected with hepatitis C virus. J. Virol. 2007, 81, 3693–3703. [Google Scholar] [CrossRef] [Green Version]
- Zou, G.; Zhang, B.; Lim, P.Y.; Yuan, Z.; Bernard, K.A.; Shi, P.Y. Exclusion of West Nile virus superinfection through RNA replication. J. Virol. 2009, 83, 11765–11776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpf, A.R.; Lenches, E.; Strauss, E.G.; Strauss, J.H.; Brown, D.T. Superinfection exclusion of alphaviruses in three mosquito cell lines persistently infected with Sindbis virus. J. Virol. 1997, 71, 7119–7123. [Google Scholar] [CrossRef] [Green Version]
- Biryukov, J.; Meyers, C. Superinfection Exclusion between Two High-Risk Human Papillomavirus Types during a Coinfection. J. Virol. 2018, 92, e01993-17. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.M.; Spear, P.G. Herpes simplex virus glycoprotein D mediates interference with herpes simplex virus infection. J. Virol. 1989, 63, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G.; Qi, S.; Avitabile, E.; Foa-Tomasi, L.; Brandimarti, R.; Roizman, B. Glycoprotein D of herpes simplex virus encodes a domain which precludes penetration of cells expressing the glycoprotein by superinfecting herpes simplex virus. J. Virol. 1990, 64, 6070–6079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloutskin, A.; Yee, M.B.; Kinchington, P.R.; Goldstein, R.S. Varicella-zoster virus and herpes simplex virus 1 can infect and replicate in the same neurons whether co- or superinfected. J. Virol. 2014, 88, 5079–5086. [Google Scholar] [CrossRef] [Green Version]
- Criddle, A.; Thornburg, T.; Kochetkova, I.; DePartee, M.; Taylor, M.P. gD-Independent Superinfection Exclusion of Alphaherpesviruses. J. Virol. 2016, 90, 4049–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meignier, B.; Norrild, B.; Roizman, B. Colonization of murine ganglia by a superinfecting strain of herpes simplex virus. Infect. Immun. 1983, 41, 702–708. [Google Scholar] [CrossRef] [Green Version]
- Roest, R.W.; Carman, W.F.; Maertzdorf, J.; Scoular, A.; Harvey, J.; Kant, M.; van der Meijden, W.; Verjans, G.M.; Osterhaus, A. Genotypic analysis of sequential genital herpes simplex virus type 1 (HSV-1) isolates of patients with recurrent HSV-1 associated genital herpes. J. Med. Virol. 2004, 73, 601–604. [Google Scholar] [CrossRef]
- Breuer, J. VZV molecular epidemiology. Curr. Top. Microbiol. Immunol. 2010, 342, 15–42. [Google Scholar] [CrossRef] [PubMed]
- Szpara, M.L.; Gatherer, D.; Ochoa, A.; Greenbaum, B.; Dolan, A.; Bowden, R.J.; Enquist, L.W.; Legendre, M.; Davison, A.J. Evolution and diversity in human herpes simplex virus genomes. J. Virol. 2014, 88, 1209–1227. [Google Scholar] [CrossRef] [Green Version]
- Norberg, P.; Depledge, D.P.; Kundu, S.; Atkinson, C.; Brown, J.; Haque, T.; Hussaini, Y.; MacMahon, E.; Molyneaux, P.; Papaevangelou, V.; et al. Recombination of Globally Circulating Varicella-Zoster Virus. J. Virol. 2015, 89, 7133–7146. [Google Scholar] [CrossRef] [Green Version]
- Bowden, R.; Sakaoka, H.; Donnelly, P.; Ward, R. High recombination rate in herpes simplex virus type 1 natural populations suggests significant co-infection. Infect. Genet. Evol. 2004, 4, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Law, G.A.; Herr, A.E.; Cwick, J.P.; Taylor, M.P. A New Approach to Assessing HSV-1 Recombination during Intercellular Spread. Viruses 2018, 10, 220. [Google Scholar] [CrossRef] [Green Version]
- Tomer, E.; Cohen, E.M.; Drayman, N.; Afriat, A.; Weitzman, M.D.; Zaritsky, A.; Kobiler, O. Coalescing replication compartments provide the opportunity for recombination between coinfecting herpesviruses. FASEB J. 2019, 33, 9388–9403. [Google Scholar] [CrossRef]
- Sallah, N.; Palser, A.L.; Watson, S.J.; Labo, N.; Asiki, G.; Marshall, V.; Newton, R.; Whitby, D.; Kellam, P.; Barroso, I. Genome-Wide Sequence Analysis of Kaposi Sarcoma-Associated Herpesvirus Shows Diversification Driven by Recombination. J. Infect. Dis. 2018, 218, 1700–1710. [Google Scholar] [CrossRef] [Green Version]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS- related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [PubMed]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 2014, 9, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Calabro, M.L.; Sarid, R. Human Herpesvirus 8 and Lymphoproliferative Disorders. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018061. [Google Scholar] [CrossRef]
- Sanchez-Ponce, Y.; Fuentes-Panana, E.M. The Role of Coinfections in the EBV-Host Broken Equilibrium. Viruses 2021, 13, 1399. [Google Scholar] [CrossRef]
- McHugh, D.; Caduff, N.; Barros, M.H.M.; Ramer, P.C.; Raykova, A.; Murer, A.; Landtwing, V.; Quast, I.; Styles, C.; Spohn, M.; et al. Persistent KSHV Infection Increases EBV-Associated Tumor Formation In Vivo via Enhanced EBV Lytic Gene Expression. Cell Host Microbe 2017, 22, 61–73.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigi, R.; Landis, J.T.; An, H.; Caro-Vegas, C.; Raab-Traub, N.; Dittmer, D.P. Epstein-Barr virus enhances genome maintenance of Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 2018, 115, E11379–E11387. [Google Scholar] [CrossRef] [Green Version]
- Faure, A.; Hayes, M.; Sugden, B. How Kaposi’s sarcoma-associated herpesvirus stably transforms peripheral B cells towards lymphomagenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 16519–16528. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.J.; Zhang, Y.J.; Deng, J.H.; Rabkin, C.S.; Flore, O.; Jenson, H.B. Molecular Polymorphism of Kaposi’s Sarcoma-Associated Herpesvirus (Human Herpesvirus 8) Latent Nuclear Antigen: Evidence for a Large Repertoire of Viral Genotypes and Dual Infection with Different Viral Genotypes. J. Infect. Dis. 1999, 180, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Judde, J.G.; Lacoste, V.; Briere, J.; Kassa-Kelembho, E.; Clyti, E.; Couppie, P.; Buchrieser, C.; Tulliez, M.; Morvan, J.; Gessain, A.; et al. Monoclonality or oligoclonality of human herpesvirus 8 terminal repeat sequences in Kaposi’s sarcoma and other diseases. J. Natl. Cancer Inst. 2000, 92, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Cornejo Castro, E.M.; Marshall, V.; Lack, J.; Lurain, K.; Immonen, T.; Labo, N.; Fisher, N.C.; Ramaswami, R.; Polizzotto, M.N.; Keele, B.F.; et al. Dual infection and recombination of Kaposi sarcoma herpesvirus revealed by whole-genome sequence analysis of effusion samples. Virus Evol. 2020, 6, veaa047. [Google Scholar] [CrossRef] [PubMed]
- Beyari, M.M.; Hodgson, T.A.; Cook, R.D.; Kondowe, W.; Molyneux, E.M.; Scully, C.M.; Teo, C.G.; Porter, S.R. Multiple human herpesvirus-8 infection. J. Infect. Dis. 2003, 188, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Ariyoshi, K.; van der Loeff, M.S.; Cook, P.; Whitby, D.; Corrah, T.; Jaffarm, S.; Cham, F.; Sabally, S.; O’Donovan, D.; Weiss, R.A.; et al. Kaposi’s sarcoma in the Gambia, West Africa is less frequent in human immunodeficiency virus type 2 than in human immunodeficiency virus type 1 infection despite a high prevalence of human herpesvirus 8. J. Hum. Virol. 1998, 1, 193–199. [Google Scholar] [PubMed]
- Sculley, T.B.; Apolloni, A.; Hurren, L.; Moss, D.J.; Cooper, D.A. Coinfection with A- and B-type Epstein-Barr virus in human immunodeficiency virus-positive subjects. J. Infect. Dis. 1990, 162, 643–648. [Google Scholar] [CrossRef]
- Yao, Q.Y.; Rowe, M.; Martin, B.; Young, L.S.; Rickinson, A.B. The Epstein-Barr virus carrier state: Dominance of a single growth-transforming isolate in the blood and in the oropharynx of healthy virus carriers. J. Gen. Virol. 1991, 72, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, A.; Sculley, T.B. Detection of A-type and B-type Epstein-Barr virus in throat washings and lymphocytes. Virology 1994, 202, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Brulois, K.F.; Chang, H.; Lee, A.S.; Ensser, A.; Wong, L.Y.; Toth, Z.; Lee, S.H.; Lee, H.-R.; Myoung, J.; Ganem, D.; et al. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 2012, 86, 9708–9720. [Google Scholar] [CrossRef] [Green Version]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [CrossRef]
- Myoung, J.; Ganem, D. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: Maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 2011, 174, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Mosca, J.D.; Pitha, P.M. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol. Cell. Biol. 1986, 6, 2279–2283. [Google Scholar] [CrossRef] [PubMed]
- Desmyter, J.; Melnick, J.L.; Rawls, W.E. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 1968, 2, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.C.; Miley, W.; Waters, D. A quantification of human cells using an ERV-3 real time PCR assay. J. Virol. Methods 2001, 91, 109–117. [Google Scholar]
- Baranovskiy, A.G.; Babayeva, N.D.; Suwa, Y.; Gu, J.; Pavlov, Y.I.; Tahirov, T.H. Structural basis for inhibition of DNA replication by aphidicolin. Nucleic Acids Res. 2014, 42, 14013–14021. [Google Scholar] [CrossRef] [Green Version]
- Pedrali-Noy, G.; Spadari, S. Mechanism of inhibition of herpes simplex virus and vaccinia virus DNA polymerases by aphidicolin, a highly specific inhibitor of DNA replication in eucaryotes. J. Virol. 1980, 36, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, A.; Wraak, M.; Oberg, B. Effect of aphidicolin on DNA synthesis in HSV-1 infected and uninfected Vero cells. Antivir. Res. 1983, 3, 87–91. [Google Scholar] [CrossRef]
- Shamay, M.; Liu, J.; Li, R.; Liao, G.; Shen, L.; Greenway, M.; Hu, S.; Zhu, J.; Xie, Z.; Ambinder, R.F.; et al. A protein array screen for Kaposi’s sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J. Virol. 2012, 86, 5179–5191. [Google Scholar] [CrossRef] [Green Version]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Xiao, B.; Jha, H.C.; Lu, J.; Banerjee, S.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded LANA can induce chromosomal instability through targeted degradation of the mitotic checkpoint kinase Bub1. J. Virol. 2014, 88, 7367–7378. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Gan, J.; Wang, C.; Zhu, C.; Cai, Q. Cell Cycle Regulatory Functions of the KSHV Oncoprotein LANA. Front. Microbiol. 2016, 7, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.L.; Knight, J.S.; Verma, S.C.; Zald, P.; Robertson, E.S. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2006, 2, e116. [Google Scholar] [CrossRef] [PubMed]
- Gelgor, A.; Gam Ze Letova, C.; Yegorov, Y.; Kalt, I.; Sarid, R. Nucleolar stress enhances lytic reactivation of the Kaposi’s sarcoma-associated herpesvirus. Oncotarget 2018, 9, 13822–13833. [Google Scholar] [CrossRef] [Green Version]
- Ellison, T.J.; Kedes, D.H. Variable episomal silencing of a recombinant herpesvirus renders its encoded GFP an unreliable marker of infection in primary cells. PLoS ONE 2014, 9, e111502. [Google Scholar] [CrossRef]
- Purushothaman, P.; Thakker, S.; Verma, S.C. Transcriptome analysis of Kaposi’s sarcoma-associated herpesvirus during de novo primary infection of human B and endothelial cells. J. Virol. 2015, 89, 3093–3111. [Google Scholar] [CrossRef] [Green Version]
- Vazquez Ede, L.; Carey, V.J.; Kaye, K.M. Identification of Kaposi’s sarcoma-associated herpesvirus LANA regions important for episome segregation, replication, and persistence. J. Virol. 2013, 87, 12270–12283. [Google Scholar] [CrossRef] [Green Version]
- Juillard, F.; Tan, M.; Li, S.; Kaye, K.M. Kaposi’s Sarcoma Herpesvirus Genome Persistence. Front. Microbiol. 2016, 7, 1149. [Google Scholar] [CrossRef]
- Verma, S.C.; Lan, K.; Robertson, E. Structure and function of latency-associated nuclear antigen. Curr. Top. Microbiol. Immunol. 2007, 312, 101–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbera, A.J.; Chodaparambil, J.V.; Kelley-Clarke, B.; Joukov, V.; Walter, J.C.; Luger, K.; Kaye, K.M. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 2006, 311, 856–861. [Google Scholar] [CrossRef]
- Kelley-Clarke, B.; De Leon-Vazquez, E.; Slain, K.; Barbera, A.J.; Kaye, K.M. Role of Kaposi’s sarcoma-associated herpesvirus C-terminal LANA chromosome binding in episome persistence. J. Virol. 2009, 83, 4326–4337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballestas, M.E.; Kaye, K.M. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi’s sarcoma-associated herpesvirus latency. Future Microbiol. 2011, 6, 1399–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.C.; Choudhuri, T.; Robertson, E.S. The minimal replicator element of the Kaposi’s sarcoma-associated herpesvirus terminal repeat supports replication in a semiconservative and cell-cycle-dependent manner. J. Virol. 2007, 81, 3402–3413. [Google Scholar] [CrossRef] [Green Version]
- Hellert, J.; Weidner-Glunde, M.; Krausze, J.; Lunsdorf, H.; Ritter, C.; Schulz, T.F.; Lührs, T. The 3D structure of Kaposi sarcoma herpesvirus LANA C-terminal domain bound to DNA. Proc. Natl. Acad. Sci. USA 2015, 112, 6694–6699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, S.; Persson, L.M.; Wong, L.; Wilson, A.C. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J. Virol. 2010, 84, 2318–2330. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.C.; Choudhuri, T.; Kaul, R.; Robertson, E.S. Latency-associated nuclear antigen (LANA) of Kaposi’s sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 2006, 80, 2243–2256. [Google Scholar] [CrossRef] [Green Version]
- Ottinger, M.; Christalla, T.; Nathan, K.; Brinkmann, M.M.; Viejo-Borbolla, A.; Schulz, T.F. Kaposi’s sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 2006, 80, 10772–10786. [Google Scholar] [CrossRef] [Green Version]
- Domsic, J.F.; Chen, H.S.; Lu, F.; Marmorstein, R.; Lieberman, P.M. Molecular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA. PLoS Pathog. 2013, 9, e1003672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Leo, A.; Deng, Z.; Vladimirova, O.; Chen, H.S.; Dheekollu, J.; Calderon, A.; Myers, K.A.; Hayden, J.; Keeney, F.; Kaufer, B.B.; et al. LANA oligomeric architecture is essential for KSHV nuclear body formation and viral genome maintenance during latency. PLoS Pathog. 2019, 15, e1007489. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, P.; Sugden, B. Identification of properties of the Kaposi’s sarcoma-associated herpesvirus latent origin of replication that are essential for the efficient establishment and maintenance of intact plasmids. J. Virol. 2014, 88, 8490–8503. [Google Scholar] [CrossRef] [Green Version]
- Boulanger, E.; Duprez, R.; Delabesse, E.; Gabarre, J.; Macintyre, E.; Gessain, A. Mono/oligoclonal pattern of Kaposi Sarcoma-associated herpesvirus (KSHV/HHV-8) episomes in primary effusion lymphoma cells. Int. J. Cancer 2005, 115, 511–518. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Gelgor, A.; Kalt, I.; Bergson, S.; Brulois, K.F.; Jung, J.U.; Sarid, R. Viral Bcl-2 Encoded by the Kaposi’s Sarcoma-Associated Herpesvirus Is Vital for Virus Reactivation. J. Virol. 2015, 89, 5298–5307. [Google Scholar] [CrossRef] [Green Version]
- Bergson, S.; Kalt, I.; Itzhak, I.; Brulois, K.F.; Jung, J.U.; Sarid, R. Fluorescent tagging and cellular distribution of the Kaposi’s sarcoma-associated herpesvirus ORF45 tegument protein. J. Virol. 2014, 88, 12839–12852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krithivas, A.; Young, D.B.; Liao, G.; Greene, D.; Hayward, S.D. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 2000, 74, 9637–9645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimuro, M.; Hayward, S.D. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J. Virol. 2003, 77, 8019–8030. [Google Scholar] [CrossRef] [PubMed] [Green Version]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gam ze Letova, C.; Kalt, I.; Shamay, M.; Sarid, R. Latently KSHV-Infected Cells Promote Further Establishment of Latency upon Superinfection with KSHV. Int. J. Mol. Sci. 2021, 22, 11994. https://doi.org/10.3390/ijms222111994
Gam ze Letova C, Kalt I, Shamay M, Sarid R. Latently KSHV-Infected Cells Promote Further Establishment of Latency upon Superinfection with KSHV. International Journal of Molecular Sciences. 2021; 22(21):11994. https://doi.org/10.3390/ijms222111994
Chicago/Turabian StyleGam ze Letova, Chen, Inna Kalt, Meir Shamay, and Ronit Sarid. 2021. "Latently KSHV-Infected Cells Promote Further Establishment of Latency upon Superinfection with KSHV" International Journal of Molecular Sciences 22, no. 21: 11994. https://doi.org/10.3390/ijms222111994
APA StyleGam ze Letova, C., Kalt, I., Shamay, M., & Sarid, R. (2021). Latently KSHV-Infected Cells Promote Further Establishment of Latency upon Superinfection with KSHV. International Journal of Molecular Sciences, 22(21), 11994. https://doi.org/10.3390/ijms222111994