
Clioquinol Decreases Levels of Phosphorylated, Truncated, and Oligomerized Tau Protein

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
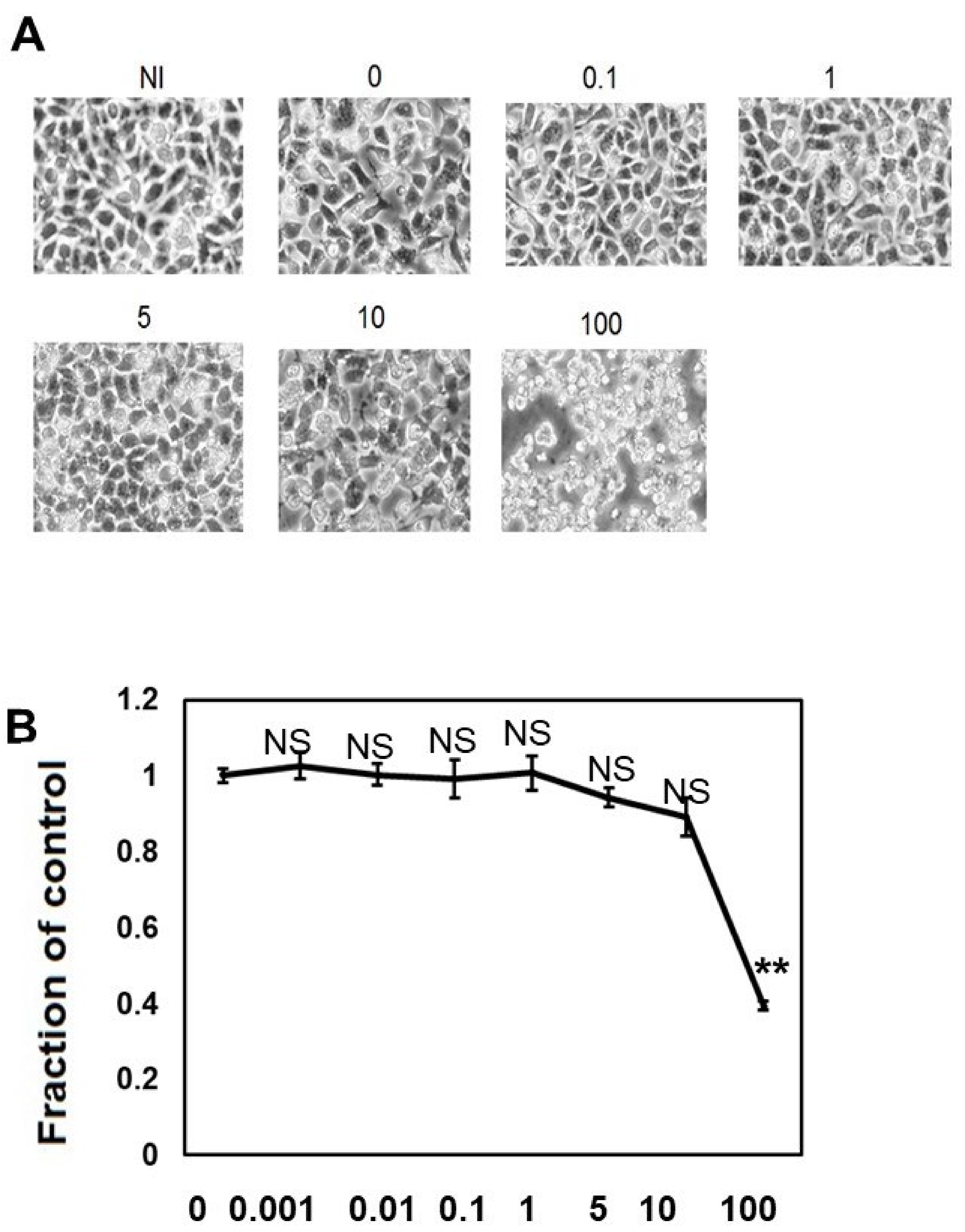
2.1. Up to 10 μM CQ Did Not Exhibit Cell Toxicity in M1C Cells
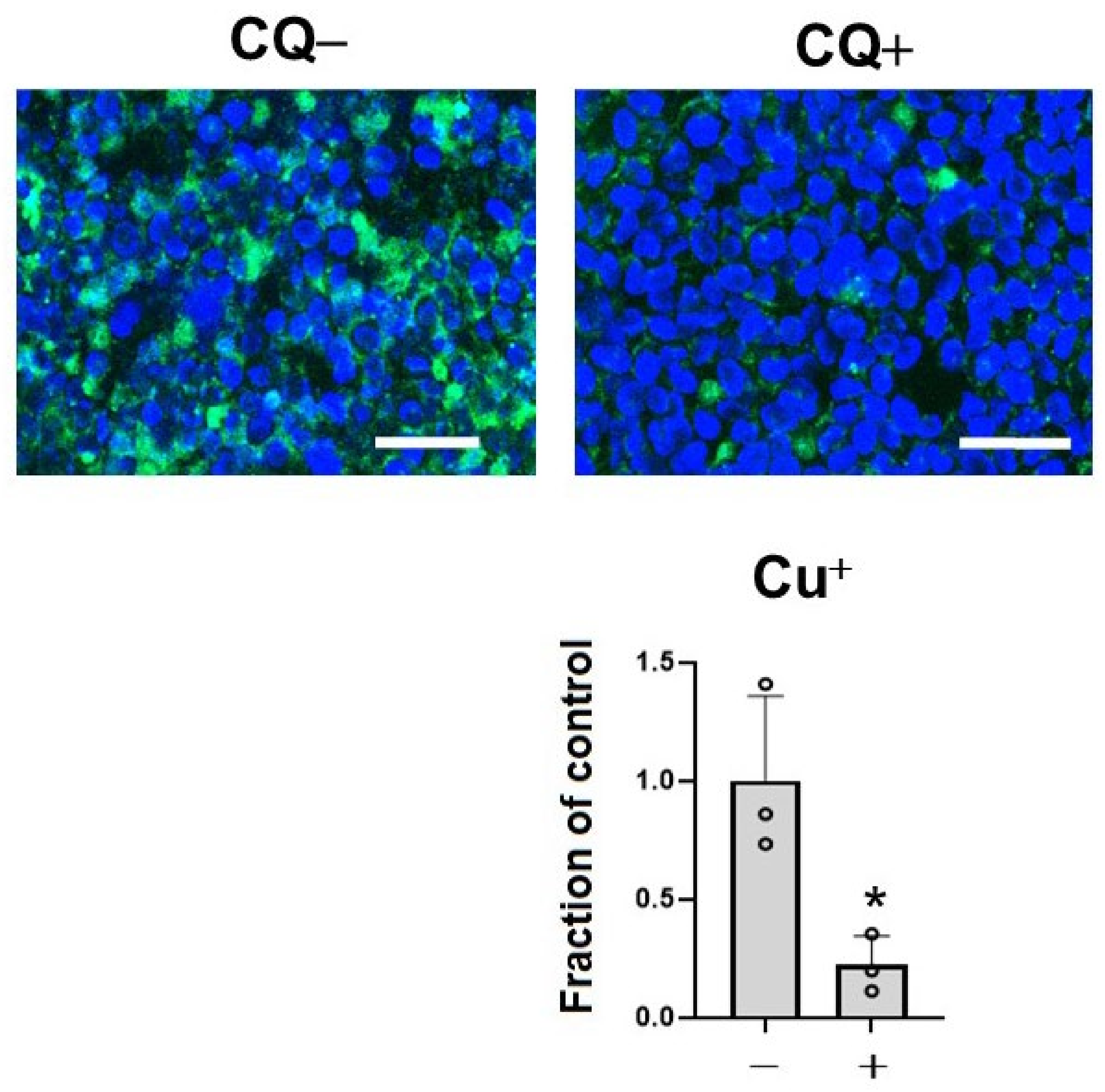
2.2. CQ Decreased Cu+ Ions in the Cell
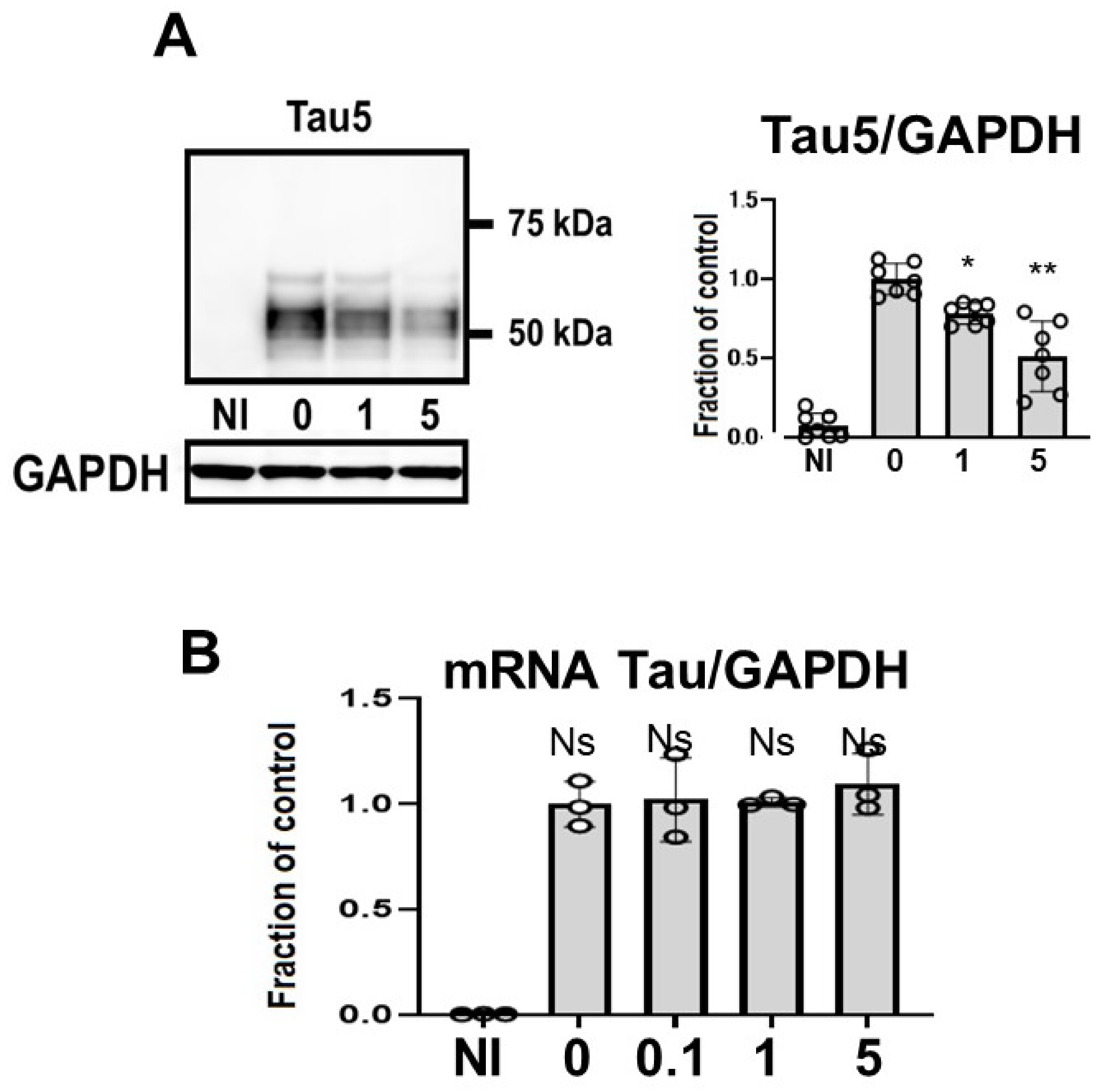
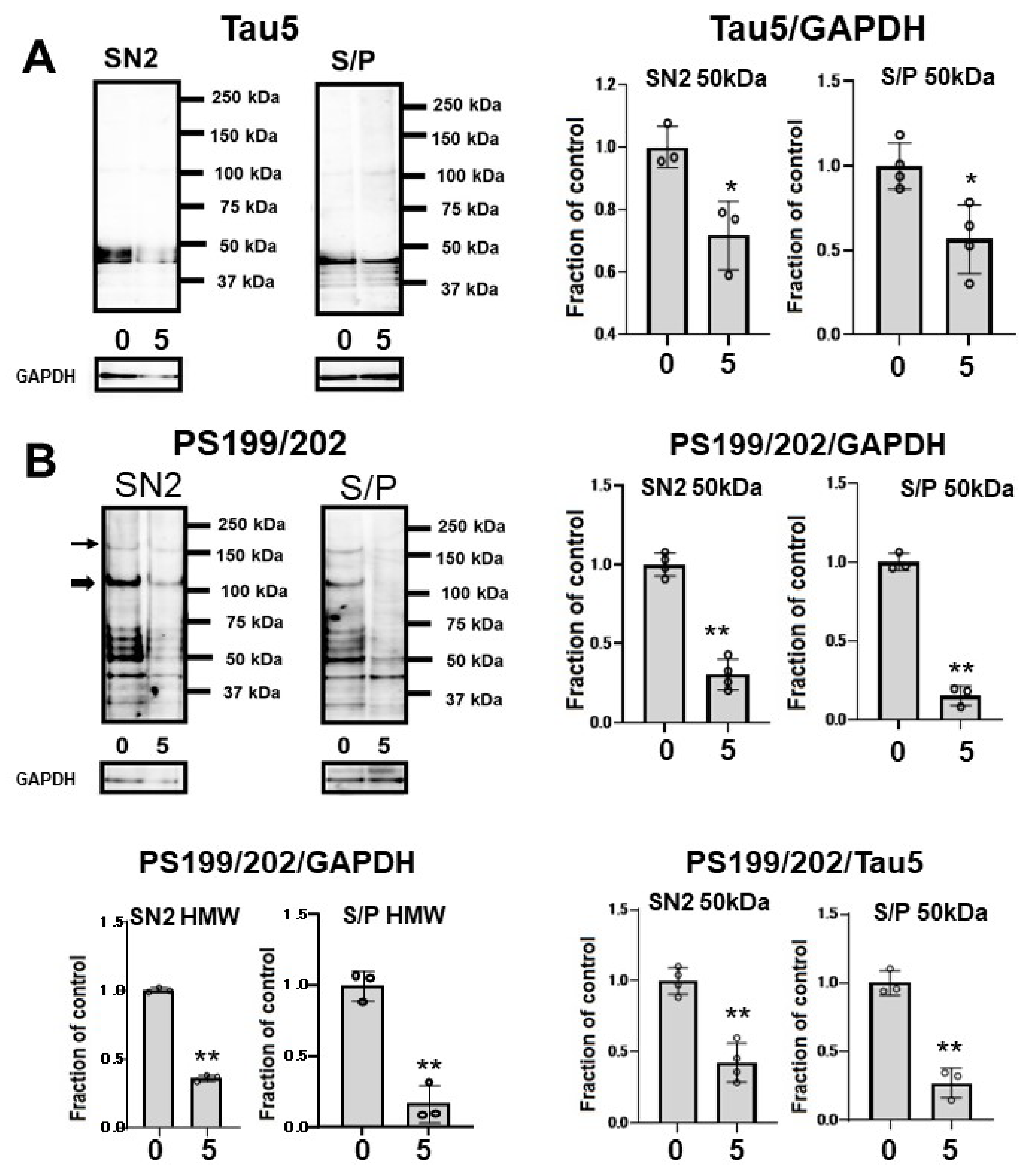
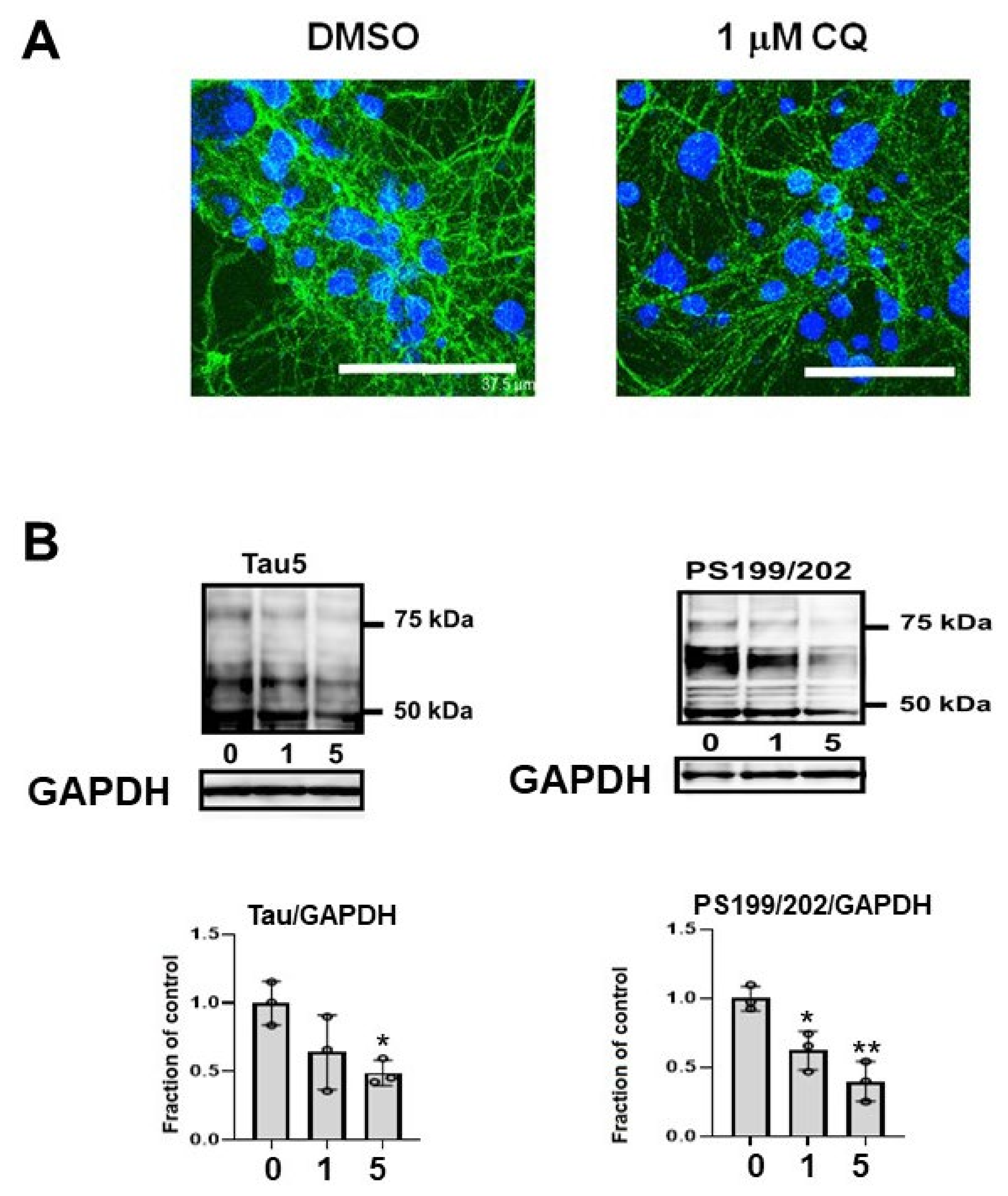
2.3. CQ Decreased the Tau Levels Detected Using Non-Phospho Dependent Tau5
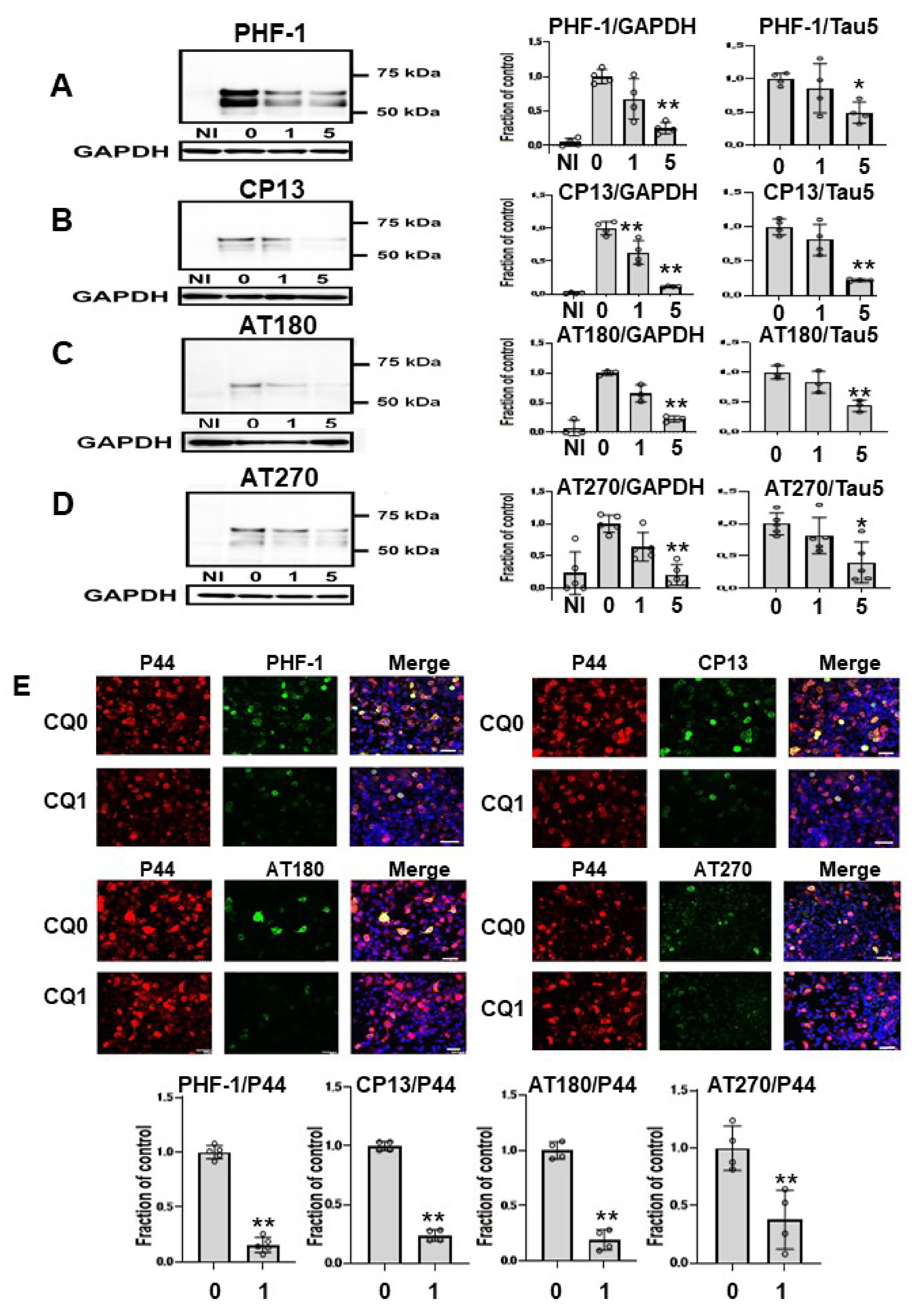
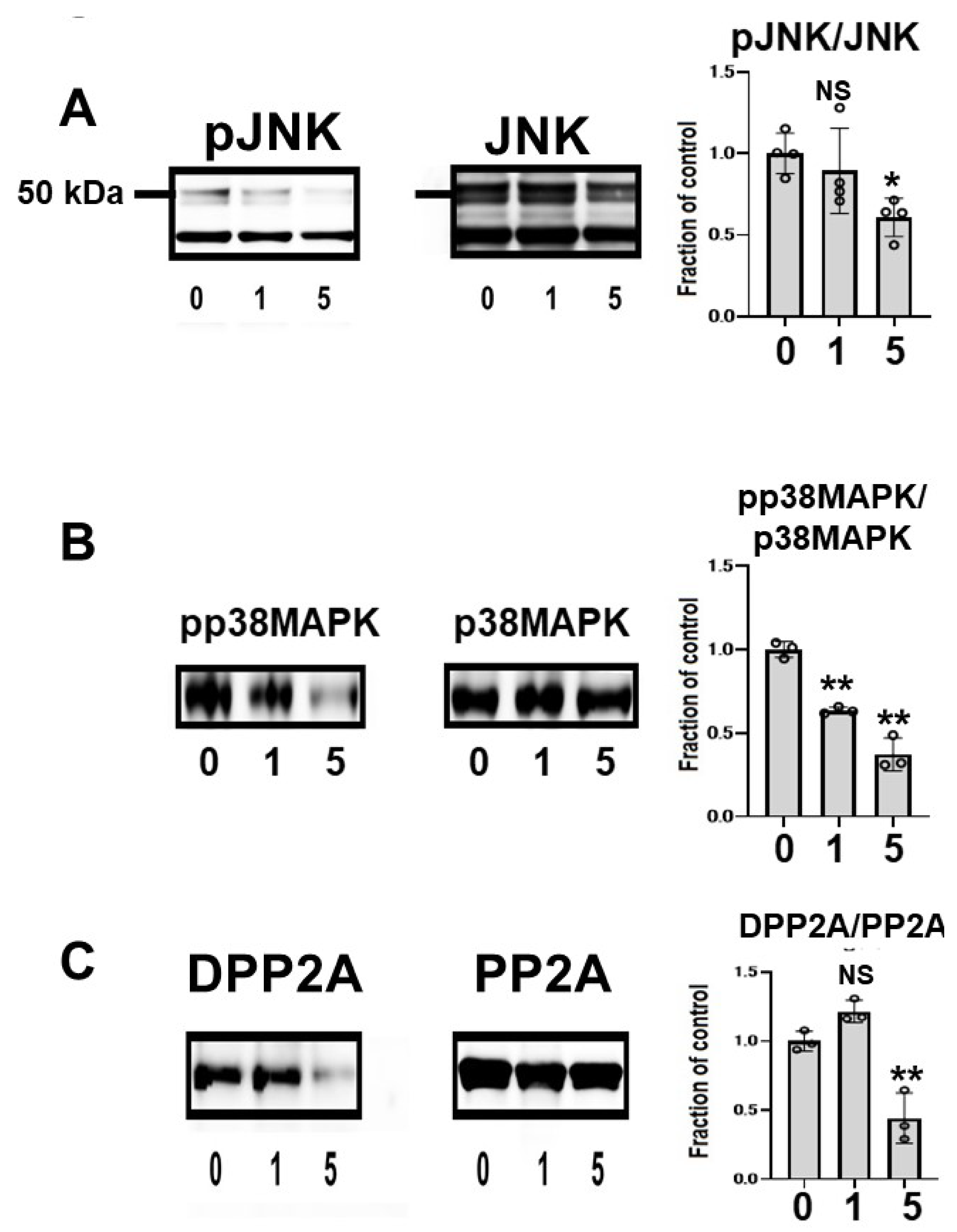
2.4. CQ Decreased Phosphorylated Tau
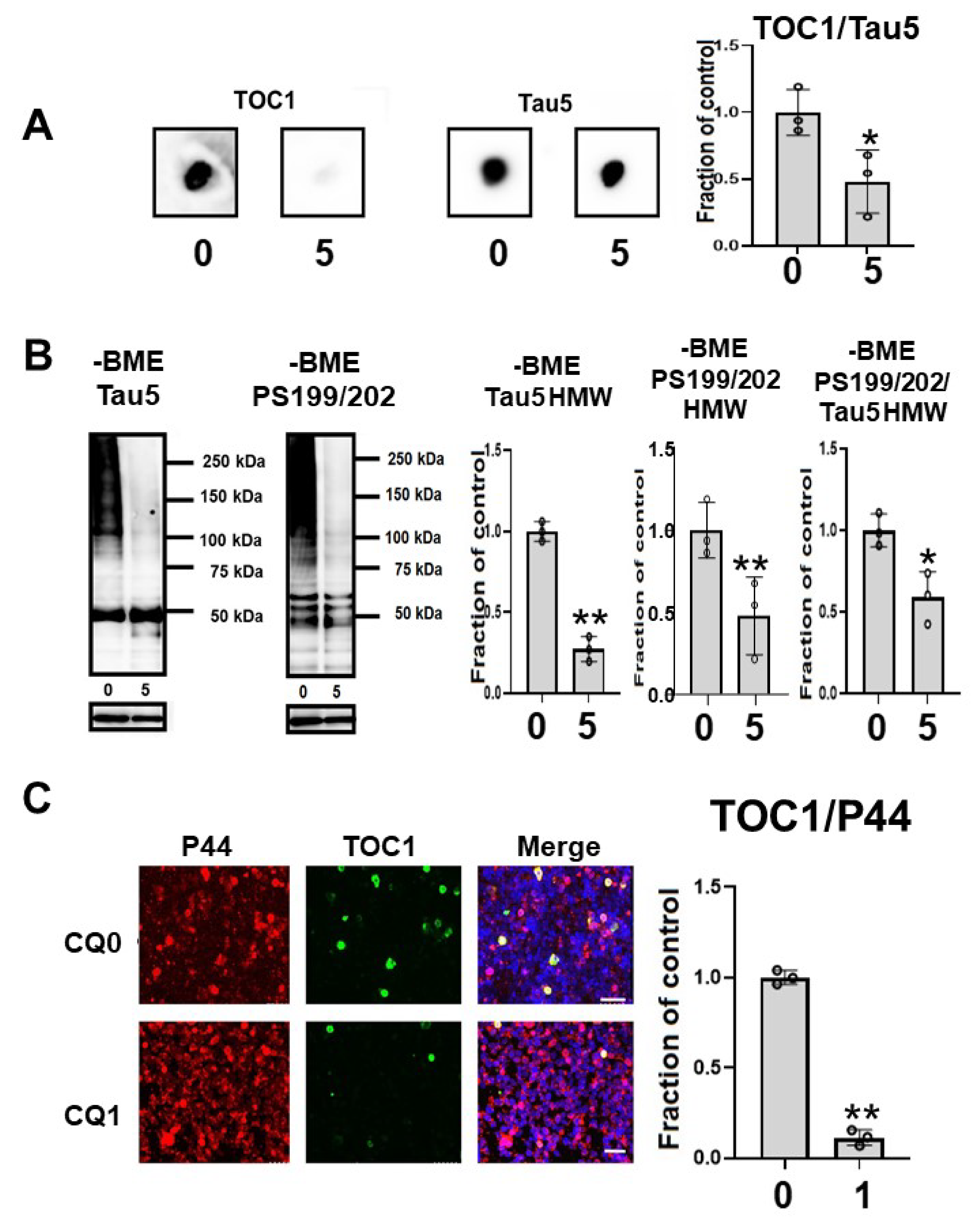
2.5. CQ Decreased Caspase Cleaved Tau
2.6. CQ Decreases Tau Oligomers in M1C cells
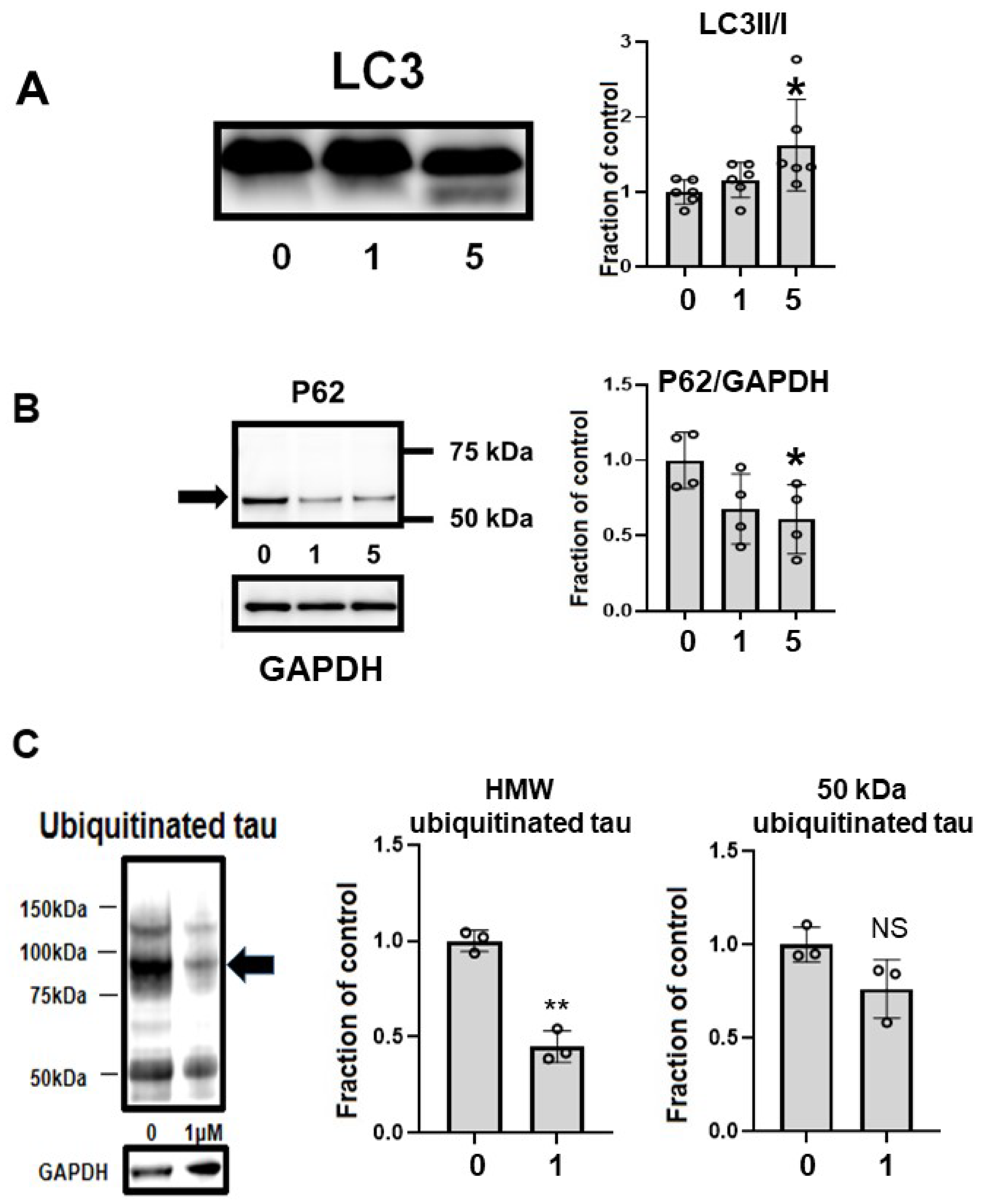
2.7. CQ Upregulated Autophagy and Activated Proteasomes
2.8. CQ Decreased Endogenous Tau
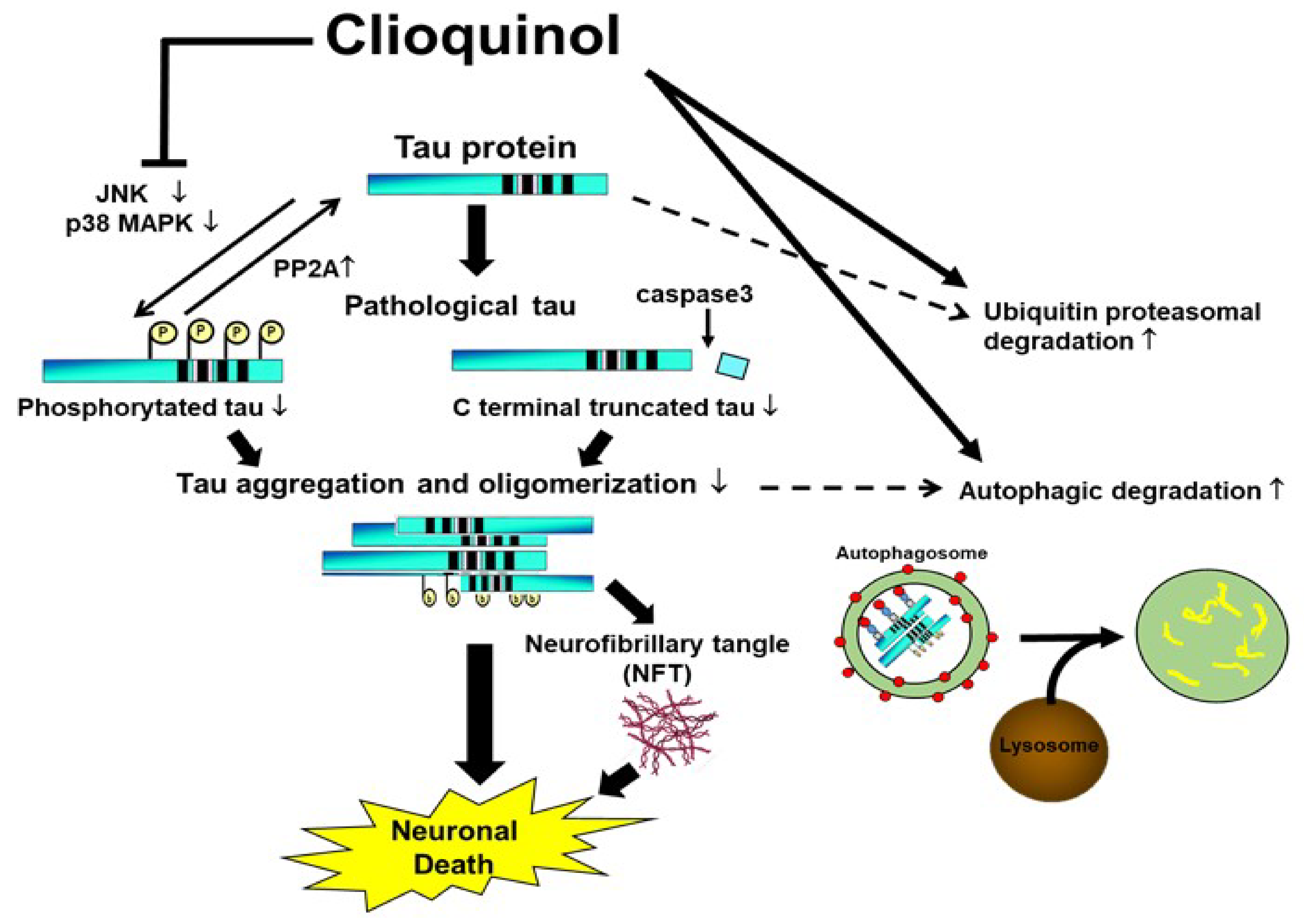
3. Discussion
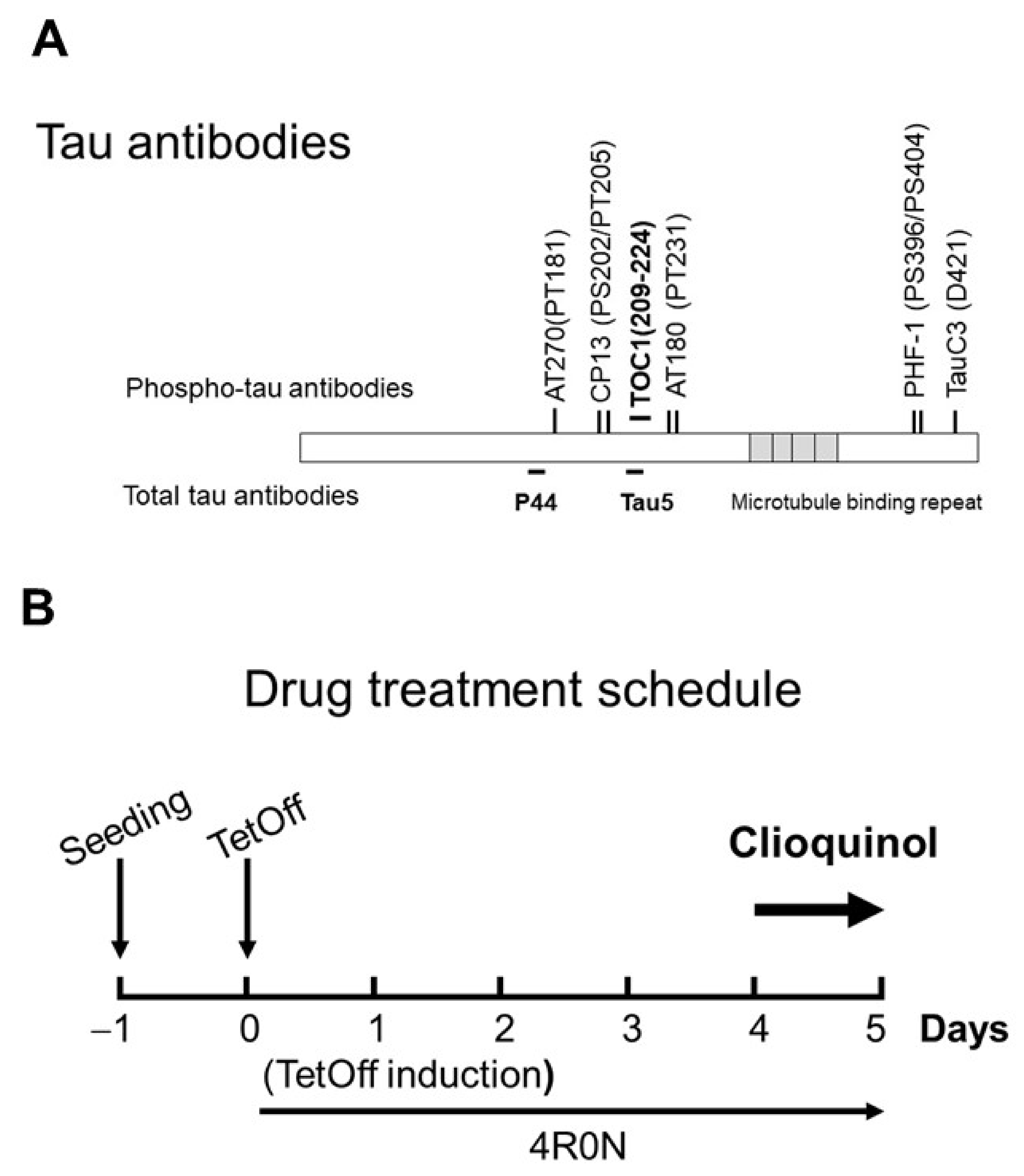
4. Materials and Methods
4.1. Materials and Antibodies
4.2. Cell Culture
4.3. Fractionation of Cell Lysates
4.4. Western Blotting
4.5. Immunocytochemical Study
4.6. Cu+ Visualization in the Cells
4.7. mRNA Expression
4.8. Immunoprecipitation of Ubiquitinated Tau
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| CQ | clioquinol |
| JNK | c-Jun N terminal kinase |
| P38MAPK | p38 MAP Kinase |
| NFT | neurofibrillary tangle |
| CC3 | cleaved caspase3 |
| PP2A | protein phosphatase 2A |
| GSK3β | glycogen synthase kinase 3β |
References
- Ihara, Y.; Nukina, N.; Miura, R.; Ogawara, M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem. 1986, 99, 1807–1810. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003, 26, 207–214. [Google Scholar] [CrossRef]
- Hager, K.; Baseman, A.S.; Nye, J.S.; Brashear, H.R.; Han, J.; Sano, M.; Davis, B.; Richards, H.M. Effects of galantamine in a 2-year, randomized, placebo-controlled study in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2014, 10, 391–401. [Google Scholar]
- Cummings, J.L.; Schneider, E.; Tariot, P.N.; Graham, S.M. Memantine MEM-MD-02 Study Group. Behavioral effects of memantine in Alzheimer disease patients receiving donepezil treatment. Neurology 2006, 67, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Leskovjan, A.C.; Lanzirotti, A.; Miller, L.M. Amyloid plaques in PSAPP mice bind less metal than plaques in human Alzheimer’s disease. Neuroimage 2009, 47, 1215–1220. [Google Scholar] [CrossRef] [Green Version]
- Roberts, B.R.; Ryan, T.M.; Bush, A.I.; Masters, C.L.; Duce, J.A. The role of metallobiology and amyloid-β peptides in Alzheimer’s disease. J. Neurochem. 2012, 120 (Suppl. S1), 149–166. [Google Scholar] [CrossRef]
- Good, P.F.; Perl, D.P.; Bierer, L.M.; Schmeidler, J. Selective accumulation of aluminum and iron in the neurofibrillary tangles of Alzheimer’s disease: A laser microprobe (LAMMA) study. Ann. Neurol. 1992, 31, 286–292. [Google Scholar] [CrossRef]
- Atwood, C.S.; Scarpa, R.C.; Huang, X.; Moir, R.D.; Jones, W.D.; Fairlie, D.P.; Tanzi, R.E.; Bush, A.I. Characterization of copper interactions with alzheimer amyloid beta peptides: Identification of an attomolar-affinity copper binding site on amyloid beta1-42. J. Neurochem. 2000, 75, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Wang, L.; Fan, Q.; Xiao, G.; Wang, X.; Zhong, Y.; Zhou, B. Genetic inhibition of solute-linked carrier 39 family transporter 1 ameliorates aβ pathology in a Drosophila model of Alzheimer’s disease. PLoS Genet. 2012, 8, e1002683. [Google Scholar] [CrossRef] [Green Version]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Crouch, P.J.; Hung, L.W.; Adlard, P.A.; Cortes, M.; Lal, V.; Filiz, G.; Perez, K.A.; Nurjono, M.; Caragounis, A.; Du, T.; et al. Increasing Cu Bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Egaña, J.T.; Zambrano, C.; Nuñez, M.T.; Gonzalez-Billault, C.; Maccioni, R.B. Iron-induced oxidative stress modify tau phosphorylation patterns in hippocampal cell cultures. Biometals 2003, 16, 215–223. [Google Scholar] [CrossRef]
- Kim, I.; Park, E.J.; Seo, J.; Ko, S.J.; Lee, J.; Kim, C.H. Zinc stimulates tau S214 phosphorylation by the activation of Raf/mitogen-activated protein kinase-kinase/extracellular signal-regulated kinase pathway. Neuroreport 2011, 22, 839–844. [Google Scholar] [CrossRef]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Köhler, A.; Glossmann, H.; et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21830–21835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.Y.; Wei, Y.P.; Xiong, Y.; Wang, X.C.; Xie, A.J.; Wang, X.L.; Yang, Y.; Wang, Q.; Lu, Y.M.; Liu, R.; et al. Synaptic released zinc promotes tau hyperphosphorylation by inhibition of protein phosphatase 2A (PP2A). J. Biol. Chem. 2012, 287, 11174–11182. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Jing, X.P.; Zhou, X.W.; Wang, X.L.; Yang, Y.; Sun, X.Y.; Qiu, M.; Cao, F.Y.; Lu, Y.M.; Liu, R.; et al. Zinc induces protein phosphatase 2A inactivation and tau hyperphosphorylation through Src dependent PP2A (tyrosine 307) phosphorylation. Neurobiol. Aging 2013, 34, 745–756. [Google Scholar] [CrossRef]
- Mo, Z.Y.; Zhu, Y.Z.; Zhu, H.L.; Fan, J.B.; Chen, J.; Liang, Y. Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J. Biol. Chem. 2009, 284, 34648–34657. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Wu, Z.; Cao, Y.; Lang, M.; Lu, B.; Zhou, B. Zinc binding directly regulates tau toxicity independent of tau hyperphosphorylation. Cell Rep. 2014, 8, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Soragni, A.; Zambelli, B.; Mukrasch, M.D.; Biernat, J.; Jeganathan, S.; Griesinger, C.; Ciurli, S.; Mandelkow, E.; Zweckstetter, M. Structural characterization of binding of Cu (II) to tau protein. Biochemistry 2008, 47, 10841–10851. [Google Scholar] [CrossRef]
- Walton, J.R. Evidence for participation of aluminum in neurofibrillary tangle formation and growth in Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, G.; Becue, I.; Smith, J.B. Clioquinol. Anal. Profiles Drug Subst. 1990, 18, 57–90. [Google Scholar]
- Bandyopadhyay, S.; Huang, X.; Lahiri, D.K.; Rogers, J.T. Novel drug targets based on metallobiology of Alzheimer’s disease. Expert Opin. Ther. Targets 2010, 14, 1177–1197. [Google Scholar] [CrossRef]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Treiber, C.; Simons, A.; Strauss, M.; Hafner, M.; Cappai, R.; Bayer, T.A.; Multhaup, G. Clioquinol mediates copper uptake and counteracts copper efflux activities of the amyloid precursor protein of Alzheimer’s disease. J. Biol. Chem. 2004, 279, 51958–51964. [Google Scholar] [CrossRef] [Green Version]
- Raman, B.; Ban, T.; Yamaguchi, K.; Sakai, M.; Kawai, T.; Naiki, H.; Goto, Y. Metal ion-dependent effects of clioquinol on the fibril growth of an amyloid {beta} peptide. J. Biol. Chem. 2005, 280, 16157–16162. [Google Scholar] [CrossRef] [Green Version]
- Regland, B.; Lehmann, W.; Abedini, I.; Blennow, K.; Jonsson, M.; Karlsson, I.; Sjögren, M.; Wallin, A.; Xilinas, M.; Gottfries, C.G. Treatment of Alzheimer’s disease with clioquinol. Dement. Geriatr. Cogn. Disord. 2001, 12, 408–414. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.X.; Tammer, A.; et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. PBT2-201-EURO study group. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, D.I.; Billings, J.L.; Adlard, P.A.; Ayton, S.; Sedjahtera, A.; Masters, C.L.; Wilkins, S.; Shackleford, D.M.; Charman, S.A.; Bal, W.; et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 53. [Google Scholar] [CrossRef] [Green Version]
- Hamano, T.; Gendron, T.F.; Causevic, E.; Yen, S.H.; Lin, W.L.; Isidoro, C.; Deture, M.; Ko, L.W. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur. J. Neurosci. 2008, 27, 1119–1130. [Google Scholar] [CrossRef]
- Hamano, T.; Gendron, T.F.; Ko, L.W.; Yen, S.H. Concentration-dependent effects of proteasomal inhibition on tau processing in a cellular model of tauopathy. Int. J. Clin. Exp. Pathol. 2009, 2, 561–573. [Google Scholar]
- Hamano, T.; Yen, S.H.; Gendron, T.; Ko, L.W.; Kuriyama, M. Pitavastatin decreases tau levels via the inactivation of rho/rock. Neurobiol. Aging 2012, 33, 2306–2320. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Shirafuji, N.; Makino, C.; Yen, S.H.; Kanaan, N.M.; Ueno, A.; Suzuki, J.; Ikawa, M.; Matsunaga, A.; Yamamura, O.; et al. Pioglitazone Prevents Tau Oligomerization. Biochem. Biophys. Res. Commun. 2016, 478, 1035–1042. [Google Scholar] [CrossRef] [Green Version]
- Hamano, T.; Shirafuji, N.; Yen, S.H.; Yoshida, H.; Kanaan, N.; Hayashi, K.; Ikawa, M.; Yamamura, O.; Fujita, Y.; Kuriyama, M.; et al. Rho-kinase ROCK inhibitors reduce oligomeric tau protein. Neurobiol. Aging 2020, 89, 41–54. [Google Scholar] [CrossRef]
- Shirafuji, N.; Hamano, T.; Yen, S.H.; Kanaan, N.M.; Yoshida, H.; Hayashi, K.; Ikawa, M.; Yamamura, O.; Kuriyama, M.; Nakamoto, Y. Homocysteine Increases Tau Phosphorylation, Truncation and Oligomerization. Int. J. Mol. Sci. 2018, 19, 891. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Jiang, M.; Xiao, Y.; Zhang, X.; Cui, S.; Huang, G. Folic acid inhibits tau phosphorylation through regulation of PP2A methylation in SH-SY5Y cells. J. Nutr. Health Aging 2015, 19, 123–129. [Google Scholar] [CrossRef]
- Sontag, E.; Nunbhakdi-Craig, V.; Lee, G.; Bloom, G.S.; Mumby, M.C. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron 1996, 17, 1201–1207. [Google Scholar] [CrossRef] [Green Version]
- Odai, T.; Terauchi, M.; Suzuki, R.; Kato, K.; Hirose, A.; Miyasaka, N. Severity of subjective forgetfulness is associated with high dietary intake of copper in Japanese senior women: A cross-sectional study. Food Sci. Nutr. 2020, 8, 4422–4431. [Google Scholar] [CrossRef]
- Gamblin, T.C.; Chen, F.; Zambrano, A.; Abraha, A.; Lagalwar, S.; Guillozet, A.L.; Lu, M.; Fu, Y.; Garcia-Sierra, F.; LaPointe, N.; et al. Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10032–10037. [Google Scholar] [CrossRef] [Green Version]
- Guillozet-Bongaarts, A.L.; Garcia-Sierra, F.; Reynolds, M.R.; Horowitz, P.M.; Fu, Y.; Wang, T.; Cahill, M.E.; Bigio, E.H.; Berry, R.W.; Binder, L.I. Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol. Aging 2005, 26, 1015–1022. [Google Scholar] [CrossRef]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I. Stress kinases involved in tau phosphorylation in Alzheimer’s disease, tauopathies and APP transgenic mice. Neurotox. Res. 2004, 6, 469–475. [Google Scholar] [CrossRef]
- Goedert, M.; Hasegawa, M.; Jakes, R.; Lawler, S.; Cuenda, A.; Cohen, P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997, 409, 57–62. [Google Scholar] [CrossRef]
- Tyagi, E.; Fiorelli, T.; Norden, M.; Padmanabhan, J. Alpha 1-Antichymotrypsin, and Inflammatory Protein Overexpressed in the Brains of Patients with Alzheimer’s Disease, Induces Tau Hyperphosphorylation through c-Jun N-Terminal Kinase Activation. Int. J. Alzheimers Dis. 2013, 2013, 606083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Hastie, C.J.; McLauchlan, H.; Cohen, P.; Goedert, M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK). J. Neurochem. 2004, 90, 352–358. [Google Scholar] [CrossRef]
- Sahara, N.; Murayama, M.; Lee, B.; Park, J.M.; Lagalwar, S.; Binder, L.I.; Takashima, A. Active c-jun N-terminal kinase induces caspase cleavage of tau and additional phosphorylation by GSK-3beta is required for tau aggregation. Eur. J. Neurosci. 2008, 27, 2897–2906. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef] [Green Version]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012, 26, 1946–1959. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Sahara, N. Characteristics of tau oligomers. Front. Neurol. 2013, 4, 102. [Google Scholar] [CrossRef] [Green Version]
- Patterson, K.R.; Remmers, C.; Fu, Y.; Brooker, S.; Kanaan, N.M.; Vana, L.; Ward, S.; Reyes, J.F.; Philibert, K.; Glucksman, M.J.; et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 2011, 286, 23063–23076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, S.M.; Himmelstein, D.S.; Ren, Y.; Fu, Y.; Yu, X.W.; Roberts, K.; Binder, L.I.; Sahara, N. TOC1: A valuable tool in assessing disease progression in the rTg4510 mouse model of tauopathy. Neurobiol. Dis. 2014, 67, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konagaya, M.; Matsumoto, A.; Takase, S.; Mizutani, T.; Sobue, G.; Konishi, T.; Hayabara, T.; Iwashita, H.; Ujihira, T.; Miyata, K.; et al. Clinical analysis of longstanding subacute myelo-optico-neuropathy: Sequelae of clioquinol at 32 years after its ban. J. Neurol. Sci. 2004, 218, 85–90. [Google Scholar] [CrossRef]
- Yassin, M.S.; Ekblom, J.; Xilinas, M.; Gottfries, C.G.; Oreland, L. Changes in uptake of vitamin b (12) and trace metals in brains of mice treated with clioquinol. J. Neurol. Sci. 2000, 173, 40–44. [Google Scholar] [CrossRef]
- Ko, L.W.; Rush, T.; Sahara, N.; Kersh, J.S.; Easson, C.; Deture, M.; Lin, W.L.; Connor, Y.D.; Yen, S.H. Assembly of filamentous tau aggregates in human neuronal cells. J. Alzheimers Dis. 2004, 6, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Dou, F.; Li, F.; Zhang, X.; Zhang, Y.W.; Zheng, H.; Lipton, S.A.; Xu, H.; Liao, F.F. Suppression of cyclin-dependent kinase 5 activation by amyloid precursor protein: A novel excitoprotective mechanism involving modulation of tau phosphorylation. J. Neurosci. 2005, 25, 11542–11552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahara, N.; Lewis, J.; DeTure, M.; McGowan, E.; Dickson, D.W.; Hutton, M.; Yen, S.H. Assembly of tau in transgenic animals expressing P301L tau: Alteration of phosphorylation and solubility. J. Neurochem. 2002, 83, 1498–1508. [Google Scholar] [CrossRef] [Green Version]
- Bunpo, P.; Dudley, A.; Cundiff, J.K.; Cavener, D.R.; Wek, R.C.; Anthony, T.G. GCN2 protein kinase is required to activate amino acid deprivation responses in mice treated with the anti-cancer agent L-asparaginase. J. Biol. Chem. 2009, 284, 32742–33249. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.W.; Nye, J.S.; Lamb, N.J.C.; Fernanez, A.; Kitzmann, M. Intracellular retention of caveolin 1 in presenilin-deficient cells. J. Biol. Chem. 2005, 280, 6663–6668. [Google Scholar] [CrossRef] [Green Version]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, G.; Zhu, F.; Kanaan, N.M.; Asano, R.; Shirafuji, N.; Sasaki, H.; Yamaguchi, T.; Enomoto, S.; Endo, Y.; Ueno, A.; et al. Clioquinol Decreases Levels of Phosphorylated, Truncated, and Oligomerized Tau Protein. Int. J. Mol. Sci. 2021, 22, 12063. https://doi.org/10.3390/ijms222112063
Lin G, Zhu F, Kanaan NM, Asano R, Shirafuji N, Sasaki H, Yamaguchi T, Enomoto S, Endo Y, Ueno A, et al. Clioquinol Decreases Levels of Phosphorylated, Truncated, and Oligomerized Tau Protein. International Journal of Molecular Sciences. 2021; 22(21):12063. https://doi.org/10.3390/ijms222112063
Chicago/Turabian StyleLin, Gaoping, Feiyan Zhu, Nicholas M. Kanaan, Rei Asano, Norimichi Shirafuji, Hirohito Sasaki, Tomohisa Yamaguchi, Soichi Enomoto, Yoshinori Endo, Asako Ueno, and et al. 2021. "Clioquinol Decreases Levels of Phosphorylated, Truncated, and Oligomerized Tau Protein" International Journal of Molecular Sciences 22, no. 21: 12063. https://doi.org/10.3390/ijms222112063
APA StyleLin, G., Zhu, F., Kanaan, N. M., Asano, R., Shirafuji, N., Sasaki, H., Yamaguchi, T., Enomoto, S., Endo, Y., Ueno, A., Ikawa, M., Hayashi, K., Yamamura, O., Yen, S. -H., Nakamoto, Y., & Hamano, T. (2021). Clioquinol Decreases Levels of Phosphorylated, Truncated, and Oligomerized Tau Protein. International Journal of Molecular Sciences, 22(21), 12063. https://doi.org/10.3390/ijms222112063