
The Effects of Vitamin D on the Expression of IL-33 and Its Receptor ST2 in Skin Cells; Potential Implication for Psoriasis

 , , ,
, , , 
 ,
,  and
and
Abstract
:1. Introduction
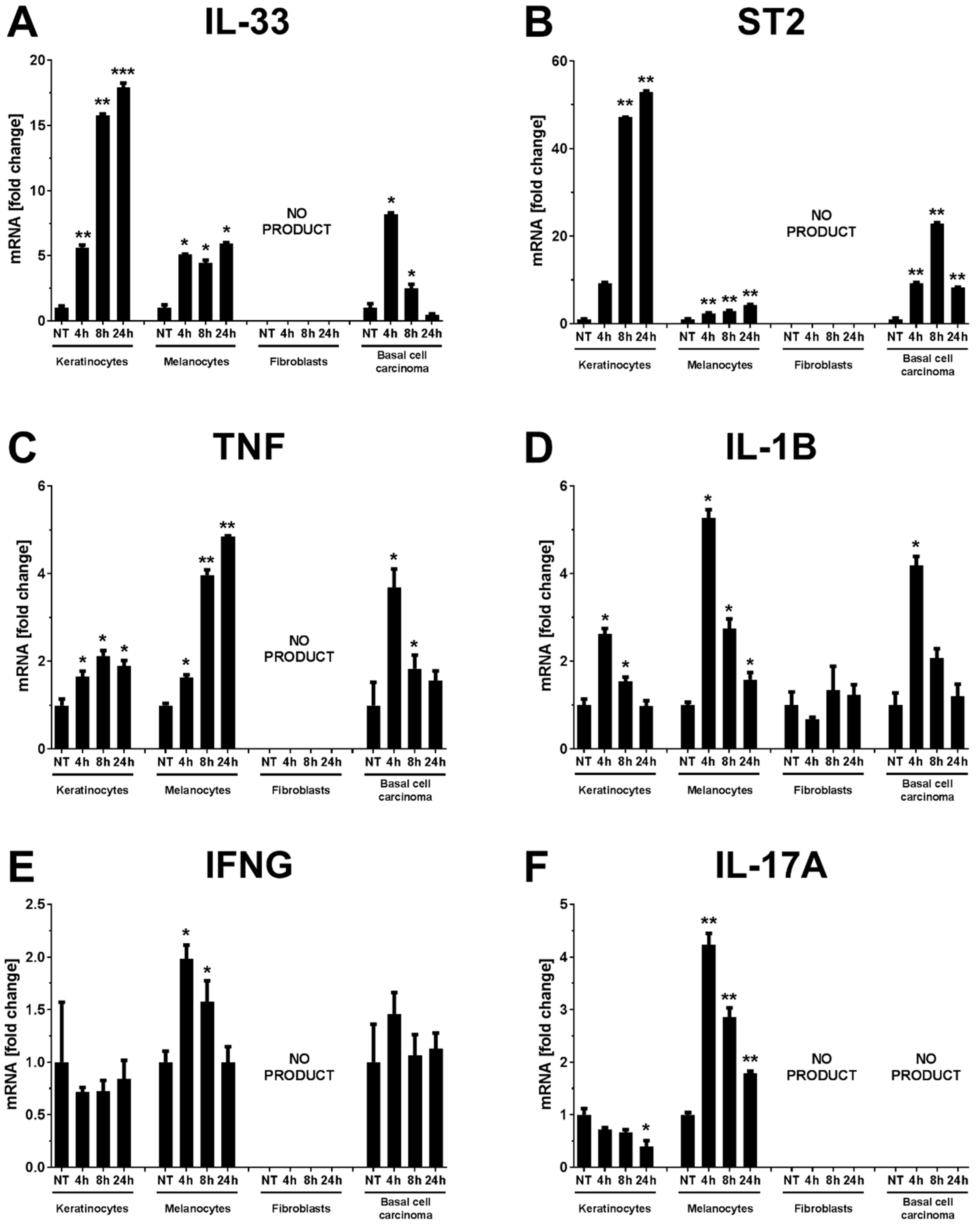
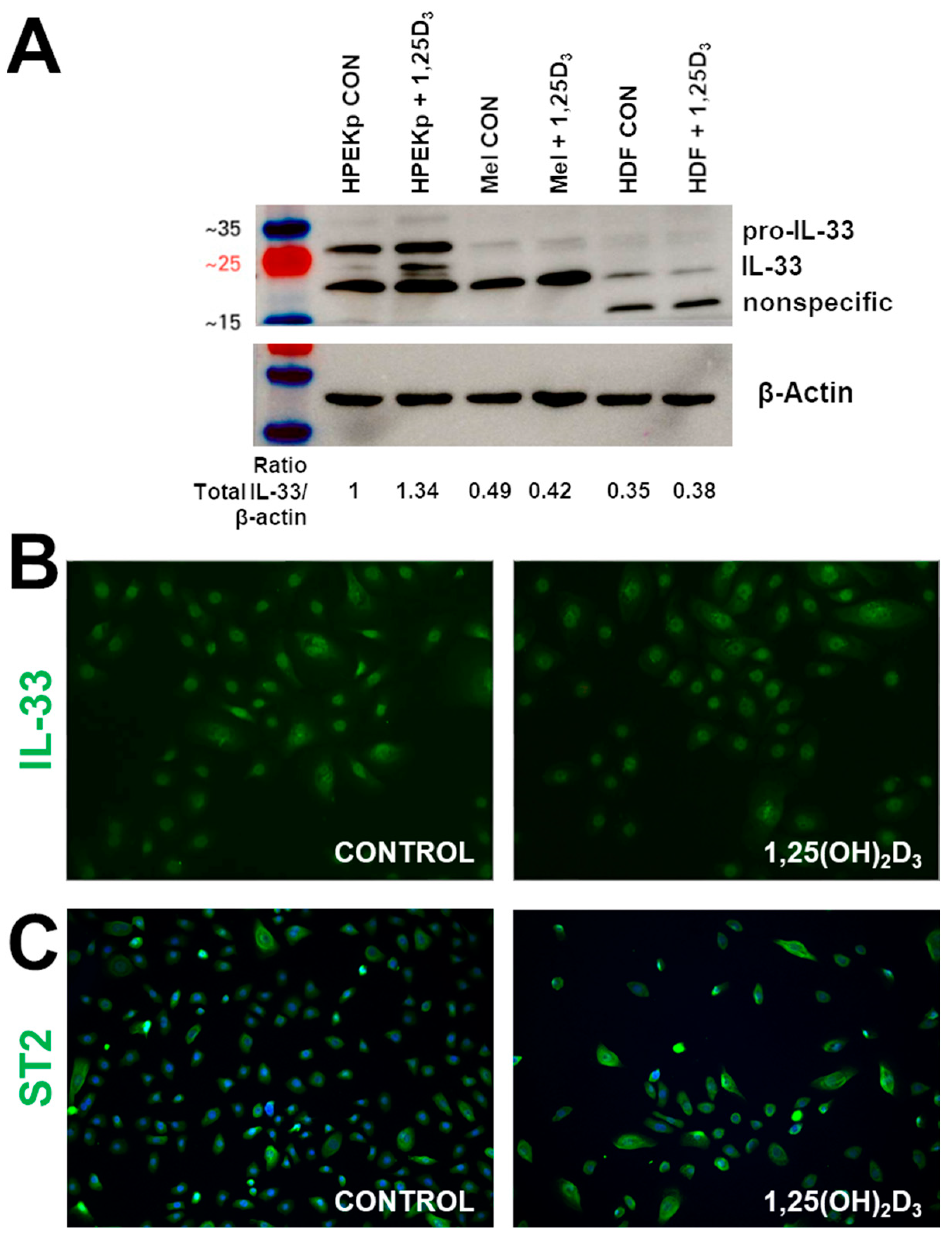
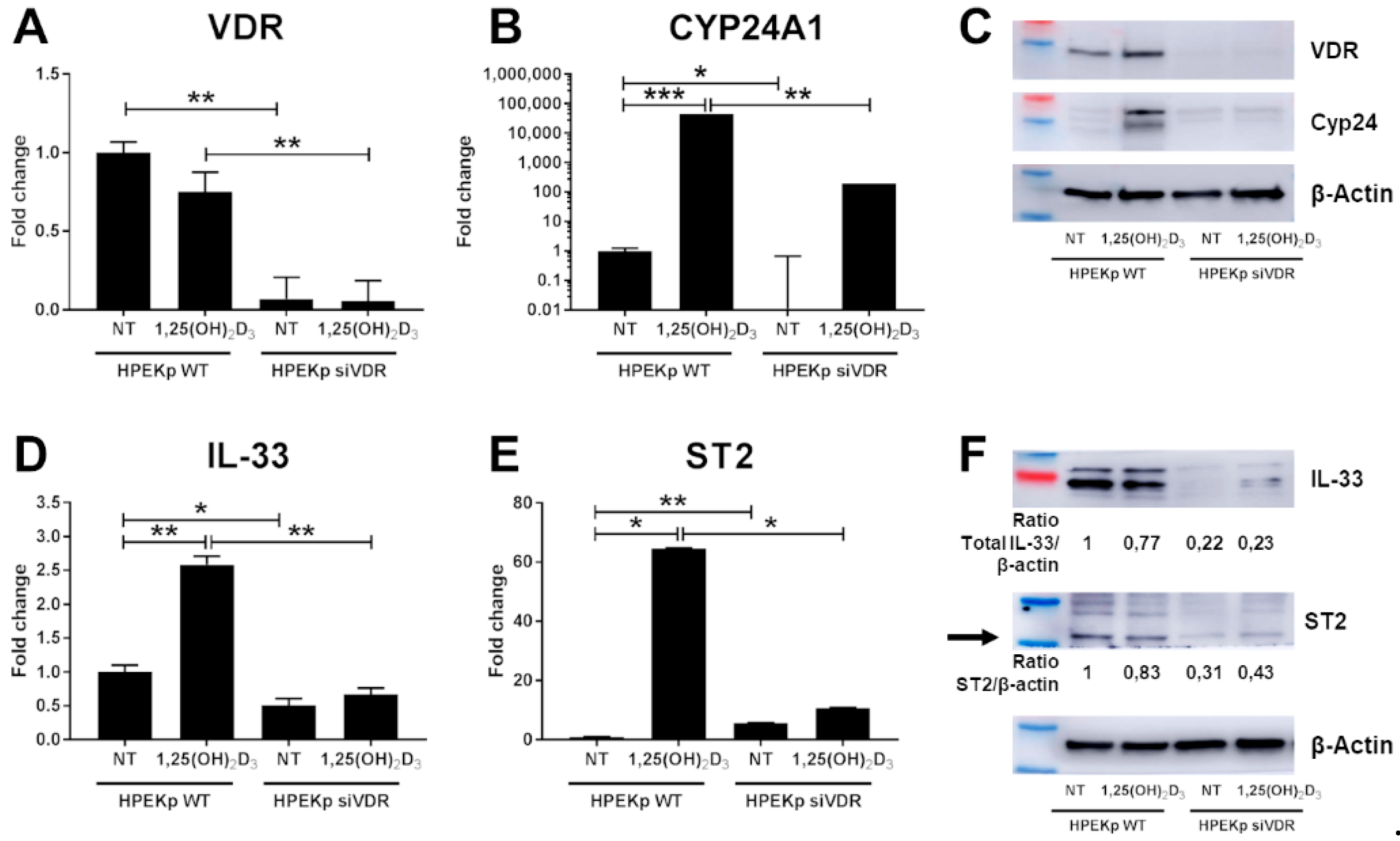
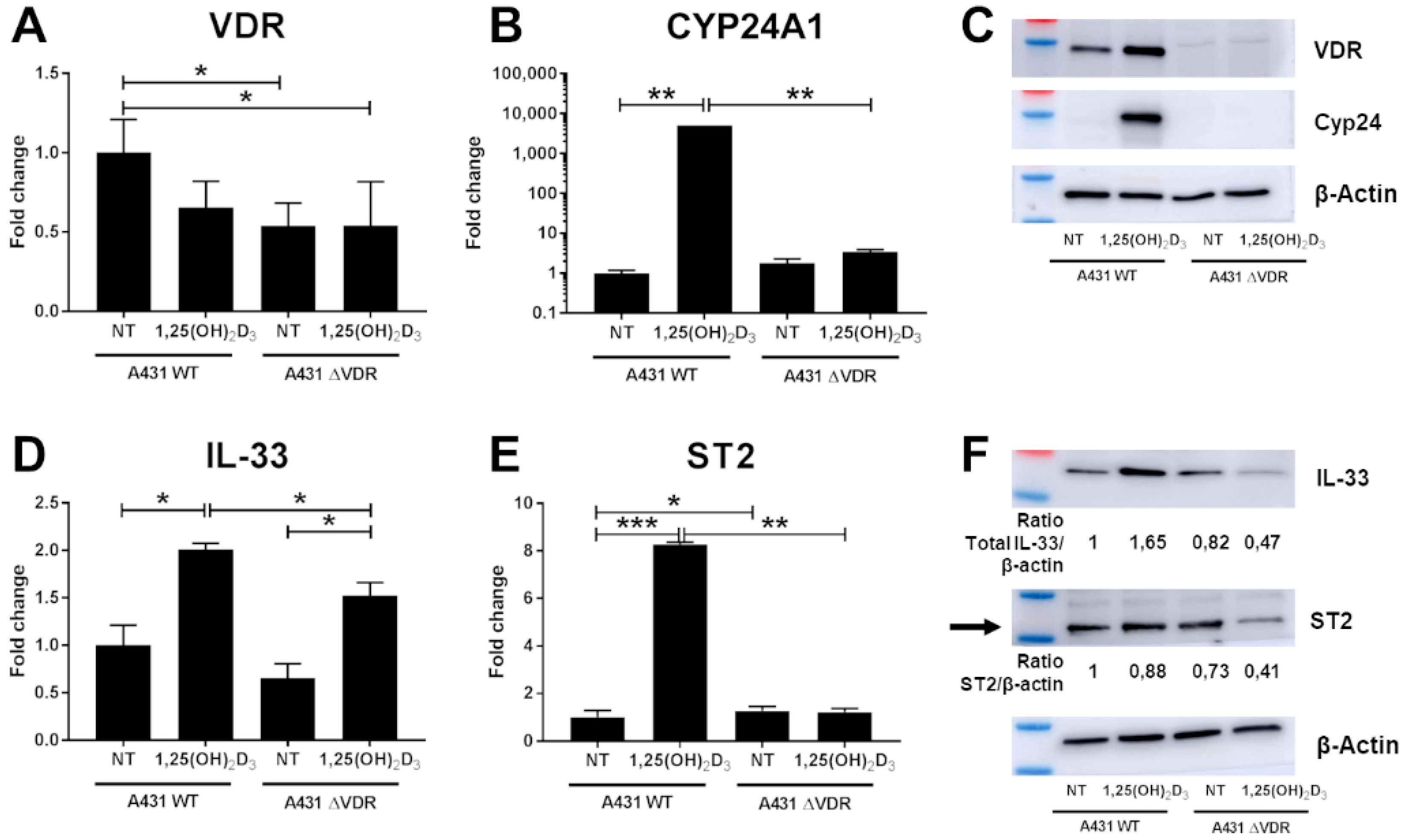
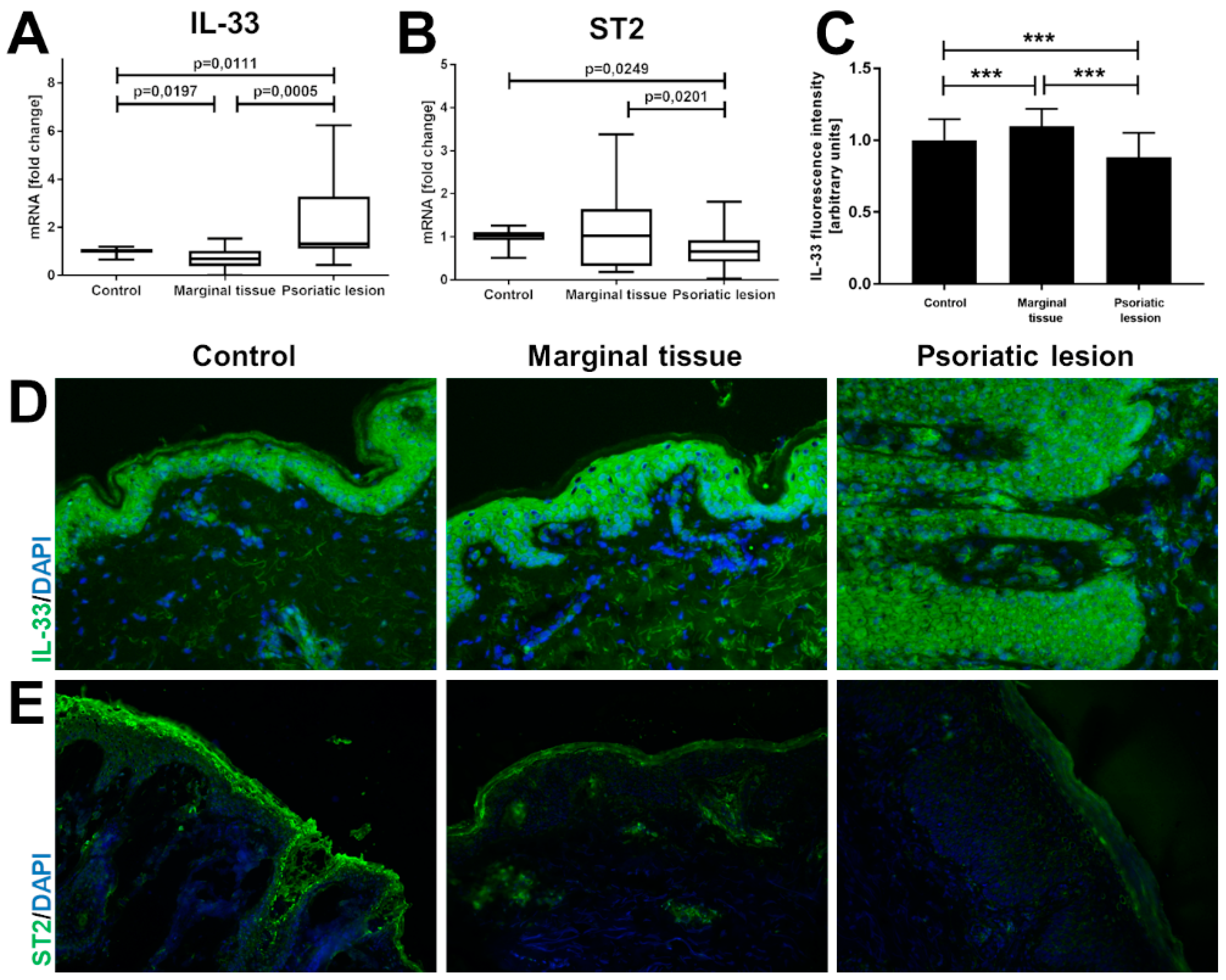
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Treatments
4.3. Patients and Controls
4.4. RNA Extraction and Real-Time PCR
4.5. VDR Silencing in the HPEKp Cell Line Based on siRNA Transfection
4.6. VDR Gene Knock-Out in the A431 Cell Line Based on CRISPR/Cas9 Technology
4.7. Immunofluorescence Staining
4.8. Immunoblotting
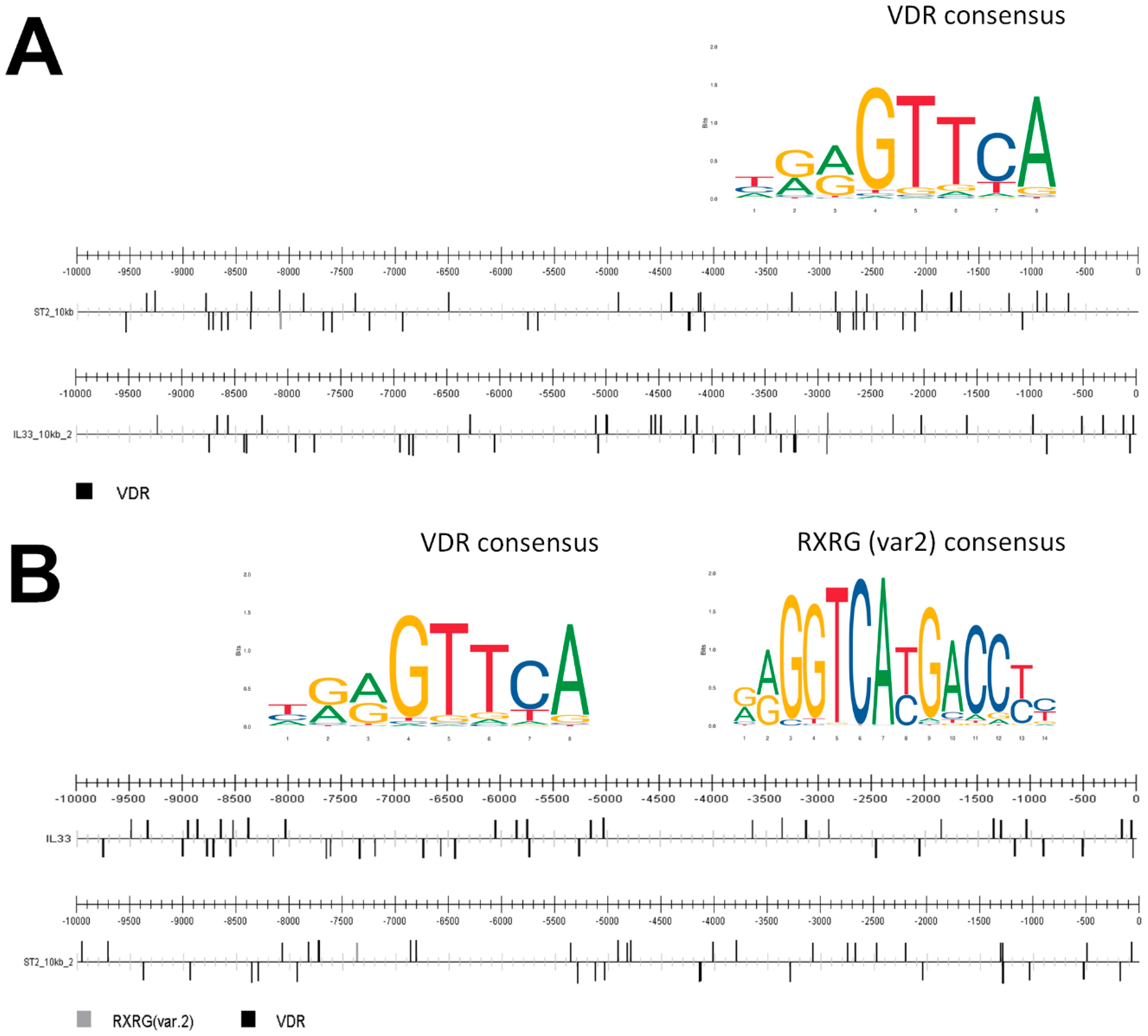
4.9. Prediction of Transcription Factor Binding Sites
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wierzbicka, J.; Piotrowska, A.; Zmijewski, M.A. The renaissance of vitamin D. Acta Biochim. Pol. 2014, 61, 679–686. [Google Scholar] [CrossRef]
- Holick, M.F.; Frommer, J.E.; McNeill, S.C.; Richtand, N.M.; Henley, J.W.; Potts, J.T., Jr. Photometabolism of 7-dehydrocholesterol to previtamin D3 in skin. Biochem. Biophys. Res. Commun. 1977, 76, 107–114. [Google Scholar] [CrossRef]
- Pillai, S.; Bikle, D.D.; Elias, P.M. Vitamin D and epidermal differentiation: Evidence for a role of endogenously produced vitamin D metabolites in keratinocyte differentiation. Skin Pharmacol. 1988, 1, 149–160. [Google Scholar] [CrossRef]
- Bikle, D.; Christakos, S. New aspects of vitamin D metabolism and action—Addressing the skin as source and target. Nat. Rev. Endocrinol. 2020, 16, 234–252. [Google Scholar] [CrossRef]
- Piotrowska, A.; Wierzbicka, J.; Zmijewski, M.A. Vitamin D in the skin physiology and pathology. Acta Biochim. Pol. 2016, 63, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Bikle, D.D. Vitamin D metabolism and function in the skin. Mol. Cell Endocrinol. 2011, 347, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Haussler, C.A.; Hsieh, D.; Hsieh, J.C.; Jurutka, P.W. Molecular mechanisms of vitamin D action. Calcif. Tissue Int. 2013, 92, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Zmijewski, M.A.; Carlberg, C. Vitamin D receptor(s): In the nucleus but also at membranes? Exp. Dermatol. 2020, 29, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Bowcock, A.M.; Krueger, J.G. Pathogenesis and therapy of psoriasis. Nature 2007, 445, 866–873. [Google Scholar] [CrossRef]
- Purzycka-Bohdan, D.; Szczerkowska-Dobosz, A.; Zablotna, M.; Wierzbicka, J.; Piotrowska, A.; Zmijewski, M.A.; Nedoszytko, B.; Nowicki, R. Assessment of Interleukin 16 Serum Levels and Skin Expression in Psoriasis Patients in Correlation with Clinical Severity of the Disease. PLoS ONE 2016, 11, e0165577. [Google Scholar] [CrossRef]
- Napolitano, M.; Caso, F.; Scarpa, R.; Megna, M.; Patrì, A.; Balato, N.; Costa, L. Psoriatic arthritis and psoriasis: Differential diagnosis. Clin. Rheumatol. 2016, 35, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Gisondi, P.; Rossini, M.; Di Cesare, A.; Idolazzi, L.; Farina, S.; Beltrami, G.; Peris, K.; Girolomoni, G. Vitamin D status in patients with chronic plaque psoriasis. Br. J. Dermatol. 2012, 166, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Maleki, M.; Nahidi, Y.; Azizahari, S.; Meibodi, N.T.; Hadianfar, A. Serum 25-OH Vitamin D Level in Psoriatic Patients and Comparison With Control Subjects. J. Cutan. Med. Surg. 2016, 20, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.B. Serum 25-hydroxyvitamin D status in individuals with psoriasis in the general population. Endocrine 2013, 44, 537–539. [Google Scholar] [CrossRef]
- Chandrashekar, L.; Kumarit, G.R.; Rajappa, M.; Revathy, G.; Munisamy, M.; Thappa, D.M. 25-hydroxy vitamin D and ischaemia-modified albumin levels in psoriasis and their association with disease severity. Br. J. Biomed. Sci. 2015, 72, 56–60. [Google Scholar] [CrossRef]
- Orgaz-Molina, J.; Magro-Checa, C.; Rosales-Alexander, J.L.; Arrabal-Polo, M.A.; Castellote-Caballero, L.; Buendía-Eisman, A.; Raya-Álvarez, E.; Arias-Santiago, S. Vitamin D insufficiency is associated with higher carotid intima-media thickness in psoriatic patients. Eur. J. Dermatol. 2014, 24, 53–62. [Google Scholar] [CrossRef]
- Atwa, M.A.; Balata, M.G.; Hussein, A.M.; Abdelrahman, N.I.; Elminshawy, H.H. Serum 25-hydroxyvitamin D concentration in patients with psoriasis and rheumatoid arthritis and its association with disease activity and serum tumor necrosis factor-alpha. Saudi Med. J. 2013, 34, 806–813. [Google Scholar]
- Ricceri, F.; Pescitelli, L.; Tripo, L.; Prignano, F. Deficiency of serum concentration of 25-hydroxyvitamin D correlates with severity of disease in chronic plaque psoriasis. J. Am. Acad. Dermatol. 2013, 68, 511–512. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D regulated keratinocyte differentiation. J. Cell Biochem. 2004, 92, 436–444. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D and the immune system: Role in protection against bacterial infection. Curr. Opin. Nephrol. Hypertens. 2008, 17, 348–352. [Google Scholar] [CrossRef]
- Szczerkowska-Dobosz, A.; Krasowska, D.; Bartosińska, J.; Stawczyk-Macieja, M.; Walczak, A.; Owczarczyk-Saczonek, A.; Reich, A.; Batycka-Baran, A.; Czajkowski, R.; Dobrucki, I.T.; et al. Pathogenesis of psoriasis in the “omic” era. Part IV. Epidemiology, genetics, immunopathogenesis, clinical manifestation and treatment of psoriatic arthritis. Postepy Dermatol. Alergol. 2020, 37, 625–634. [Google Scholar] [CrossRef]
- Fassio, A.; Matzneller, P.; Idolazzi, L. Recent Advances in Imaging for Diagnosis, Monitoring, and Prognosis of Psoriatic Arthritis. Front. Med. 2020, 7, 551684. [Google Scholar] [CrossRef]
- Nedoszytko, B.; Szczerkowska-Dobosz, A.; Stawczyk-Macieja, M.; Owczarczyk-Saczonek, A.; Reich, A.; Bartosiñska, J.; Batycka-Baran, A.; Czajkowski, R.; Dobrucki, I.T.; Dobrucki, L.W.; et al. Pathogenesis of psoriasis in the “omic” era. Part II. Genetic, genomic and epigenetic changes in psoriasis. Postepy Dermatol. Alergol. 2020, 37, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Owczarczyk-Saczonek, A.; Purzycka-Bohdan, D.; Nedoszytko, B.; Reich, A.; Szczerkowska-Dobosz, A.; Bartosiñska, J.; Batycka-Baran, A.; Czajkowski, R.; Dobrucki, I.T.; Dobrucki, L.W.; et al. Pathogenesis of psoriasis in the “omic” era. Part III. Metabolic disorders, metabolomics, nutrigenomics in psoriasis. Postepy Dermatol. Alergol. 2020, 37, 452–467. [Google Scholar] [CrossRef] [PubMed]
- Samotij, D.; Nedoszytko, B.; Bartosińska, J.; Batycka-Baran, A.; Czajkowski, R.; Dobrucki, I.T.; Dobrucki, L.W.; Górecka-Sokołowska, M.; Janaszak-Jasienicka, A.; Krasowska, D.; et al. Pathogenesis of psoriasis in the “omic” era. Part I. Epidemiology, clinical manifestation, immunological and neuroendocrine disturbances. Postepy Dermatol. Alergol. 2020, 37, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Purzycka-Bohdan, D.; Kisielnicka, A.; Bohdan, M.; Szczerkowska-Dobosz, A.; Sobalska-Kwapis, M.; Nedoszytko, B.; Nowicki, R.J. Analysis of the Potential Genetic Links between Psoriasis and Cardiovascular Risk Factors. Int. J. Mol. Sci. 2021, 22, 9063. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, J.; Grewal, S.; Langan, S.M.; Mehta, N.N.; Ogdie, A.; Van Voorhees, A.S.; Gelfand, J.M. Psoriasis and comorbid diseases: Epidemiology. J. Am. Acad. Dermatol. 2017, 76, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.W.; Harskamp, C.T.; Armstrong, E.J. Psoriasis and metabolic syndrome: A systematic review and meta-analysis of observational studies. J. Am. Acad. Dermatol. 2013, 68, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Damiani, G.; Bragazzi, N.L.; Garbarino, S.; Chattu, V.K.; Shapiro, C.M.; Pacifico, A.; Malagoli, P.; Pigatto, P.D.M.; Conic, R.R.Z.; Tiodorovic, D.; et al. Psoriatic and psoriatic arthritis patients with and without jet-lag: Does it matter for disease severity scores? Insights and implications from a pilot, prospective study. Chronobiol. Int. 2019, 36, 1733–1740. [Google Scholar] [CrossRef]
- Koo, J.; Marangell, L.B.; Nakamura, M.; Armstrong, A.; Jeon, C.; Bhutani, T.; Wu, J.J. Depression and suicidality in psoriasis: Review of the literature including the cytokine theory of depression. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1999–2009. [Google Scholar] [CrossRef]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA 2020, 323, 1945–1960. [Google Scholar] [CrossRef]
- Balato, A.; Raimondo, A.; Balato, N.; Ayala, F.; Lembo, S. Interleukin-33: Increasing role in dermatological conditions. Arch. Dermatol. Res. 2016, 308, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, G.; Gabay, C. Interleukin-33 biology with potential insights into human diseases. Nat. Rev. Rheumatol. 2011, 7, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, U.; Hvid, M.; Johansen, C.; Buchner, M.; Fölster-Holst, R.; Deleuran, M.; Vestergaard, C. TSLP, IL-31, IL-33 and sST2 are new biomarkers in endophenotypic profiling of adult and childhood atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; Park, J.H.; Kim, J.N.; Park, Y.H.; Shin, S.Y.; Lee, Y.H.; Kye, Y.C.; Son, S.W. Up-regulation of TNF-alpha secretion by cigarette smoke is mediated by Egr-1 in HaCaT human keratinocytes. Exp. Dermatol. 2010, 19, e206–e212. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Zhang, B.; Kempuraj, D.; Tagen, M.; Vasiadi, M.; Angelidou, A.; Alysandratos, K.D.; Kalogeromitros, D.; Asadi, S.; Stavrianeas, N.; et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc. Natl. Acad. Sci. USA 2010, 107, 4448–4453. [Google Scholar] [CrossRef] [Green Version]
- Balato, A.; Lembo, S.; Mattii, M.; Schiattarella, M.; Marino, R.; De Paulis, A.; Balato, N.; Ayala, F. IL-33 is secreted by psoriatic keratinocytes and induces pro-inflammatory cytokines via keratinocyte and mast cell activation. Exp. Dermatol. 2012, 21, 892–894. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Chen, H.; Chen, L.; Mao, J.; Cai, S.; Xiao, Y.; Li, J.; Shi, J.; Li, B.; Xu, Y.; et al. An Autocrine Circuit of IL-33 in Keratinocytes Is Involved in the Progression of Psoriasis. J. Investig. Dermatol. 2021, 141, 596–606.e7. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E. HMGB1, IL-1alpha, IL-33 and S100 proteins: Dual-function alarmins. Cell Mol. Immunol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Cayrol, C.; Girard, J.P. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, H.; Komine, M.; Karakawa, M.; Etoh, T.; Tominaga, S.; Ohtsuki, M. Novel splice variants of IL-33: Differential expression in normal and transformed cells. J. Investig. Dermatol. 2012, 132, 2661–2664. [Google Scholar] [CrossRef] [Green Version]
- Roussel, L.; Erard, M.; Cayrol, C.; Girard, J.P. Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Rep. 2008, 9, 1006–1012. [Google Scholar] [CrossRef] [Green Version]
- Bessa, J.; Meyer, C.A.; de Vera Mudry, M.C.; Schlicht, S.; Smith, S.H.; Iglesias, A.; Cote-Sierra, J. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J. Autoimmun. 2014, 55, 33–41. [Google Scholar] [CrossRef]
- Villarreal, D.O.; Weiner, D.B. Interleukin 33: A switch-hitting cytokine. Curr. Opin. Immunol. 2014, 28, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Luthi, A.U.; Cullen, S.P.; McNeela, E.A.; Duriez, P.J.; Afonina, I.S.; Sheridan, C.; Brumatti, G.; Taylor, R.C.; Kersse, K.; Vandenabeele, P.; et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009, 31, 84–98. [Google Scholar] [CrossRef]
- Bae, S.; Kang, T.; Hong, J.; Lee, S.; Choi, J.; Jhun, H.; Kwak, A.; Hong, K.; Kim, E.; Jo, S.; et al. Contradictory functions (activation/termination) of neutrophil proteinase 3 enzyme (PR3) in interleukin-33 biological activity. J. Biol. Chem. 2012, 287, 8205–8213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefrancais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Meephansan, J.; Komine, M.; Tsuda, H.; Karakawa, M.; Tominaga, S.; Ohtsuki, M. Expression of IL-33 in the epidermis: The mechanism of induction by IL-17. J. Dermatol. Sci. 2013, 71, 107–114. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008, 226, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Sundnes, O.; Pietka, W.; Loos, T.; Sponheim, J.; Rankin, A.L.; Pflanz, S.; Bertelsen, V.; Sitek, J.C.; Hol, J.; Haraldsen, G.; et al. Epidermal Expression and Regulation of Interleukin-33 during Homeostasis and Inflammation: Strong Species Differences. J. Investig. Dermatol. 2015, 135, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Lefrancais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.P. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef] [Green Version]
- Gearing, L.J.; Cumming, H.E.; Chapman, R.; Finkel, A.M.; Woodhouse, I.B.; Luu, K.; Gould, J.A.; Forster, S.C.; Hertzog, P.J. CiiiDER: A tool for predicting and analysing transcription factor binding sites. PLoS ONE 2019, 14, e0215495. [Google Scholar] [CrossRef] [Green Version]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Mohs, A.; Thomas, M.; Klare, J.; Ross, R.; Schmitz, M.L.; Martin, M.U. The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription. J. Immunol. 2011, 187, 1609–1616. [Google Scholar] [CrossRef] [Green Version]
- Balato, A.; Di Caprio, R.; Canta, L.; Mattii, M.; Lembo, S.; Raimondo, A.; Schiattarella, M.; Balato, N.; Ayala, F. IL-33 is regulated by TNF-α in normal and psoriatic skin. Arch. Dermatol. Res. 2014, 306, 299–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meephansan, J.; Tsuda, H.; Komine, M.; Tominaga, S.; Ohtsuki, M. Regulation of IL-33 expression by IFN-γ and tumor necrosis factor-α in normal human epidermal keratinocytes. J. Investig. Dermatol. 2012, 132, 2593–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soleymani, T.; Hung, T.; Soung, J. The role of vitamin D in psoriasis: A review. Int. J. Dermatol. 2015, 54, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Kechichian, E.; Ezzedine, K. Vitamin D and the Skin: An Update for Dermatologists. Am. J. Clin. Dermatol. 2018, 19, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Priyadarssini, M.; Divya Priya, D.; Indhumathi, S.; Rajappa, M.; Chandrashekar, L.; Thappa, D.M. Immunophenotyping of T cells in the peripheral circulation in psoriasis. Br. J. Biomed. Sci. 2016, 73, 174–179. [Google Scholar] [CrossRef]
- Bulek, K.; Swaidani, S.; Qin, J.; Lu, Y.; Gulen, M.F.; Herjan, T.; Min, B.; Kastelein, R.A.; Aronica, M.; Kosz-Vnenchak, M.; et al. The essential role of single Ig IL-1 receptor-related molecule/Toll IL-1R8 in regulation of Th2 immune response. J. Immunol. 2009, 182, 2601–2609. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, P.E.; Chen, Y.H.; Woszczek, G.; Matthews, N.C.; Chevretton, E.; Gupta, A.; Saglani, S.; Bush, A.; Corrigan, C.; Cousins, D.J.; et al. Vitamin D enhances production of soluble ST2, inhibiting the action of IL-33. J. Allergy Clin. Immunol. 2015, 135, 824–827.e823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrobot, A.; Demkow, U.; Wachowska, M. Immunomodulatory Role of Vitamin D: A Review. Adv. Exp. Med. Biol. 2018, 1108, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Lovato, P.; Norsgaard, H.; Tokura, Y.; Røpke, M.A. Calcipotriol and betamethasone dipropionate exert additive inhibitory effects on the cytokine expression of inflammatory dendritic cell-Th17 cell axis in psoriasis. J. Dermatol. Sci. 2016, 81, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusuba, N.; Kitoh, A.; Dainichi, T.; Honda, T.; Otsuka, A.; Egawa, G.; Nakajima, S.; Miyachi, Y.; Kabashima, K. Inhibition of IL-17-committed T cells in a murine psoriasis model by a vitamin D analogue. J. Allergy Clin. Immunol. 2018, 141, 972–981.e910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiani, G.; Kridin, K.; Pacifico, A.; Malagoli, P.; Pigatto, P.D.M.; Finelli, R.; Taccone, F.S.; Peluso, L.; Conic, R.R.Z.; Bragazzi, N.L.; et al. Antihistamines-refractory chronic pruritus in psoriatic patients undergoing biologics: Aprepitant vs antihistamine double dosage, a real-world data. J. Dermatol. Treat. 2020, 1–4. [Google Scholar] [CrossRef]
- Damiani, G.; Cazzaniga, S.; Conic, R.R.; Naldi, L. Pruritus Characteristics In A Large Italian Cohort Of Psoriatic Patients. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1316–1324. [Google Scholar] [CrossRef]
- Kircik, L.H.; Schlesinger, T.E.; Tanghetti, E. Efficacy and Safety of Calcipotriene 0.005%/Betamethasone Dipropionate 0.064% Foam With Apremilast for Moderate Plaque Psoriasis. J. Drugs Dermatol. 2020, 19, 874–880. [Google Scholar] [CrossRef]
- Sehat, M.; Talaei, R.; Dadgostar, E.; Nikoueinejad, H.; Akbari, H. Evaluating Serum Levels of IL-33, IL-36, IL-37 and Gene Expression of IL-37 in Patients with Psoriasis Vulgaris. Iran J. Allergy Asthma Immunol. 2018, 17, 179–187. [Google Scholar] [PubMed]
- Duan, Y.; Dong, Y.; Hu, H.; Wang, Q.; Guo, S.; Fu, D.; Song, X.; Kalvakolanu, D.V.; Tian, Z. IL-33 contributes to disease severity in Psoriasis-like models of mouse. Cytokine 2019, 119, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicka, J.M.; Zmijewski, M.A.; Antoniewicz, J.; Sobjanek, M.; Slominski, A.T. Differentiation of Keratinocytes Modulates Expression of Epidermal Elements of the Cutaneous Analog of Hypothalamus-Pituitary-Adrenal Axis. J. Cell Physiol. 2016. [Google Scholar] [CrossRef]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018, 19, e46255. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| RPL37 | TTCTGATGGCGGACTTTACC | CACTTGCTCTTTCTGTGGCA |
| IL-33 | GTGGAAGAACACAGCAAGCA | GTGGAAGAACACAGCAAGCA |
| ST2 | CAGCACCTCTTGAGTGGTTTA | TCGCCGTCACACTATAATTGG |
| TNF | CCAGGGACCTCTCTCTAATCA | TCAGCTTGAGGGTTTGCTAC |
| IL-1B | ATCTCCGACCACCACTACA | GAAGGTGCTCAGGTCATTCTC |
| IFNG | GGAAAGAGGAGAGTGACAGAAA | TCATGTCTTCCTTGATGGTCTC |
| IL-17A | GGTTAAATGGACCCTGGCCT | ATAGTTGGTGTGCGGCTGAA |
| VDR | CTGTGGCAACCAAGACTACA | CATGCAAGTTCAGCTTCTTCAG |
| CYP24A1 | GCAGCCTAGTGCAGATTT | ATTCACCCAGAACTGTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wierzbicka, J.M.; Piotrowska, A.; Purzycka-Bohdan, D.; Olszewska, A.; Nowak, J.I.; Szczerkowska-Dobosz, A.; Nedoszytko, B.; Nowicki, R.J.; Żmijewski, M.A. The Effects of Vitamin D on the Expression of IL-33 and Its Receptor ST2 in Skin Cells; Potential Implication for Psoriasis. Int. J. Mol. Sci. 2021, 22, 12907. https://doi.org/10.3390/ijms222312907
Wierzbicka JM, Piotrowska A, Purzycka-Bohdan D, Olszewska A, Nowak JI, Szczerkowska-Dobosz A, Nedoszytko B, Nowicki RJ, Żmijewski MA. The Effects of Vitamin D on the Expression of IL-33 and Its Receptor ST2 in Skin Cells; Potential Implication for Psoriasis. International Journal of Molecular Sciences. 2021; 22(23):12907. https://doi.org/10.3390/ijms222312907
Chicago/Turabian StyleWierzbicka, Justyna M., Anna Piotrowska, Dorota Purzycka-Bohdan, Anna Olszewska, Joanna I. Nowak, Aneta Szczerkowska-Dobosz, Bogusław Nedoszytko, Roman J. Nowicki, and Michał A. Żmijewski. 2021. "The Effects of Vitamin D on the Expression of IL-33 and Its Receptor ST2 in Skin Cells; Potential Implication for Psoriasis" International Journal of Molecular Sciences 22, no. 23: 12907. https://doi.org/10.3390/ijms222312907
APA StyleWierzbicka, J. M., Piotrowska, A., Purzycka-Bohdan, D., Olszewska, A., Nowak, J. I., Szczerkowska-Dobosz, A., Nedoszytko, B., Nowicki, R. J., & Żmijewski, M. A. (2021). The Effects of Vitamin D on the Expression of IL-33 and Its Receptor ST2 in Skin Cells; Potential Implication for Psoriasis. International Journal of Molecular Sciences, 22(23), 12907. https://doi.org/10.3390/ijms222312907