
Rho-Kinase as a Target for Cancer Therapy and Its Immunotherapeutic Potential

Abstract
:1. Introduction
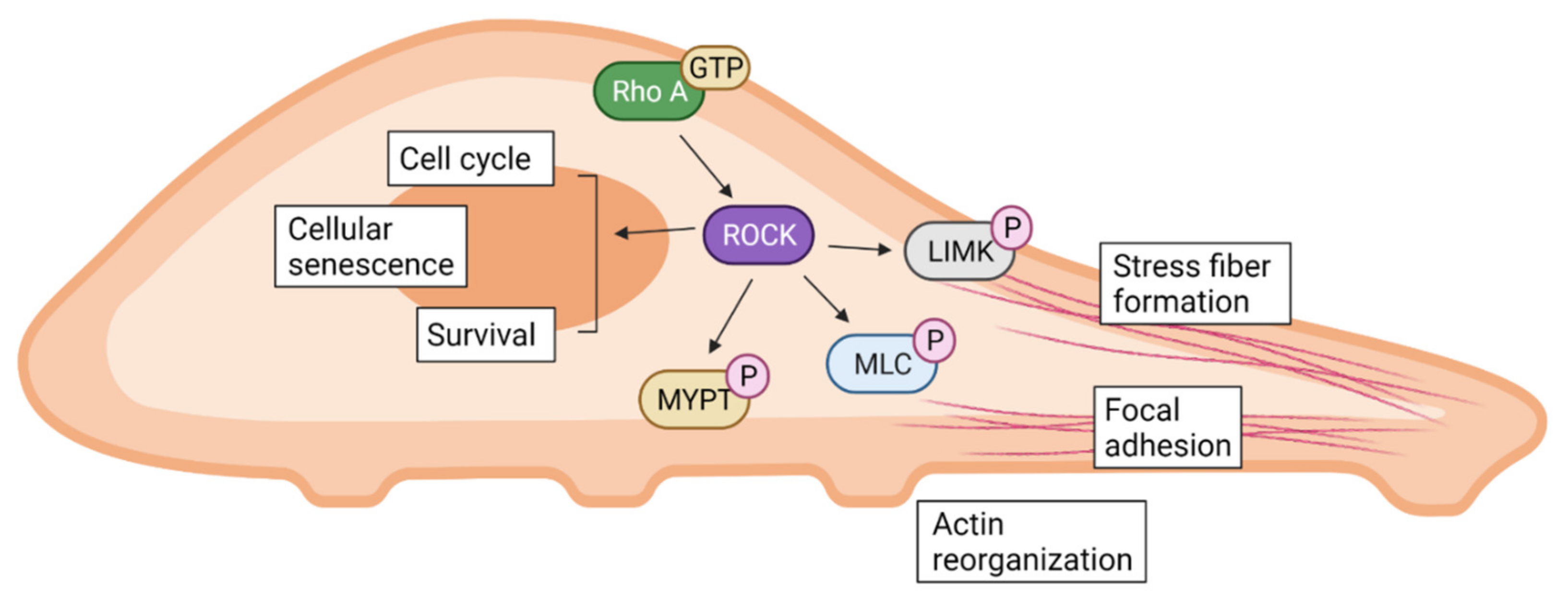
2. Rho-Kinase (ROCK)
3. Targeting ROCK for Cancer Treatment
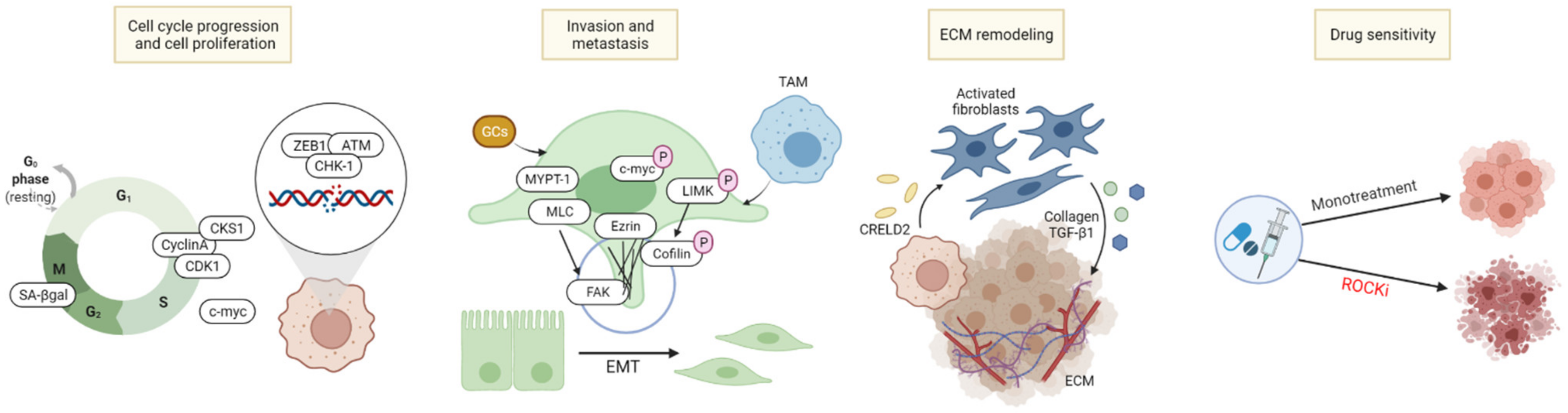
3.1. Cell Cycle Arrest/Cell Death Induction
3.2. Inhibition of Tumor Metastasis, Invasion, and Migration
3.3. ECM Remodeling
3.4. Combinatory Effects with Other Chemotherapeutic Agents
4. ROCK Regulates Immune Cells
4.1. Dendritic Cells and Macrophages
4.2. T-Cells
4.3. Natural Killer Cells (NK Cells)
4.4. Other Immune Cells
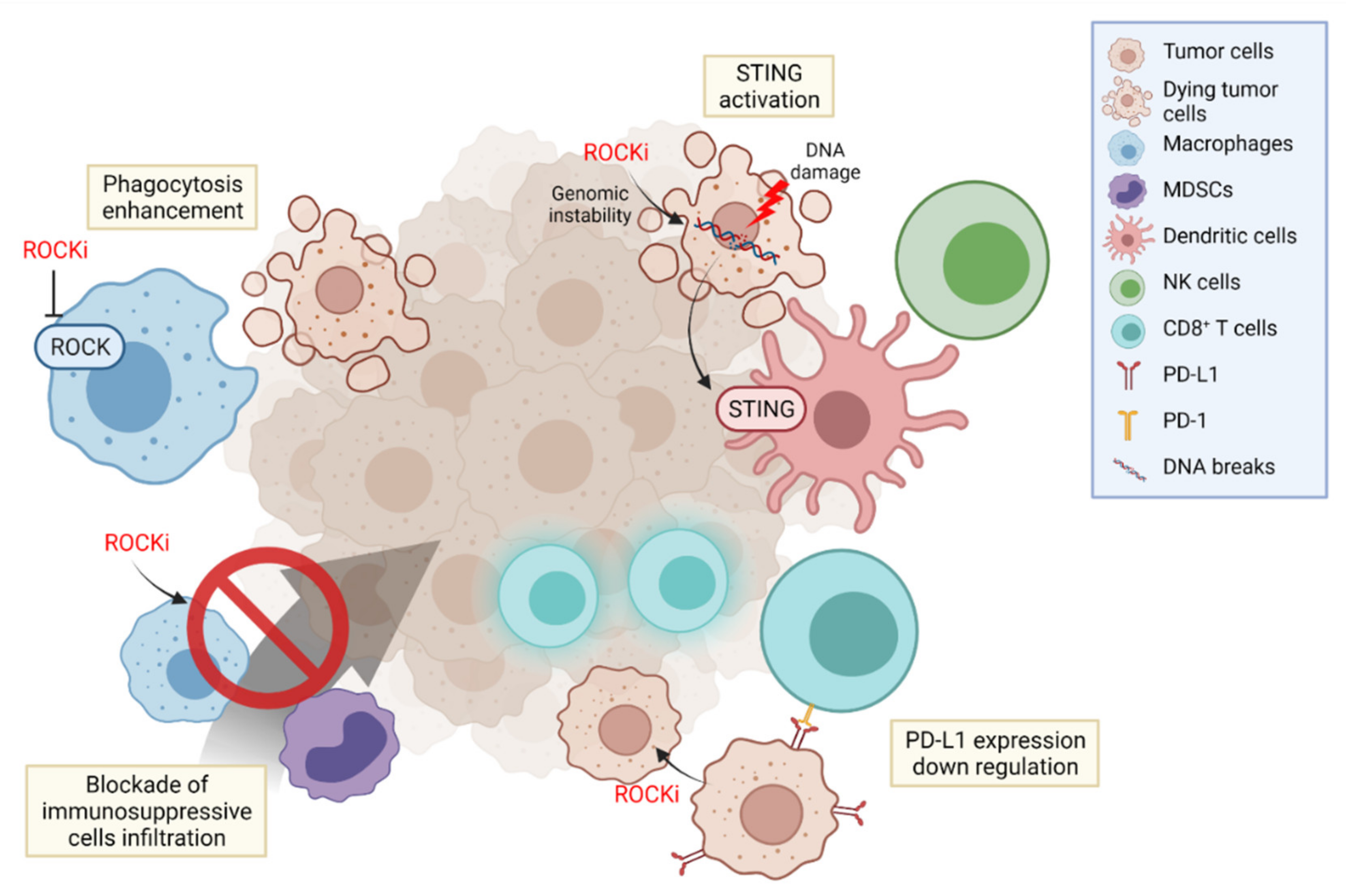
5. ROCK and Cancer Immunotherapy
5.1. Regulation of Phagocytosis
5.2. Activating Innate Immune System
5.3. PD-L1 Depletion
5.4. Overcoming Resistance to Immunotherapy
5.5. YAP Inhibition
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Munhoz, R.R.; Postow, M.A. Clinical development of PD-1 in advanced Melanoma. Cancer J. 2018, 24, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M. PD-1 Blockade in classic Hodgkin Lymphoma. JCO Oncol. Pr. 2021, 17, 72–73. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.; Bhatia, S.; Kaufman, H.L.; Lipson, E.J. Immunotherapy for Merkel cell carcinoma: A turning point in patient care. J. Immunother. Cancer 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Gajewski, T.F. Mechanisms of tumor cell–intrinsic immune evasion. Annu. Rev. Cancer Biol. 2018, 2, 213–228. [Google Scholar] [CrossRef]
- Yang, Y.; Nam, G.-H.; Kim, G.B.; Kim, Y.K.; Kim, I.-S. Intrinsic cancer vaccination. Adv. Drug Deliv. Rev. 2019, 151, 2–22. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Saha, B.C.; Kumari, R.; Kushumesh, R.; Ambasta, A.; Sinha, B.P. Status of Rho kinase inhibitors in glaucoma therapeutics—An overview. Int. Ophthalmol. 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Niftullayev, S.; Lamarche-Vane, N. Regulators of Rho GTPases in the nervous system: Molecular implication in axon guidance and neurological disorders. Int. J. Mol. Sci. 2019, 20, 1497. [Google Scholar] [CrossRef] [Green Version]
- Pernis, A.B.; Ricker, E.; Weng, C.-H.; Rozo, C.; Yi, W. Rho Kinases in autoimmune diseases. Annu. Rev. Med. 2016, 67, 355–374. [Google Scholar] [CrossRef]
- Clayton, N.S.; Ridley, A.J. Targeting Rho GTPase signaling networks in cancer. Front. Cell Dev. Biol. 2020, 8, 222. [Google Scholar] [CrossRef]
- Defert, O.; Boland, S. Rho kinase inhibitors: A patent review (2014–2016). Expert Opin. Ther. Pat. 2017, 27, 507–515. [Google Scholar] [CrossRef]
- Tanihara, H.; Kakuda, T.; Sano, T.; Kanno, T.; Imada, R.; Shingaki, W.; Gunji, R. Safety and efficacy of Ripasudil in Japanese patients with Glaucoma or Ocular Hypertension: 3-month interim analysis of ROCK-J, a post-marketing surveillance study. Adv. Ther. 2019, 36, 333–343. [Google Scholar] [CrossRef]
- Zhao, J.; Zhou, D.; Guo, J.; Ren, Z.; Zhou, L.; Wang, S.; Xu, B.; Wang, R. Effect of Fasudil Hydrochloride, a protein kinase inhibitor, on cerebral Vasospasm and delayed cerebral Ischemic symptoms after aneurysmal Subarachnoid Hemorrhage-results of a randomized trial of Fasudil Hydrochloride versus Nimodipine-. Neurol. Med. Chir. 2006, 46, 421–428. [Google Scholar] [CrossRef] [Green Version]
- McLeod, R.; Kumar, R.; Papadatos-Pastos, D.; Mateo, J.; Brown, J.S.; Garces, A.H.I.; Ruddle, R.; Decordova, S.; Jueliger, S.; Ferraldeschi, R.; et al. First-in-human study of AT13148, a dual ROCK-AKT inhibitor in Patients with Solid Tumors. Clin. Cancer Res. 2020, 26, 4777–4784. [Google Scholar] [CrossRef]
- Leung, T.; Manser, E.; Tan, L.; Lim, L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J. Biol. Chem. 1995, 270, 29051–29054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Amano, M.; Yamamoto, T.; Chihara, K.; Nakafuku, M.; Ito, M.; Nakano, T.; Okawa, K.; Iwamatsu, A.; Kaibuchi, K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996, 15, 2208–2216. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Amano, M.; Chihara, K.; Nakamura, N.; Kaneko, T.; Matsuura, Y.; Kaibuchi, K. The COOH terminus of Rho-kinase negatively regulates Rho-kinase activity. J. Biol. Chem. 1999, 274, 32418–32424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, O.; Fujisawa, K.; Ishizaki, T.; Saito, Y.; Nakao, K.; Narumiya, S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996, 392, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, A.P.; Ridley, A. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp. Cell Res. 2004, 301, 43–49. [Google Scholar] [CrossRef]
- Sapet, C.; Simoncini, S.; Loriod, B.; Puthier, D.; Sampol, J.; Nguyen, C.; Dignat-George, F.; Anfosso, F. Thrombin-induced endothelial microparticle generation: Identification of a novel pathway involving ROCK-II activation by caspase-2. Blood 2006, 108, 1868–1876. [Google Scholar] [CrossRef]
- Sebbagh, M.; Renvoizé, C.; Hamelin, J.; Riché, N.; Bertoglio, J.; Bréard, J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 2001, 3, 346–352. [Google Scholar] [CrossRef]
- Sebbagh, M.; Hamelin, J.; Bertoglio, J.; Solary, E.; Bréard, J. Direct cleavage of ROCK II by granzyme B induces target cell membrane blebbing in a caspase-independent manner. J. Exp. Med. 2005, 201, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Julian, L.; Olson, M.F. Rho-associated coiled-coil containing kinases (ROCK). Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Fukata, Y.; Kaibuchi, K.; Amano, M. Rho–Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol. Sci. 2001, 22, 32–39. [Google Scholar] [CrossRef]
- Ishizaki, T.; Naito, M.; Fujisawa, K.; Maekawa, M.; Watanabe, N.; Saito, Y.; Narumiya, S. p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett. 1997, 404, 118–124. [Google Scholar] [CrossRef] [Green Version]
- Leung, T.; Chen, X.Q.; Manser, E.; Lim, L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 1996, 16, 5313–5327. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wu, X.; Surma, M.; Vemula, S.; Zhang, L.; Yang, Y.; Kapur, R.; Wei, L. Distinct roles for ROCK1 and ROCK2 in the regulation of cell detachment. Cell Death Dis. 2013, 4, e483. [Google Scholar] [CrossRef] [Green Version]
- Lock, F.E.; Ryan, K.R.; Poulter, N.S.; Parsons, M.; Hotchin, N.A. Differential regulation of adhesion complex turnover by ROCK1 and ROCK2. PLoS ONE 2012, 7, e31423. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, A.; Ushakov, D.; Multhaupt, H.A.; Couchman, J.R. Fibronectin matrix assembly requires distinct contributions from Rho Kinases I and -II. Mol. Biol. Cell 2007, 18, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, A.J. Rho GTPases and cell migration. J. Cell Sci. 2001, 114, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Singleton, P.A.; Bourguignon, L.Y. CD44v10 interaction with Rho-kinase (ROK) activates inositol 1,4,5-triphosphate (IP3) receptor-mediated Ca2+ signaling during hyaluronan (HA)-induced endothelial cell migration. Cell Motil. Cytoskelet. 2002, 53, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Niggli, V. Rho-kinase in human neutrophils: A role in signalling for myosin light chain phosphorylation and cell migration. FEBS Lett. 1999, 445, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Ai, S.; Kuzuya, M.; Koike, T.; Asai, T.; Kanda, S.; Maeda, K.; Shibata, T.; Iguchi, A. Rho–Rho kinase is involved in smooth muscle cell migration through myosin light chain phosphorylation-dependent and independent pathways. Atherosclerosis 2001, 155, 321–327. [Google Scholar] [CrossRef]
- Shi, J.; Wei, L. Rho kinase in the regulation of cell death and survival. Arch. Immunol. Ther. Exp. 2007, 55, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, M.; Petit, M.; Bertoglio, J. Cell cycle regulation of Rho signaling pathways. Cell Cycle 2012, 11, 3003–3010. [Google Scholar] [CrossRef] [Green Version]
- Kümper, S.; Mardakheh, F.; McCarthy, A.; Yeo, M.; Stamp, G.W.; Paul, A.; Worboys, J.; Sadok, A.; Jorgensen, C.; Guichard, S.; et al. Rho-associated kinase (ROCK) function is essential for cell cycle progression, senescence and tumorigenesis. eLife 2016, 5, e12203. [Google Scholar] [CrossRef]
- Wei, L.; Surma, M.; Shi, S.; Lambert-Cheatham, N.; Shi, J. Novel insights into the roles of Rho Kinase in cancer. Arch. Immunol. Ther. Exp. 2016, 64, 259–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gökmen-Polar, Y. Roles of Rho/ROCK in cancer signaling. In Predictive Biomarkers in Oncology; Springer: Cham, Switzerland, 2018; pp. 207–212. [Google Scholar] [CrossRef]
- Lee, M.-H.; Kundu, J.K.; Chae, J.-I.; Shim, J.-H. Targeting ROCK/LIMK/cofilin signaling pathway in cancer. Arch. Pharmacal Res. 2019, 42, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Bottino, J.; Gelaleti, G.B.; Maschio, L.B.; Jardim-Perassi, B.V.; de Campos Zuccari, D.A.P. Immunoexpression of ROCK-1 and MMP-9 as prognostic markers in breast cancer. Acta Histochem. 2014, 116, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Vennin, C.; Rath, N.; Pajic, M.; Olson, M.F.; Timpson, P. Targeting ROCK activity to disrupt and prime pancreatic cancer for chemotherapy. Small GTPases 2020, 11, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sari, I.; Berberoglu, B.; Ozkara, E.; Oztuzcu, S.; Camci, C.; Demiryürek, A.T. Role of Rho-Kinase gene Polymorphisms and protein expressions in colorectal cancer development. Pathobiology 2013, 80, 138–145. [Google Scholar] [CrossRef]
- Wong, C.C.-L.; Wong, C.-M.; Tung, E.K.-K.; Man, K.; Ng, I.O.-L. Rho-kinase 2 is frequently overexpressed in hepatocellular carcinoma and involved in tumor invasion. Hepatology 2009, 49, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Dyberg, C.; Andonova, T.; Olsen, T.K.; Brodin, B.; Kool, M.; Kogner, P.; Johnsen, J.I.; Wickström, M. Inhibition of Rho-associated Kinase suppresses Medulloblastoma growth. Cancers 2019, 12, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyberg, C.; Fransson, S.; Andonova, T.; Sveinbjörnsson, B.; Lännerholm-Palm, J.; Olsen, T.K.; Forsberg, D.; Herlenius, E.; Martinsson, T.; Brodin, B.; et al. Rho-associated kinase is a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. USA 2017, 114, E6603–E6612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaya, C.N.; Mitchell, D.C.; Bryan, B.A. Rho kinase proteins display aberrant upregulation in vascular tumors and contribute to vascular tumor growth. BMC Cancer 2017, 17, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Biroc, S.L.; Li, W.-W.; Alicke, B.; Xuan, J.-A.; Pagila, R.; Ohashi, Y.; Okada, T.; Kamata, Y.; Dinter, H. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol. Cancer Ther. 2006, 5, 2158–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, N.; Munro, J.; Cutiongco, M.F.; Jagiełło, A.; Gadegaard, N.; McGarry, L.; Unbekandt, M.; Michalopoulou, E.; Kamphorst, J.J.; Sumpton, D.; et al. Rho kinase inhibition by AT13148 blocks pancreatic ductal Adenocarcinoma invasion and tumor growth. Cancer Res. 2018, 78, 3321–3336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routhier, A.; Astuccio, M.; Lahey, D.; Monfredo, N.; Johnson, A.; Callahan, W.; Partington, A.; Fellows, K.; Ouellette, L.; Zhidro, S.; et al. Pharmacological inhibition of Rho-kinase signaling with Y-27632 blocks melanoma tumor growth. Oncol. Rep. 2010, 23, 861–867. [Google Scholar] [PubMed]
- Somlyo, A.V.; Bradshaw, D.; Ramos, S.; Murphy, C.; Myers, C.E.; Somlyo, A.P. Rho-Kinase inhibitor retards migration and in vivo dissemination of human prostate cancer cells. Biochem. Biophys. Res. Commun. 2000, 269, 652–659. [Google Scholar] [CrossRef]
- Abe, H.; Kamai, T.; Hayashi, K.; Anzai, N.; Shirataki, H.; Mizuno, T.; Yamaguchi, Y.; Masuda, A.; Yuki, H.; Betsunoh, H.; et al. The Rho-kinase inhibitor HA-1077 suppresses proliferation/migration and induces apoptosis of urothelial cancer cells. BMC Cancer 2014, 14, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chircop, M. Rho GTPases as regulators of mitosis and cytokinesis in mammalian cells. Small GTPases 2014, 5, e29770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosako, H.; Yoshida, T.; Matsumura, F.; Ishizaki, T.; Narumiya, S.; Inagaki, M. Rho-kinase/ROCK is involved in cytokinesis through the phosphorylation of myosin light chain and not ezrin/radixin/moesin proteins at the cleavage furrow. Oncogene 2000, 19, 6059–6064. [Google Scholar] [CrossRef] [PubMed]
- Madaule, P.; Eda, M.; Watanabe, N.; Fujisawa, K.; Matsuoka, T.; Bito, H.; Ishizaki, T.; Narumiya, S. Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nat. Cell Biol. 1998, 394, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, Y.; Mehdiabad, M.V.; Zhou, K.; Chen, Y.; Li, L.; Guo, J.; Xu, C. Enhanced anti-tumor effect of liposomal Fasudil on hepatocellular carcinoma in vitro and in vivo. PLoS ONE 2019, 14, e0223232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Li, G.; Li, R.; Liu, Q.; He, Q.; Zhang, J. Rho-kinase inhibitor, fasudil, suppresses glioblastoma cell line progression in vitro and in vivo. Cancer Biol. Ther. 2010, 9, 875–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Z.; Su, Y.; Dong, Y.; Zheng, Y.; Zhang, Q.; Duan, Y.; Wang, G. Rho-kinase inhibition by Fasudil promotes tumor maturation and apoptosis in small-cell lung cancer. Am. J. Transl. Res. 2020, 12, 4354–4370. [Google Scholar] [PubMed]
- He, M.; Luo, M.; Liu, Q.; Chen, J.; Li, K.; Zheng, M.; Weng, Y.; Ouyang, L.; Liu, A. Combination treatment with fasudil and clioquinol produces synergistic anti-tumor effects in U87 glioblastoma cells by activating apoptosis and autophagy. J. NeuroOncology 2016, 127, 261–270. [Google Scholar] [CrossRef]
- Xie, F.-J.; Zheng, Q.-Q.; Qin, J.; Zhang, L.-L.; Han, N.; Mao, W.-M. Autophagy inhibition stimulates apoptosis in Oesophageal Squamous cell Carcinoma treated with Fasudil. J. Cancer 2018, 9, 1050–1056. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, Y.T.; Farias, J.O.; Silva, L.E.; Forti, F.L. GTPases, genome, actin: A hidden story in DNA damage response and repair mechanisms. DNA Repair 2021, 100, 103070. [Google Scholar] [CrossRef]
- Herraiz, C.; Calvo, F.; Pandya, P.; Cantelli, G.; Rodriguez-Hernandez, I.; Orgaz, J.; Kang, N.; Chu, T.; Sahai, E.; Sanz-Moreno, V. Reactivation of p53 by a cytoskeletal sensor to control the balance between DNA damage and tumor dissemination. J. Natl. Cancer Inst. 2016, 108, djv289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-F.; Takenaka, K.; Nakanishi, A.; Miki, Y. BRCA2 and Nucleophosmin coregulate centrosome amplification and form a complex with the Rho effector kinase ROCK2. Cancer Res. 2011, 71, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, Y.; Wang, K.; Li, T.; Zhang, M.; Yang, Y.; Wang, R.; Hu, R. ROCK2 Confers acquired gemcitabine resistance in pancreatic cancer cells by Upregulating transcription factor ZEB1. Cancers 2019, 11, 1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pranatharthi, A.; Thomas, P.; Udayashankar, A.H.; Bhavani, C.; Suresh, S.B.; Krishna, S.; Thatte, J.; Srikantia, N.; Ross, C.R.; Srivastava, S. RhoC regulates radioresistance via crosstalk of ROCK2 with the DNA repair machinery in cervical cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Wolf, K. Plasticity of cell migration: A multiscale tuning model. J. Cell Biol. 2010, 188, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadok, A.; McCarthy, A.; Caldwell, J.; Collins, I.; Garrett, M.D.; Yeo, M.; Hooper, S.; Sahai, E.; Kümper, S.; Mardakheh, F.; et al. Rho Kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res. 2015, 75, 2272–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genda, T.; Sakamoto, M.; Ichida, T.; Asakura, H.; Kojiro, M.; Narumiya, S.; Hirohashi, S. Cell motility mediated by rho and rho-associated protein kinase plays a critical role in intrahepatic metastasis of human hepatocellular carcinoma. Hepatology 1999, 30, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, D.; Guo, Z.; Zhao, J.; Wu, B.; Deng, H.; Zhou, T.; Xiang, H.; Gao, F.; Yu, X.; et al. Rho Kinase phosphorylation promotes ezrin-mediated metastasis in hepatocellular Carcinoma. Cancer Res. 2011, 71, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Goldstein, R.H.; Scepansky, E.M.; Rosenblatt, M. Inhibition of Rho-associated kinase signaling prevents breast cancer metastasis to human bone. Cancer Res. 2009, 69, 8742–8751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhang, S.; Zhang, Z.; He, J.; Xu, Y.; Liu, S. ROCK has a crucial role in regulating prostate tumor growth through interaction with c-Myc. Oncogene 2013, 33, 5582–5591. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Choi, P.; Casey, S.C.; Dill, D.L.; Felsher, D.W. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell 2014, 26, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosla, J.; Paňková, D.; Plachý, J.; Tolde, O.; Bicanová, K.; Dvořák, M.; Rösel, D.; Brábek, J. Metastasis of aggressive amoeboid sarcoma cells is dependent on Rho/ROCK/MLC signaling. Cell Commun. Signal. 2013, 11, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilkes, D.M.; Xiang, L.; Lee, S.J.; Chaturvedi, P.; Hubbi, M.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signaling in breast cancer cells. Proc. Natl. Acad. Sci. USA 2014, 111, E384–E393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semprucci, E.; Tocci, P.; Cianfrocca, R.; Sestito, R.; Caprara, V.; Veglione, M.; di Castro, V.; Spadaro, F.; Ferrandina, M.G.; Bagnato, A.; et al. Endothelin A receptor drives invadopodia function and cell motility through the β-arrestin/PDZ-RhoGEF pathway in ovarian carcinoma. Oncogene 2016, 35, 3432–3442. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.; Chen, Y.-C.; Madden, J.M.; Fournier, C.L.; Altemus, M.A.; Hiziroglu, A.B.; Cheng, Y.-H.; Wu, Z.F.; Bao, L.; Yates, J.; et al. Macrophages enhance migration in inflammatory breast cancer cells via RhoC GTPase signaling. Sci. Rep. 2016, 6, 39190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, A.C.; Pathanjeli, P.; Wu, Z.; Bao, L.; Goo, L.E.; Yates, J.; Oliver, C.R.; Soellner, M.B.; Merajver, S.D. IL-4/IL-13 Stimulated macrophages enhance breast cancer invasion Via Rho-GTPase regulation of synergistic VEGF/CCL-18 signaling. Front. Oncol. 2019, 9, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obradovic, M.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.-M.; Muenst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids promote breast cancer metastasis. Nat. Cell Biol. 2019, 567, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-X.; Wang, Y.; Su, J.; Zhou, P.; Li, B.; Yin, L.-J.; Lu, J. Up-regulation of Rho-associated kinase 1/2 by glucocorticoids promotes migration, invasion and metastasis of melanoma. Cancer Lett. 2017, 410, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Gosens, R.; Wieland, T.; Schmidt, M. Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacol. Ther. 2018, 183, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Deng, S.; Gopal, V.; Yap, K.; Halim, C.; Lye, M.; Ong, M.; Tan, T.; Sethi, G.; Hooi, S.; et al. Cytoskeletal dynamics in Epithelial-Mesenchymal transition: Insights into therapeutic targets for cancer metastasis. Cancers 2021, 13, 1882. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Rong, G.; Li, X.; Geng, L.; Zeng, Z.; Jiang, D.; Yang, J.; Wei, Y. Diosgenin and GSK126 produce synergistic effects on Epithelial–Mesenchymal transition in gastric cancer cells by mediating EZH2 via the Rho/ROCK signaling pathway. OncoTargets Ther. 2020, 13, 5057–5067. [Google Scholar] [CrossRef]
- Jung, J.; Yang, K.; Kim, H.-J.; Lee, Y.-J.; Kim, M.; Choi, Y.-H.; Kang, J.L. RhoA-dependent HGF and c-Met mediate Gas6-Induced inhibition of Epithelial–Mesenchymal transition, migration, and invasion of lung alveolar epithelial cells. Biomololecules 2019, 9, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, X.; Liu, Y.; Yi, B.; Yu, X. Epithelial–mesenchymal transition of rat peritoneal mesothelial cells via Rhoa/Rock pathway. Vitr. Cell. Dev. Biol. Anim. 2010, 47, 165–172. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-β1 mediates Epithelial to Mesenchymal Transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Witte, D.; Lehnert, H. The role of small GTPases of the Rho/Rac family in TGF-β-induced EMT and cell motility in cancer. Dev. Dyn. 2018, 247, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, S.; Wang, Z.; Wang, F.; Cao, X.; Wu, Q.; Zhao, C.; Ma, H.; Ye, F.; Wang, H.; et al. Supervillin promotes epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma in hypoxia via activation of the RhoA/ROCK-ERK/p38 pathway. J. Exp. Clin. Cancer Res. 2018, 37, 128. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Kümper, S.; Marshall, C.J. ROCK-driven Actomyosin contractility induces tissue stiffness and tumor growth. Cancer Cell 2011, 19, 695–697. [Google Scholar] [CrossRef] [Green Version]
- Wyckoff, J.B.; Pinner, S.E.; Gschmeissner, S.; Condeelis, J.S.; Sahai, E. ROCK- and Myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr. Biol. 2006, 16, 1515–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, M.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Munro, J.; Schröder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and β-Catenin activation to induce epidermal Hyperplasia and tumor growth. Cancer Cell 2011, 19, 776–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Street, C.A.; Bryan, B.A. Rho kinase proteins--pleiotropic modulators of cell survival and apoptosis. Anticancer. Res. 2011, 31, 3645–3657. [Google Scholar] [PubMed]
- Whatcott, C.J.; Ng, S.; Barrett, M.T.; Hostetter, G.; Von Hoff, D.D.; Han, H. Inhibition of ROCK1 kinase modulates both tumor cells and stromal fibroblasts in pancreatic cancer. PLoS ONE 2017, 12, e0183871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vennin, C.; Chin, V.T.; Warren, S.C.; Lucas, M.C.; Herrmann, D.; Magenau, A.; Melenec, P.; Walters, S.N.; del Monte-Nieto, G.; Conway, J.R.W.; et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, S.; Poltavets, V.; Kular, J.; Pyne, N.T.; Sandow, J.J.; Lewis, A.C.; Murphy, K.J.; Kolesnikoff, N.; Moretti, P.A.B.; Tea, M.N.; et al. ROCK-mediated selective activation of PERK signalling causes fibroblast reprogramming and tumour progression through a CRELD2-dependent mechanism. Nat. Cell Biol. 2020, 22, 1–14. [Google Scholar] [CrossRef]
- Azab, A.K.; Azab, F.; Blotta, S.; Pitsillides, C.M.; Thompson, B.; Runnels, J.M.; Roccaro, A.M.; Ngo, H.T.; Melhem, M.R.; Sacco, A.; et al. RhoA and Rac1 GTPases play major and differential roles in stromal cell–derived factor-1–induced cell adhesion and chemotaxis in multiple myeloma. Blood 2009, 114, 619–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federico, C.; Alhallak, K.; Sun, J.; Duncan, K.; Azab, F.; Sudlow, G.P.; De La Puente, P.; Muz, B.; Kapoor, V.; Zhang, L.; et al. Tumor microenvironment-targeted nanoparticles loaded with bortezomib and ROCK inhibitor improve efficacy in multiple myeloma. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Gao, L.; Huang, S.; Zhang, H.; Hua, W.; Xin, S.; Cheng, L.; Guan, W.; Yu, Y.; Mao, Y.; Pei, G. Suppression of glioblastoma by a drug cocktail reprogramming tumor cells into neuronal like cells. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Okada, M.; Suzuki, S.; Kuramoto, K.; Sakaki, H.; Watarai, H.; Sanomachi, T.; Seino, S.; Yoshioka, T.; Kitanaka, C. Rho-associated protein kinase (ROCK) inhibitors inhibit survivin expression and sensitize pancreatic cancer stem cells to Gemcitabine. Anticancer. Res. 2016, 36, 6311–6318. [Google Scholar] [CrossRef] [Green Version]
- Iskit, S.; Lieftink, C.; Halonen, P.; Shahrabi, A.; Possik, P.A.; Beijersbergen, R.L.; Peeper, D.S. Integrated in vivo genetic and pharmacologic screening identifies co-inhibition of EGRF and ROCK as a potential treatment regimen for triple-negative breast cancer. Oncotarget 2016, 7, 42859–42872. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hu, K.; Guo, J.; Cheng, F.; Lv, J.; Jiang, W.; Lu, W.; Liu, J.; Pang, X.; Liu, M. Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat. Commun. 2016, 7, 11363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.J.; Smit, M.A.; Maddalo, G.; Possik, P.A.; Sparidans, R.W.; Van Der Burg, S.H.; Verdegaal, E.M.; Heck, A.J.R.; Samatar, A.A.; Beijnen, J.H.; et al. Cooperative induction of apoptosis in NRAS mutant melanoma by inhibition of MEK and ROCK. Pigment. Cell Melanoma Res. 2015, 28, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Howard, G.A.; Pittman, K.; Boykin, C.; Herring, L.E.; Wilkerson, E.M.; Verbanac, K.; Lu, Q. Therapeutic effect of Y-27632 on Tumorigenesis and Cisplatin-induced peripheral sensory loss through RhoA-NF-κB. Mol. Cancer Res. 2019, 17, 1910–1919. [Google Scholar] [CrossRef]
- Park, S.-Y.; Kim, I.-S. Harnessing immune checkpoints in myeloid lineage cells for cancer immunotherapy. Cancer Lett. 2019, 452, 51–58. [Google Scholar] [CrossRef]
- Tosello-Trampont, A.-C.; Nakada-Tsukui, K.; Ravichandran, K.S. Engulfment of Apoptotic cells is negatively regulated by Rho-mediated signaling. J. Biol. Chem. 2003, 278, 49911–49919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-Y.; Kim, S.; Bae, D.-J.; Park, S.-Y.; Lee, G.-Y.; Park, G.-M.; Kim, I.-S. Coordinated balance of Rac1 and RhoA plays key roles in determining phagocytic appetite. PLoS ONE 2017, 12, e0174603. [Google Scholar] [CrossRef]
- Hanley, P.J.; Xu, Y.; Kronlage, M.; Grobe, K.; Schon, P.; Song, J.; Sorokin, L.; Schwab, A.; Bahler, M. Motorized RhoGAP myosin IXb (Myo9b) controls cell shape and motility. Proc. Natl. Acad. Sci. USA 2010, 107, 12145–12150. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Pektor, S.; Balkow, S.; Hemkemeyer, S.A.; Liu, Z.; Grobe, K.; Hanley, P.J.; Shen, L.; Bros, M.; Schmidt, T.; et al. Dendritic cell motility and T cell activation requires regulation of Rho-cofilin signaling by the Rho-GTPase activating protein myosin IXb. J. Immunol. 2014, 192, 3559–3568. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Azuma, E.; Ido, M.; Hirayama, M.; Jiang, Q.; Iwamoto, S.; Kumamoto, T.; Yamamoto, H.; Sakurai, M.; Komada, Y. A Pivotal role of Rho GTPase in the regulation of morphology and function of Dendritic cells. J. Immunol. 2001, 167, 3585–3591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Lee, S.W.; Lee, W.S.; Rhim, B.Y.; Lee, S.J.; Kwon, S.M.; Hong, K.W.; Kim, C.D. RhoA/ROCK-dependent pathway is required for TLR2-mediated IL-23 production in human synovial macrophages: Suppression by cilostazol. Biochem. Pharmacol. 2013, 86, 1320–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandi, S.; Nakao, S.; Chun, K.-H.; Fiorina, P.; Sun, D.; Arita, R.; Zhao, M.; Kim, E.; Schueller, O.; Campbell, S.; et al. ROCK-Isoform-specific polarization of Macrophages associated with age-related macular degeneration. Cell Rep. 2015, 10, 1173–1186. [Google Scholar] [CrossRef] [Green Version]
- Hasan, Z.; Palani, K.S.H.; Zhang, S.; Lepsenyi, M.; Hwaiz, R.; Rahman, M.; Syk, I.; Jeppsson, B.; Thorlacius, H. Rho Kinase regulates induction of T-cell immune dysfunction in abdominal Sepsis. Infect. Immun. 2013, 81, 2499–2506. [Google Scholar] [CrossRef] [Green Version]
- Thauland, T.J.; Hu, K.H.; Bruce, M.A.; Butte, M.J. Cytoskeletal adaptivity regulates T cell receptor signaling. Sci. Signal. 2017, 10, eaah3737. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.; Bracke, M.; Leitinger, B.; Porter, J.; Hogg, N. LFA-1-induced T cell migration on ICAM-1 involves regulation of MLCK-mediated attachment and ROCK-dependent detachment. J. Cell Sci. 2003, 116, 3123–3133. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Yang, Y.; Mei, C.; Dong, P.; Mu, S.; Wu, H.; Zhou, Y.; Zheng, Y.; Guo, F.; Yang, J.-Q. Inhibition of Rho-Kinase downregulates Th17 cells and ameliorates hepatic fibrosis by Schistosoma japonicum infection. Cells 2019, 8, 1262. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Nyuydzefe, M.S.; Weiss, J.M.; Zhang, J.; Waksal, S.D.; Zanin-Zhorov, A. ROCK2, but not ROCK1 interacts with phosphorylated STAT3 and co-occupies TH17/TFH gene promoters in TH17-activated human T cells. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.; Paz, K.; Du, J.; Reichenbach, D.K.; Taylor, P.A.; Panoskaltsis-Mortari, A.; Vulic, A.; Luznik, L.; MacDonald, K.; Hill, G.; et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood 2016, 127, 2144–2154. [Google Scholar] [CrossRef] [Green Version]
- Jagasia, M.; Lazaryan, A.; Bachier, C.R.; Salhotra, A.; Weisdorf, D.J.; Zoghi, B.; Essell, J.; Green, L.; Schueller, O.; Patel, J.; et al. ROCK2 inhibition with Belumosudil (KD025) for the treatment of chronic graft-versus-host disease. J. Clin. Oncol. 2021, 39, 1888–1898. [Google Scholar] [CrossRef]
- Zanin-Zhorov, A.; Weiss, J.M.; Nyuydzefe, M.S.; Chen, W.; Scher, J.U.; Mo, R.; Depoil, D.; Rao, N.; Liu, B.; Wei, J.; et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 16814–16819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, H.; Wilson, B.; Caswell, P.T. Control of adhesion and protrusion in cell migration by Rho GTPases. Curr. Opin. Cell Biol. 2019, 56, 64–70. [Google Scholar] [CrossRef]
- Zhu, Y.; Xie, J.; Shi, J. Rac1/ROCK-driven membrane dynamics promote natural killer cell cytotoxicity via granzyme-induced necroptosis. BMC Biol. 2021, 19, 1–14. [Google Scholar] [CrossRef]
- Lou, Z.; Billadeau, D.D.; Savoy, D.N.; Schoon, R.A.; Leibson, P.J. A role for a RhoA/ROCK/LIM-Kinase pathway in the regulation of Cytotoxic Lymphocytes. J. Immunol. 2001, 167, 5749–5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Karunanithi, S.; Jackson, Z.; Wald, D. Small molecule screening identifies Rho-Associate protein kinase (ROCK) as a regulator of NK Cell Cytotoxicity against cancer. Blood 2019, 134, 3607. [Google Scholar] [CrossRef]
- Kumar, V.; Dasoveanu, D.C.; Chyou, S.; Tzeng, T.-C.; Rozo, C.; Liang, Y.; Stohl, W.; Fu, Y.-X.; Ruddle, N.H.; Lu, T.T. A dendritic-cell-stromal axis maintains immune responses in Lymph Nodes. Immunity 2015, 42, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Matoba, K.; Kawanami, D.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. Rho-kinase mediates TNF-α-induced MCP-1 expression via p38 MAPK signaling pathway in mesangial cells. Biochem. Biophys. Res. Commun. 2010, 402, 725–730. [Google Scholar] [CrossRef]
- Takeda, Y.; Matoba, K.; Kawanami, D.; Nagai, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. ROCK2 Regulates monocyte migration and cell to cell adhesion in vascular endothelial cells. Int. J. Mol. Sci. 2019, 20, 1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.; Jung, H.; Nam, G.-H.; Kim, I.-S. The right Timing, right combination, right sequence, and right delivery for Cancer immunotherapy. J. Control. Release 2021, 331, 321–334. [Google Scholar] [CrossRef]
- Nam, G.-H.; Lee, E.J.; Kim, Y.K.; Hong, Y.; Choi, Y.; Ryu, M.-J.; Woo, J.; Cho, Y.; Ahn, D.J.; Yang, Y.; et al. Combined Rho-kinase inhibition and immunogenic cell death triggers and propagates immunity against cancer. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Kim, S.; Kim, S.A.; Nam, G.-H.; Hong, Y.; Kim, G.B.; Choi, Y.; Lee, S.; Cho, Y.; Kwon, M.; Jeong, C.; et al. In situ immunogenic clearance induced by a combination of photodynamic therapy and rho-kinase inhibition sensitizes immune checkpoint blockade response to elicit systemic antitumor immunity against intraocular melanoma and its metastasis. J. Immunother. Cancer 2021, 9, e001481. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisländer, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and cancer immunotherapy: A STING in the tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef]
- Nicolai, C.J.; Wolf, N.; Chang, I.-C.; Kirn, G.; Marcus, A.; Ndubaku, C.O.; McWhirter, S.M.; Raulet, D.H. NK cells mediate clearance of CD8 + T cell–resistant tumors in response to STING agonists. Sci. Immunol. 2020, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Seen, D.; Zheng, C.; Zeng, R.; Li, E. Role of small GTPase RhoA in DNA damage response. Biomolecules 2021, 11, 212. [Google Scholar] [CrossRef]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.-C.; Ho, Y.-J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell–mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Su, Y.; Xu, B. Rho-associated protein kinase-dependent moesin phosphorylation is required for PD-L1 stabilization in breast cancer. Mol. Oncol. 2020, 14, 2701–2712. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Qi, X.; Zhang, A.; Sui, F.; Wang, X.; Proud, C.G.; Lin, C.; Fan, X.; Li, J. MRTF-A-NF-κB/p65 axis-mediated PDL1 transcription and expression contributes to immune evasion of non-small-cell lung cancer via TGF-β. Exp. Mol. Med. 2021, 53, 1366–1378. [Google Scholar] [CrossRef]
- Orgaz, J.L.; Sanz-Moreno, V. What does not kill you makes you stronger: Surviving anti-cancer therapies by cytoskeletal remodeling and Myosin II reactivation. Mol. Cell. Oncol. 2020, 7, 1735911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgouli, M.; Herraiz, C.; Molist, E.C.; Fanshawe, B.; Maiques, O.; Perdrix, A.; Pandya, P.; Rodriguez-Hernandez, I.; Ilieva, K.M.; Cantelli, G.; et al. Regional activation of Myosin II in cancer cells drives tumor progression via a secretory cross-talk with the immune microenvironment. Cell 2019, 176, 757–774.e23. [Google Scholar] [CrossRef]
- Orgaz, J.; Molist, E.C.; Sadok, A.; Perdrix-Rosell, A.; Maiques, O.; Rodriguez-Hernandez, I.; Monger, J.; Mele, S.; Georgouli, M.; Bridgeman, V.; et al. Myosin II reactivation and Cytoskeletal remodeling as a Hallmark and a vulnerability in melanoma therapy resistance. Cancer Cell 2020, 37, 85–103.e9. [Google Scholar] [CrossRef]
- Teiti, I.; Florie, B.; Pich-Bavastro, C.; Gence, R.; Lajoie-Mazenc, I.; Rochaix, P.; Favre, G.; Tilkin-Mariamé, A.-F. In vivo effects in melanoma of ROCK inhibition-induced FasL overexpression. Front. Oncol. 2015, 5, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y. Analysis of the role of the Hippo pathway in cancer. J. Transl. Med. 2019, 17, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Ohgushi, M.; Minaguchi, M.; Sasai, Y. Rho-signaling-directed YAP/TAZ activity underlies the long-term survival and expansion of human embryonic stem cells. Cell Stem Cell 2015, 17, 448–461. [Google Scholar] [CrossRef] [Green Version]
- Zucchini, C.; Manara, M.C.; Cristalli, C.; Carrabotta, M.; Greco, S.; Pinca, R.S.; Ferrari, C.; Landuzzi, L.; Pasello, M.; Lollini, P.-L.; et al. ROCK2 deprivation leads to the inhibition of tumor growth and metastatic potential in osteosarcoma cells through the modulation of YAP activity. J. Exp. Clin. Cancer Res. 2019, 38, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Thaventhiran, J.E.D.; Hoffmann, A.; Magiera, L.; de la Roche, M.; Lingel, H.; Brunner-Weinzierl, M.; Fearon, D.T. Activation of the Hippo pathway by CTLA-4 regulates the expression of Blimp-1 in the CD8+ T cell. Proc. Natl. Acad. Sci. USA 2012, 109, E2223–E2229. [Google Scholar] [CrossRef] [Green Version]
- Lebid, A.; Chung, L.; Pardoll, D.M.; Pan, F. YAP Attenuates CD8 T cell-mediated anti-tumor response. Front. Immunol. 2020, 11, 580. [Google Scholar] [CrossRef] [Green Version]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is essential for treg-mediated suppression of antitumor immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Gao, Y.; Rao, J.; Wang, K.; Zhang, F.; Zhang, C. YAP-1 Promotes Tregs differentiation in Hepatocellular Carcinoma by enhancing TGFBR2 transcription. Cell. Physiol. Biochem. 2017, 41, 1189–1198. [Google Scholar] [CrossRef]
- Stampouloglou, E.; Cheng, N.; Federico, A.; Slaby, E.; Monti, S.; Szeto, G.L.; Varelas, X. Yap suppresses T-cell function and infiltration in the tumor microenvironment. PLoS Biol. 2020, 18, e3000591. [Google Scholar] [CrossRef]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-J.; Yang, C.-K.; Wei, P.-L.; Huynh, T.-T.; Whang-Peng, J.; Meng, T.-C.; Hsiao, M.; Tzeng, Y.-M.; Wu, A.T.; Yen, Y. Ovatodiolide suppresses colon tumorigenesis and prevents polarization of M2 tumor-associated macrophages through YAP oncogenic pathways. J. Hematol. Oncol. 2017, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Miao, J.; Hsu, P.C.; Yang, Y.-L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, C.G.; Kim, S.K.; Shin, S.J.; Choe, E.-A.; Park, S.-H.; Shin, E.-C.; Kim, J. YAP-Induced PD-L1 Expression drives immune evasion in BRAFi-resistant melanoma. Cancer Immunol. Res. 2018, 6, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.S.; Park, D.I.; Lee, D.H.; Lee, J.E.; Yeo, M.K.; Park, Y.H.; Lim, D.S.; Choi, W.; Yoo, G.; Kim, H.-B.; et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2017, 491, 493–499. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | NCT Number | Indication | Phase | Status | Note |
|---|---|---|---|---|---|
| AR-12286 (Verosudil) | NCT01330979 | Open Angle Glaucoma, Ocular Hypertension (OH) | 2 | Completed | |
| NCT01699464 | 2 | Completed | |||
| NCT01936389 | Exfoliation Syndrome, OH, Open Angle Glaucoma | 2 | Unknown | ||
| NCT02174991 | Glaucoma | 2 | Unknown | ||
| NCT01060579 | 2 | Completed | |||
| NCT02173223 | Advanced Glaucoma | 2 | Unknown | ||
| NCT02152774 | Chronic Angle-closure Glaucoma | 2 | Unknown | ||
| NCT00902200 | Elevated Intraocular Pressure | 2 | Completed | ||
| NCT01302249 | Glaucoma, OH | 2 | Completed | Latanoprost | |
| NCT01474135 | 2 | Completed | Travoprost | ||
| Fasudil | Cerebral Vasospasm, Cerebral Ischemic Symptoms | Approved in Japan (June, 1995) | |||
| NCT01935518 | Amyotrophic Lateral Sclerosis | 2 | Unknown | ||
| NCT03792490 | 2 | Recruiting | |||
| NCT00670202 | Carotid Stenosis | 2 | Terminated | ||
| NCT04734379 | Progressive Supranuclear Palsy, Corticobasal Syndrome | 2 | Recruiting | ||
| NCT04191954 | Retinopathy of Prematurity | 2/3 | Recruiting | ||
| NCT04793659 | Dementia | 2 | Active, not recruiting | ||
| NCT00498615 | Raynaud’s Disease, Scleroderma | 3 | Completed | ||
| NCT01823081 | Diabetic Macular Edema | 3 | Completed | ||
| NCT03753269 | ST Segment Elevation, Myocardial Infarction | 4 | Not yet recruiting | ||
| NCT00120718 | Atherosclerosis, Hypercholesterolemia | 2 | Completed | ||
| NCT03404843 | Cardiovascular Diseases | 2 | Completed | ||
| NCT03391219 | Retinal Vein Occlusion | 2/3 | Unknown | bevacizumab | |
| NCT04734379 | Progressive Supranuclear Palsy, Sorticobasal Syndrome | 2 | Recruiting | ||
| Ripasudil | Glaucoma, OH | Approved in Japan (September, 2014) | |||
| NCT03575130 | Fuchs’ Endothelial Dystrophy, Fuchs Dystrophy, Corneal Endothelial Dystrophy, Corneal Endothelial Cell Loss, Cornea Guttata | 2 | Unknown | ||
| NCT03813056 | Fuchs’ Endothelial Dystrophy | 2 | Recruiting | ||
| NCT03249337 | 4 | Recruiting | |||
| NCT04621136 | Retinopathy of Prematurity | 1/2 | Recruiting | ||
| Netarsudil | Open-Angle glaucoma, OH | Approved (2019) | Latanoprost | ||
| NCT04057053 | Fuchs’ Endothelial Dystrophy, Cataract | Early 1 | Completed | ||
| NCT04752020 | Fuchs’ Endothelial Dystrophy | Early 1 | Recruiting | ||
| NCT03248037 | Fuchs’ Endothelial Dystrophy, Bullous Keratopathy | 3 | Completed | ||
| NCT04620135 | Primary Open Angle Glaucoma, OH | 3 | Completed | ||
| NCT03233308 | 2 | Completed | |||
| NCT04064918 | Open-Angle Glaucoma, OH | Not applicable | Not yet recruiting | ||
| NCT02558374 | 3 | Completed | |||
| NCT02874846 | 2 | Completed | |||
| NCT02246764 | OH, Glaucoma | 3 | Completed | ||
| NCT04498169 | Corneal Edema | 2 | Completed | ||
| KD025 (Belumosudil) | Chronic Graft-Versus-Host Disease (cGVHD) | Approved (2021) | |||
| NCT03640481 | cGVHD | 2 | Recruiting | ||
| NCT02841995 | GVHD | 2 | Active, not recruiting | ||
| NCT02317627 | Psoriasis Vulgaris | 2 | Completed | ||
| NCT02106195 | 2 | Completed | |||
| NCT03907540 | Autoimmune Diseases, Fibrosis | 1 | Completed | ||
| NCT02688647 | Idiopathic Pulmonary Fibrosis | 2 | Completed | ||
| NCT03919799 | Systemic Sclerosis, Diffuse Cutaneous Systemic Sclerosis | 2 | Recruiting | ||
| NCT04166942 | Hepatic Impairment | 1 | Recruiting | ||
| AT13148 2 | NCT01585701 | Advanced Solid Tumor | 1 | Completed | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Kim, S.A.; Han, J.; Kim, I.-S. Rho-Kinase as a Target for Cancer Therapy and Its Immunotherapeutic Potential. Int. J. Mol. Sci. 2021, 22, 12916. https://doi.org/10.3390/ijms222312916
Kim S, Kim SA, Han J, Kim I-S. Rho-Kinase as a Target for Cancer Therapy and Its Immunotherapeutic Potential. International Journal of Molecular Sciences. 2021; 22(23):12916. https://doi.org/10.3390/ijms222312916
Chicago/Turabian StyleKim, Seohyun, Seong A. Kim, Jihoon Han, and In-San Kim. 2021. "Rho-Kinase as a Target for Cancer Therapy and Its Immunotherapeutic Potential" International Journal of Molecular Sciences 22, no. 23: 12916. https://doi.org/10.3390/ijms222312916
APA StyleKim, S., Kim, S. A., Han, J., & Kim, I. -S. (2021). Rho-Kinase as a Target for Cancer Therapy and Its Immunotherapeutic Potential. International Journal of Molecular Sciences, 22(23), 12916. https://doi.org/10.3390/ijms222312916