
Regulation of Hedgehog Signal Transduction by Ubiquitination and Deubiquitination


Abstract
:1. General Introduction
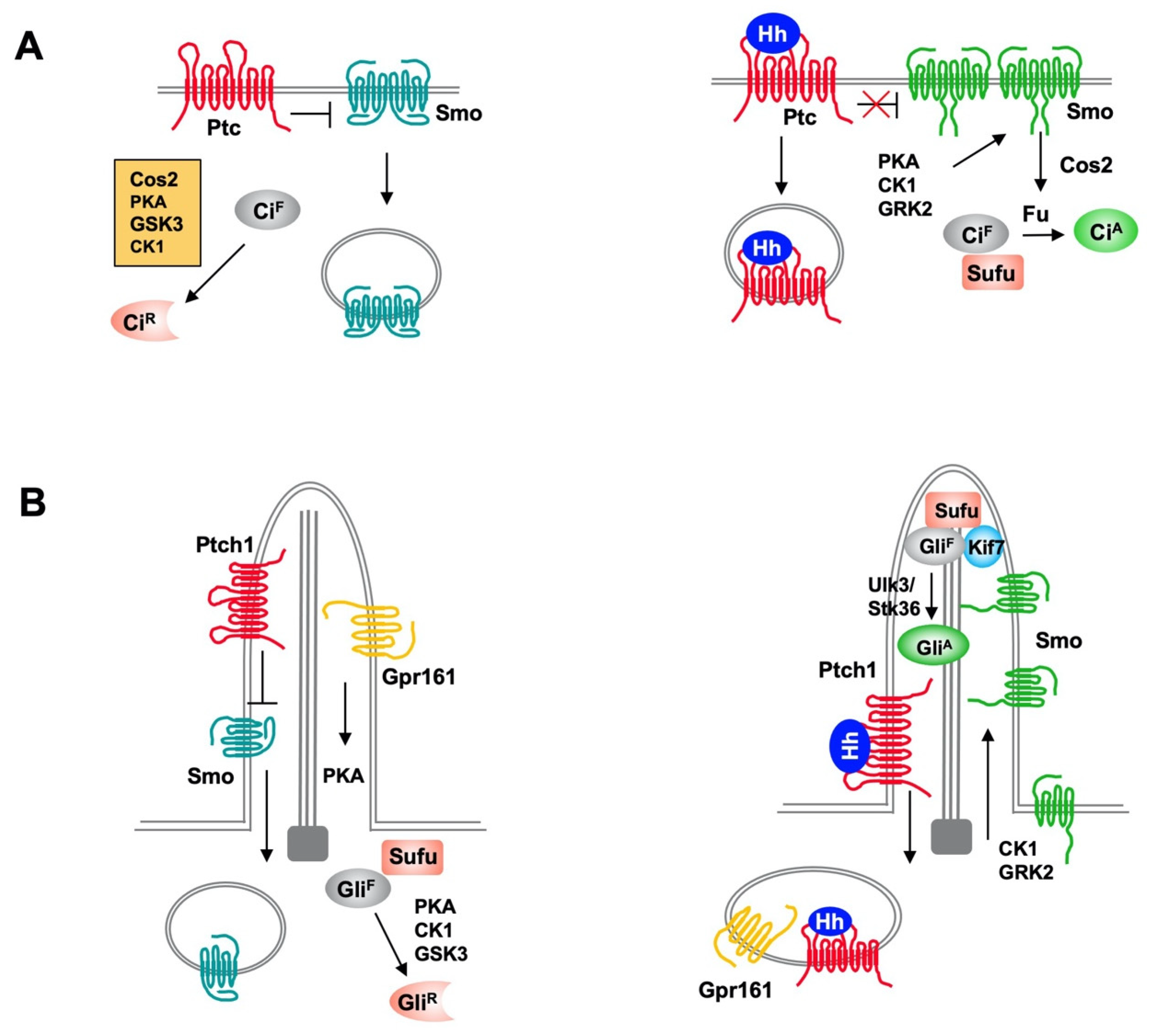
2. Overview of Hh Signaling Pathway
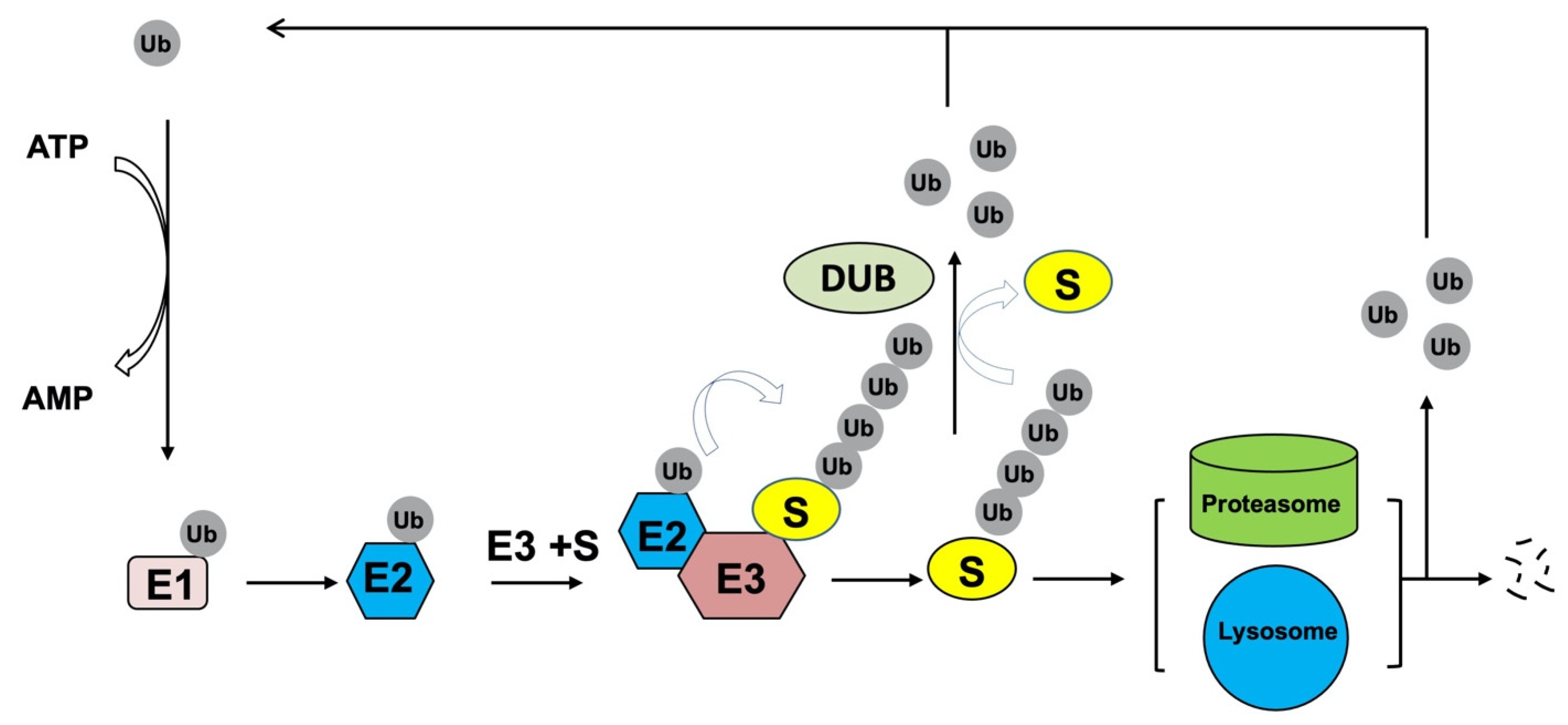
3. Ubiquitination and Deubiquitination
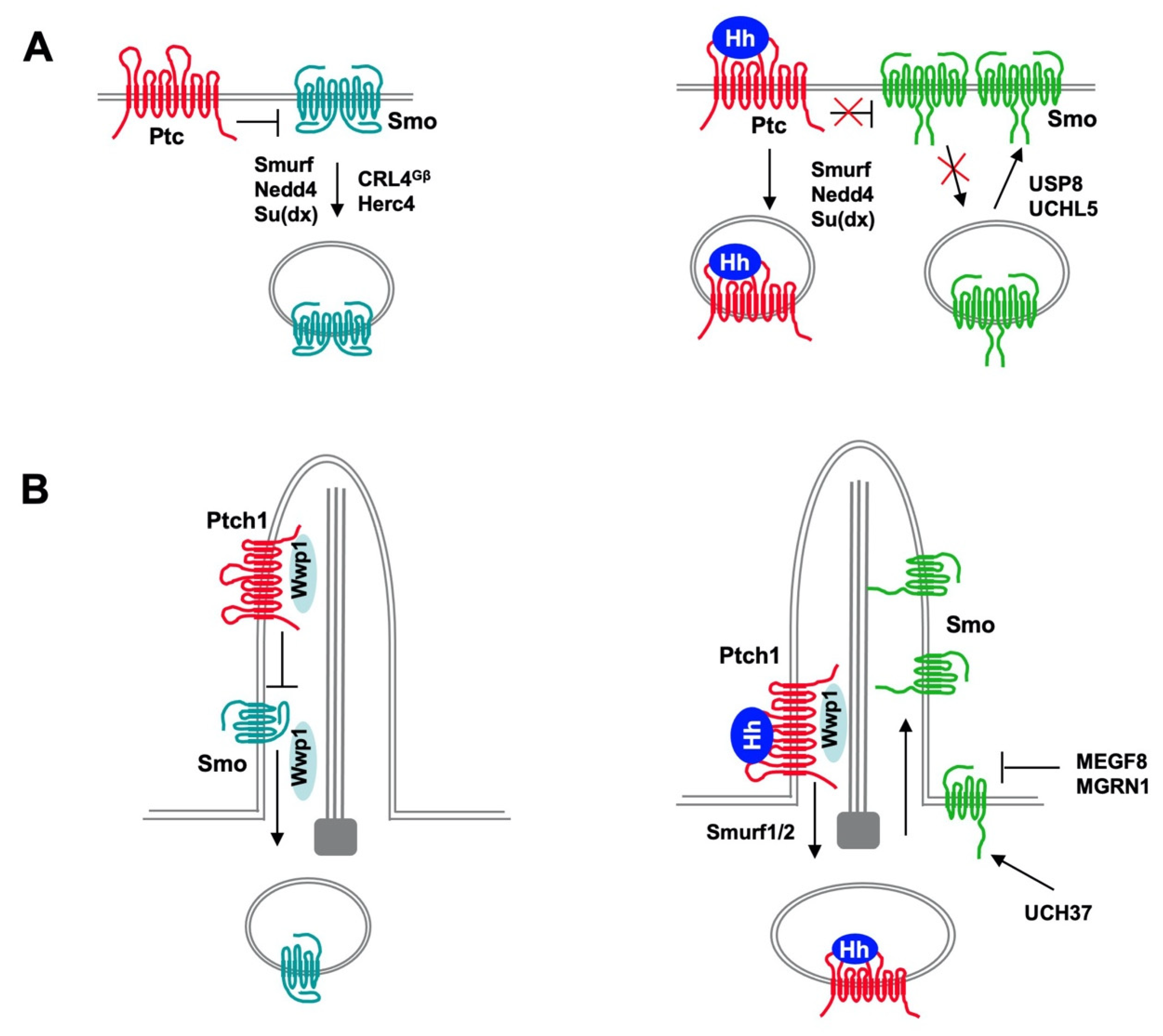
4. Regulation of Ptc and Smo by Ubiquitination
4.1. Regulation of Ptc/Ptch1 by Nedd4 Family of E3s
4.2. Regulation of Ptch1 Ubiquitination by TSPAN8 and ATXN3
4.3. Regulation of Drosophila Smo Trafficking and Turnover by Ubiquitination
4.4. Regulation of Mammalian Smo Ciliary Trafficking by Ubiquitination
4.5. Regulation of Smo by Other E3s and DUBs
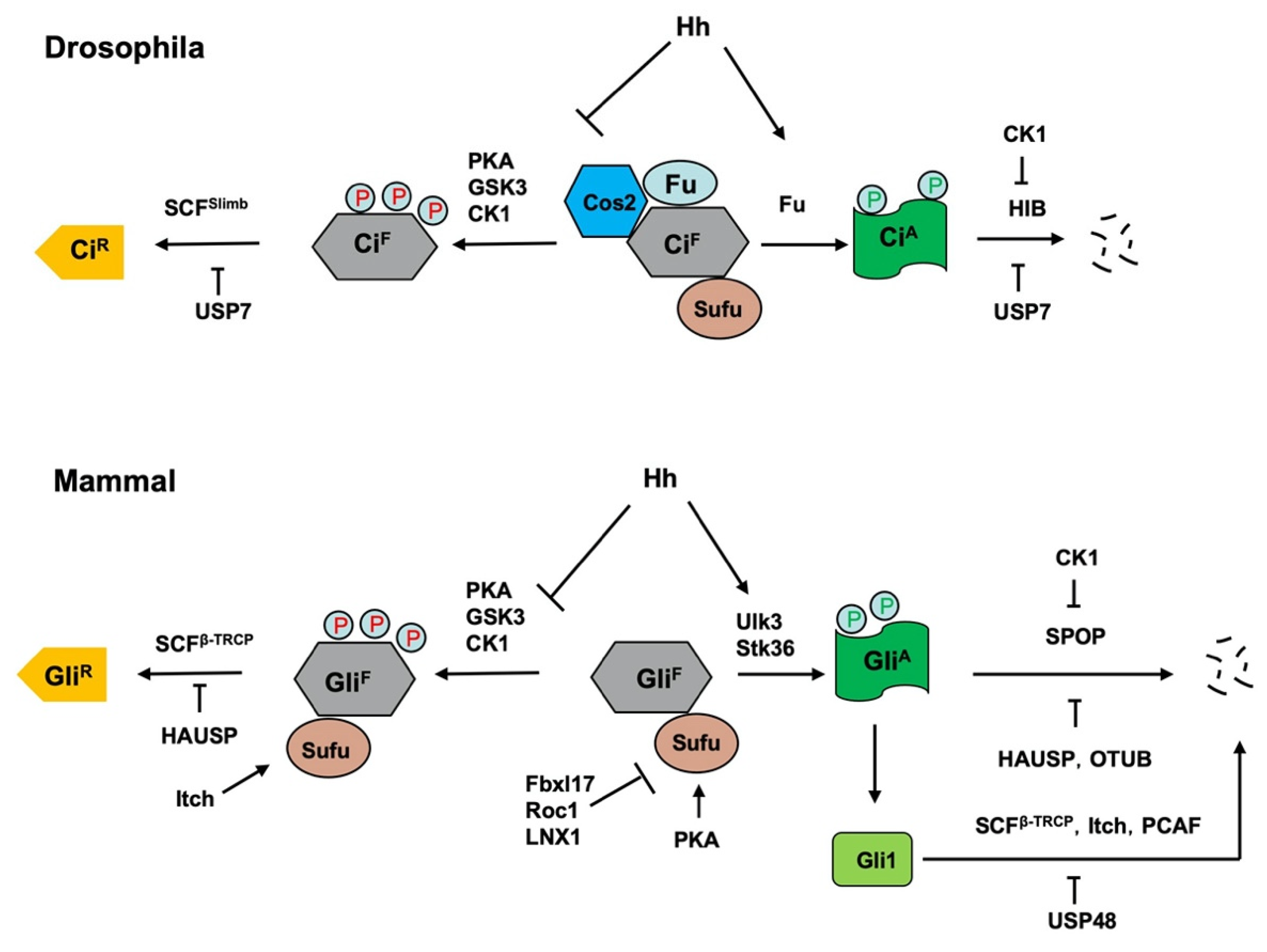
5. Regulation of Hh Pathway Transcription Factors Ci/Gli by Ubiquitination and Deubiquitination
5.1. Regulation of the Production of CiR/GliR by Slimb/β-TRCP
5.2. Regulation of Ci/Gli Degradation by HIB/SPOP
5.3. Proteolytic and Non-Proteolytic Regulation of Gli by Other E3s
5.4. Regulation of Ci/Gli Processing and Degradation by DUBs
6. Regulation of Hh Intracellular Signaling Components by Ubiquitination
6.1. Regulation of Cos2/Kif7 by UBR3
6.2. Regulation of Sufu by Multiple E3s
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J. Hedgehog signaling mechanism and role in cancer. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Iriana, S.; Asha, K.; Repak, M.; Sharma-Walia, N. Hedgehog Signaling: Implications in Cancers and Viral Infections. Int. J. Mol. Sci. 2021, 22, 1042. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, J. Decoding the phosphorylation code in Hedgehog signal transduction. Cell Res. 2013, 23, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Li, S.; Han, Y.; Li, S.; Yue, T.; Wang, B.; Jiang, J. Regulation of Smoothened Trafficking and Hedgehog Signaling by the SUMO Pathway. Dev. Cell 2016, 39, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, S.; Han, Y.; Tong, C.; Wang, B.; Chen, Y.; Jiang, J. Regulation of Smoothened Phosphorylation and High-Level Hedgehog Signaling Activity by a Plasma Membrane Associated Kinase. PLoS Biol. 2016, 14, e1002481. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J. Regulation of Hh/Gli signaling by dual ubiquitin pathways. Cell Cycle 2006, 5, 2457–2463. [Google Scholar] [CrossRef]
- Liu, A. Proteostasis in the Hedgehog signaling pathway. Semin. Cell Dev. Biol. 2019, 93, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Bufalieri, F.; Lospinoso Severini, L.; Caimano, M.; Infante, P.; Di Marcotullio, L. DUBs Activating the Hedgehog Signaling Pathway: A Promising Therapeutic Target in Cancer. Cancers 2020, 12, 1518. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.; Jia, J. Mechanisms of Smoothened Regulation in Hedgehog Signaling. Cells 2021, 10, 2138. [Google Scholar] [CrossRef]
- Wilson, C.W.; Chuang, P.T. Mechanism and evolution of cytosolic Hedgehog signal transduction. Development 2010, 137, 2079–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhao, Y.; Tong, C.; Wang, G.; Wang, B.; Jia, J.; Jiang, J. Hedgehog-regulated costal2-kinase complexes control phosphorylation and proteolytic processing of cubitus interruptus. Dev. Cell 2005, 8, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhang, L.; Zhang, Q.; Tong, C.; Wang, B.; Hou, F.; Amanai, K.; Jiang, J. Phosphorylation by double-time/CKIepsilon and CKIalpha targets cubitus interruptus for Slimb/beta-TRCP-mediated proteolytic processing. Dev. Cell 2005, 9, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Smelkinson, M.G.; Kalderon, D. Processing of the Drosophila hedgehog signaling effector Ci-155 to the repressor Ci-75 is mediated by direct binding to the SCF component Slimb. Curr. Biol. 2006, 16, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Schmiege, P.; Coutavas, E.; Wang, J.; Li, X. Structures of human Patched and its complex with native palmitoylated sonic hedgehog. Nature 2018, 560, 128–132. [Google Scholar] [CrossRef]
- Han, Y.; Wang, B.; Cho, Y.S.; Zhu, J.; Wu, J.; Chen, Y.; Jiang, J. Phosphorylation of Ci/Gli by Fused Family Kinases Promotes Hedgehog Signaling. Dev. Cell 2019, 50, 610–626. [Google Scholar] [CrossRef]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef]
- Kong, J.H.; Siebold, C.; Rohatgi, R. Biochemical mechanisms of vertebrate hedgehog signaling. Development 2019, 146, dev166892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu Rev Biochem 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Walden, H.; Rittinger, K. RBR ligase-mediated ubiquitin transfer: A tale with many twists and turns. Nat. Struct. Mol. Biol. 2018, 25, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbe, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Eguether, T.; Ermolaeva, M.A.; Zhao, Y.; Bonnet, M.C.; Jain, A.; Pasparakis, M.; Courtois, G.; Tassin, A.M. The deubiquitinating enzyme CYLD controls apical docking of basal bodies in ciliated epithelial cells. Nat. Commun. 2014, 5, 4585. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ran, J.; Liu, M.; Li, D.; Li, Y.; Shi, X.; Meng, D.; Pan, J.; Ou, G.; Aneja, R.; et al. CYLD mediates ciliogenesis in multiple organs by deubiquitinating Cep70 and inactivating HDAC6. Cell Res. 2014, 24, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.L.; Yuan, J.F.; Qin, X.H.; Song, G.P.; Hu, H.B.; Tu, H.Q.; Song, Z.Q.; Li, P.Y.; Xu, Y.L.; Li, S.; et al. LUBAC regulates ciliogenesis by promoting CP110 removal from the mother centriole. J. Cell Biol. 2021, 221, e202105092. [Google Scholar] [CrossRef]
- Goncalves, A.B.; Hasselbalch, S.K.; Joensen, B.B.; Patzke, S.; Martens, P.; Ohlsen, S.K.; Quinodoz, M.; Nikopoulos, K.; Suleiman, R.; Damso Jeppesen, M.P.; et al. CEP78 functions downstream of CEP350 to control biogenesis of primary cilia by negatively regulating CP110 levels. eLife 2021, 10, e63731. [Google Scholar] [CrossRef]
- Lu, X.; Liu, S.; Kornberg, T.B. The C-terminal tail of the Hedgehog receptor Patched regulates both localization and turnover. Genes Dev. 2006, 20, 2539–2551. [Google Scholar] [CrossRef] [Green Version]
- Brigui, A.; Hofmann, L.; Arguelles, C.; Sanial, M.; Holmgren, R.A.; Plessis, A. Control of the dynamics and homeostasis of the Drosophila Hedgehog receptor Patched by two C2-WW-HECT-E3 Ubiquitin ligases. Open Biol. 2015, 5, 150112. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Zhang, Z.; Zhang, C.; Lv, X.; Zheng, X.; Chen, Z.; Sun, L.; Wang, H.; Zhu, Y.; Zhang, J.; et al. Activation of Smurf E3 ligase promoted by smoothened regulates hedgehog signaling through targeting patched turnover. PLoS Biol. 2013, 11, e1001721. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Tang, L.Y.; Tang, Y.; Tang, Y.; Shen, Q.H.; Ding, J.; Chen, Y.; Zhang, Z.; Yu, T.T.; Zhang, Y.E.; et al. Requirement of Smurf-mediated endocytosis of Patched1 in sonic hedgehog signal reception. eLife 2014, 3, e02555. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Chinchilla, P.; Fombonne, J.; Ho, L.; Guix, C.; Keen, J.H.; Mehlen, P.; Riobo, N.A. Patched-1 proapoptotic activity is downregulated by modification of K1413 by the E3 ubiquitin-protein ligase Itchy homolog. Mol. Cell Biol. 2014, 34, 3855–3866. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Gires, O.; Zhu, L.; Liu, J.; Li, J.; Yang, H.; Ju, G.; Huang, J.; Ge, W.; Chen, Y.; et al. TSPAN8 promotes cancer cell stemness via activation of sonic Hedgehog signaling. Nat. Commun. 2019, 10, 2863. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, S.; Wang, B.; Jiang, J. Hedgehog reciprocally controls trafficking of Smo and Ptc through the Smurf family of E3 ubiquitin ligases. Sci. Signal. 2018, 11, eaan8660. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Cho, Y.S.; Wang, B.; Li, S.; Jiang, J. Regulation of Smoothened ubiquitylation and cell surface expression through a Cul4-DDB1-Gbeta E3 ubiquitin ligase complex. J. Cell Sci. 2018, 131, jcs218016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Yao, X.; Shan, Z.; Li, W.; Gao, Y.; Zhang, Q. E3 ligase Herc4 regulates Hedgehog signalling through promoting Smoothened degradation. J. Mol. Cell Biol. 2019, 11, 791–803. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Chen, Y.; Shi, Q.; Yue, T.; Wang, B.; Jiang, J. Hedgehog-regulated ubiquitination controls smoothened trafficking and cell surface expression in Drosophila. PLoS Biol. 2012, 10, e1001239. [Google Scholar] [CrossRef] [Green Version]
- Xia, R.; Jia, H.; Fan, J.; Liu, Y.; Jia, J. USP8 promotes smoothened signaling by preventing its ubiquitination and changing its subcellular localization. PLoS Biol. 2012, 10, e1001238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Yao, X.; Pang, S.; Chen, P.; Jiang, W.; Shan, Z.; Zhang, Q. The deubiquitinase UCHL5/UCH37 positively regulates Hedgehog signaling by deubiquitinating Smoothened. J. Mol. Cell. Biol. 2018, 10, 243–257. [Google Scholar] [CrossRef]
- Lv, B.; Stuck, M.W.; Desai, P.B.; Cabrera, O.A.; Pazour, G.J. E3 ubiquitin ligase Wwp1 regulates ciliary dynamics of the Hedgehog receptor Smoothened. J. Cell Biol. 2021, 220, e201912104. [Google Scholar] [CrossRef]
- Shinde, S.R.; Nager, A.R.; Nachury, M.V. Ubiquitin chains earmark GPCRs for BBSome-mediated removal from cilia. J. Cell Biol. 2020, 219, e202010177. [Google Scholar] [CrossRef]
- Sun, X.; Sun, B.; Cui, M.; Zhou, Z. HERC4 exerts an anti-tumor role through destabilizing the oncoprotein Smo. Biochem. Biophys Res. Commun. 2019, 513, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Smelkinson, M.G.; Zhou, Q.; Kalderon, D. Regulation of Ci-SCFSlimb binding, Ci proteolysis, and hedgehog pathway activity by Ci phosphorylation. Dev. Cell 2007, 13, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, L.; Wang, B.; Ou, C.Y.; Chien, C.T.; Jiang, J. A hedgehog-induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Dev. Cell 2006, 10, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, D.; Bush, E.W.; Hooper, J.E. Roadkill attenuates Hedgehog responses through degradation of Cubitus interruptus. Development 2006, 133, 2001–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Yao, X.; Li, S.; Xiong, Y.; Dong, X.; Zhao, Y.; Jiang, J.; Zhang, Q. Deubiquitination of Ci/Gli by Usp7/HAUSP Regulates Hedgehog Signaling. Dev. Cell 2015, 34, 58–72. [Google Scholar] [CrossRef] [Green Version]
- Huntzicker, E.G.; Estay, I.S.; Zhen, H.; Lokteva, L.A.; Jackson, P.K.; Oro, A.E. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006, 20, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.H.; Wilson, C.W.; Li, Y.J.; Law, K.K.; Lu, C.S.; Gacayan, R.; Zhang, X.; Hui, C.C.; Chuang, P.T. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Lai, C.K.; Evangelista, M.; Hongo, J.A.; de Sauvage, F.J.; Scales, S.J. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell. Biol. 2010, 30, 1910–1922. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Pan, Y.; Wang, B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010, 137, 2001–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Liu, A. Spop promotes skeletal development and homeostasis by positively regulating Ihh signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 14751–14756. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Liu, A. Spop regulates Gli3 activity and Shh signaling in dorsoventral patterning of the mouse spinal cord. Dev. Biol. 2017, 432, 72–85. [Google Scholar] [CrossRef]
- Coquenlorge, S.; Yin, W.C.; Yung, T.; Pan, J.; Zhang, X.; Mo, R.; Belik, J.; Hui, C.C.; Kim, T.H. GLI2 Modulated by SUFU and SPOP Induces Intestinal Stem Cell Niche Signals in Development and Tumorigenesis. Cell Rep. 2019, 27, 3006–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, W.C.; Satkunendran, T.; Mo, R.; Morrissy, S.; Zhang, X.; Huang, E.S.; Uuskula-Reimand, L.; Hou, H.; Son, J.E.; Liu, W.; et al. Dual Regulatory Functions of SUFU and Targetome of GLI2 in SHH Subgroup Medulloblastoma. Dev. Cell 2019, 48, 167–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marcotullio, L.; Ferretti, E.; Greco, A.; De Smaele, E.; Po, A.; Sico, M.A.; Alimandi, M.; Giannini, G.; Maroder, M.; Screpanti, I.; et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat. Cell Biol. 2006, 8, 1415–1423. [Google Scholar] [CrossRef]
- Ma, P.; Song, N.N.; Li, Y.; Zhang, Q.; Zhang, L.; Zhang, L.; Kong, Q.; Ma, L.; Yang, X.; Ren, B.; et al. Fine-Tuning of Shh/Gli Signaling Gradient by Non-proteolytic Ubiquitination during Neural Patterning. Cell Rep. 2019, 28, 541–553.e544. [Google Scholar] [CrossRef]
- Mazza, D.; Infante, P.; Colicchia, V.; Greco, A.; Alfonsi, R.; Siler, M.; Antonucci, L.; Po, A.; De Smaele, E.; Ferretti, E.; et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013, 20, 1688–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Gong, W.; Wu, H.; Wang, J.; Jin, Q.; Lin, C.; Xu, S.; Bao, W.; Wang, Y.; Wu, J.; et al. DKK1 suppresses WWP2 to enhance bortezomib resistance in multiple myeloma via regulating GLI2 ubiquitination. Carcinogenesis 2021, 42, 1223–1231. [Google Scholar] [CrossRef]
- Li, X.Y.; Mao, X.F.; Tang, X.Q.; Han, Q.Q.; Jiang, L.X.; Qiu, Y.M.; Dai, J.; Wang, Y.X. Regulation of Gli2 stability by deubiquitinase OTUB2. Biochem. Biophys Res. Commun. 2018, 505, 113–118. [Google Scholar] [CrossRef]
- Zhou, A.; Lin, K.; Zhang, S.; Ma, L.; Xue, J.; Morris, S.A.; Aldape, K.D.; Huang, S. Gli1-induced deubiquitinase USP48 aids glioblastoma tumorigenesis by stabilizing Gli1. EMBO Rep. 2017, 18, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, J.; Blanco-Sanchez, B.; Giagtzoglou, N.; Lin, G.; Yamamoto, S.; Jaiswal, M.; Chen, K.; Zhang, J.; Wei, W.; et al. Ubr3, a Novel Modulator of Hh Signaling Affects the Degradation of Costal-2 and Kif7 through Poly-ubiquitination. PLoS Genet. 2016, 12, e1006054. [Google Scholar] [CrossRef] [Green Version]
- Raducu, M.; Fung, E.; Serres, S.; Infante, P.; Barberis, A.; Fischer, R.; Bristow, C.; Thezenas, M.L.; Finta, C.; Christianson, J.C.; et al. SCF (Fbxl17) ubiquitylation of Sufu regulates Hedgehog signaling and medulloblastoma development. EMBO J. 2016, 35, 1400–1416. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Faedda, R.; Bernardi, F.; Bufalieri, F.; Lospinoso Severini, L.; Alfonsi, R.; Mazza, D.; Siler, M.; Coni, S.; Po, A.; et al. Itch/beta-arrestin2-dependent non-proteolytic ubiquitylation of SuFu controls Hedgehog signalling and medulloblastoma tumorigenesis. Nat. Commun. 2018, 9, 976. [Google Scholar] [CrossRef]
- Wang, W.; Qiu, J.; Qu, P.; Chen, H.; Lan, J.; Chen, H.; Li, L.; Gu, M. Regulator of cullins-1 (ROC1) negatively regulates the Gli2 regulator SUFU to activate the hedgehog pathway in bladder cancer. Cancer Cell Int. 2021, 21, 75. [Google Scholar] [CrossRef]
- Yan, Z.; Cheng, M.; Hu, G.; Wang, Y.; Zeng, S.; Huang, A.; Xu, L.; Liu, Y.; Shi, C.; Deng, L.; et al. Positive feedback of SuFu negating protein 1 on Hedgehog signaling promotes colorectal tumor growth. Cell Death Dis. 2021, 12, 199. [Google Scholar] [CrossRef] [PubMed]
- Myers, B.R.; Neahring, L.; Zhang, Y.; Roberts, K.J.; Beachy, P.A. Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proc. Natl. Acad. Sci. USA 2017, 114, E11141–E11150. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Qian, H.; Cao, P.; Zhao, X.; Zhou, Q.; Lei, J.; Yan, N. Structural basis for the recognition of Sonic Hedgehog by human Patched1. Science 2018, 361, eaas8935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Bulkley, D.P.; Xin, Y.; Roberts, K.J.; Asarnow, D.E.; Sharma, A.; Myers, B.R.; Cho, W.; Cheng, Y.; Beachy, P.A. Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 2018, 175, 1352–1364.e1314. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Cao, P.; Hu, M.; Gao, S.; Yan, N.; Gong, X. Inhibition of tetrameric Patched1 by Sonic Hedgehog through an asymmetric paradigm. Nat. Commun. 2019, 10, 2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, C.; Di Minin, G.; Vercellino, I.; Wutz, A.; Korkhov, V.M. Structural basis of sterol recognition by human hedgehog receptor PTCH1. Sci. Adv. 2019, 5, eaaw6490. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, A.F.; Kinnebrew, M.; Kowatsch, C.; Ansell, T.B.; El Omari, K.; Bishop, B.; Pardon, E.; Schwab, R.A.; Malinauskas, T.; Qian, M.; et al. The morphogen Sonic hedgehog inhibits its receptor Patched by a pincer grasp mechanism. Nat. Chem. Biol. 2019, 15, 975–982. [Google Scholar] [CrossRef]
- Petrov, K.; Wierbowski, B.M.; Liu, J.; Salic, A. Distinct Cation Gradients Power Cholesterol Transport at Different Key Points in the Hedgehog Signaling Pathway. Dev. Cell 2020, 55, 314–327.e317. [Google Scholar] [CrossRef]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187.e1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Zheng, S.; Wierbowski, B.M.; Kim, Y.; Nedelcu, D.; Aravena, L.; Liu, J.; Kruse, A.C.; Salic, A. Structural Basis of Smoothened Activation in Hedgehog Signaling. Cell 2018, 174, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Denef, N.; Neubuser, D.; Perez, L.; Cohen, S.M. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 2000, 102, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arveseth, C.D.; Happ, J.T.; Hedeen, D.S.; Zhu, J.F.; Capener, J.L.; Klatt Shaw, D.; Deshpande, I.; Liang, J.; Xu, J.; Stubben, S.L.; et al. Smoothened transduces Hedgehog signals via activity-dependent sequestration of PKA catalytic subunits. PLoS Biol. 2021, 19, e3001191. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Tong, C.; Wang, B.; Luo, L.; Jiang, J. Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 2004, 432, 1045–1050. [Google Scholar] [CrossRef]
- Zhao, Y.; Tong, C.; Jiang, J. Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 2007, 450, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Li, S.; Tong, C.; Zhao, Y.; Wang, B.; Liu, Y.; Jia, J.; Jiang, J. G protein-coupled receptor kinase 2 promotes high-level Hedgehog signaling by regulating the active state of Smo through kinase-dependent and kinase-independent mechanisms in Drosophila. Genes Dev. 2010, 24, 2054–2067. [Google Scholar] [CrossRef] [Green Version]
- Eguether, T.; San Agustin, J.T.; Keady, B.T.; Jonassen, J.A.; Liang, Y.; Francis, R.; Tobita, K.; Johnson, C.A.; Abdelhamed, Z.A.; Lo, C.W.; et al. IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev. Cell 2014, 31, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.B.; Stuck, M.W.; Lv, B.; Pazour, G.J. Ubiquitin links smoothened to intraflagellar transport to regulate Hedgehog signaling. J. Cell Biol. 2020, 219, 279–290. [Google Scholar] [CrossRef]
- French, M.E.; Klosowiak, J.L.; Aslanian, A.; Reed, S.I.; Yates, J.R., 3rd; Hunter, T. Mechanism of ubiquitin chain synthesis employed by a HECT domain ubiquitin ligase. J. Biol. Chem. 2017, 292, 10398–10413. [Google Scholar] [CrossRef] [Green Version]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Kong, J.H.; Young, C.B.; Pusapati, G.V.; Patel, C.B.; Ho, S.; Krishnan, A.; Lin, J.I.; Devine, W.; Moreau de Bellaing, A.; Athni, T.S.; et al. A Membrane-Tethered Ubiquitination Pathway Regulates Hedgehog Signaling and Heart Development. Dev. Cell 2020, 55, 432–449.e412. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J. Degrading Ci: Who is Cul-pable? Genes Dev. 2002, 16, 2315–2321. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Struhl, G. Protein kinase A and hedgehog signaling in Drosophila limb development. Cell 1995, 80, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Struhl, G. Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature 1998, 391, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Price, M.A.; Kalderon, D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 2002, 108, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Amanai, K.; Wang, G.; Tang, J.; Wang, B.; Jiang, J. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature 2002, 416, 548–552. [Google Scholar] [CrossRef]
- Tempe, D.; Casas, M.; Karaz, S.; Blanchet-Tournier, M.F.; Concordet, J.P. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol. Cell. Biol. 2006, 26, 4316–4326. [Google Scholar] [CrossRef] [Green Version]
- Methot, N.; Basler, K. Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell 1999, 96, 819–831. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Wang, B. A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J. Biol. Chem. 2007, 282, 10846–10852. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lv, X.; Yin, W.C.; Zhang, X.; Feng, J.; Wu, W.; Hui, C.C.; Zhang, L.; Zhao, Y. Ter94 ATPase complex targets k11-linked ubiquitinated ci to proteasomes for partial degradation. Dev. Cell 2013, 25, 636–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, J.; Tong, C.; Jiang, J. Smoothened transduces Hedgehog signal by physically interacting with Costal2/Fused complex through its C-terminal tail. Genes Dev. 2003, 17, 2709–2720. [Google Scholar] [CrossRef] [Green Version]
- Ruel, L.; Gallet, A.; Raisin, S.; Truchi, A.; Staccini-Lavenant, L.; Cervantes, A.; Therond, P.P. Phosphorylation of the atypical kinesin Costal2 by the kinase Fused induces the partial disassembly of the Smoothened-Fused-Costal2-Cubitus interruptus complex in Hedgehog signalling. Development 2007, 134, 3677–3689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Li, S.; Jia, J.; Jiang, J. The Hedgehog-induced Smoothened conformational switch assembles a signaling complex that activates Fused by promoting its dimerization and phosphorylation. Development 2011, 138, 4219–4231. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Ma, G.; Wang, B.; Jiang, J. Hedgehog induces formation of PKA-Smoothened complexes to promote Smoothened phosphorylation and pathway activation. Sci. Signal. 2014, 7, ra62. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, N.; Therond, P.P.; Ruel, L. Switch of PKA substrates from Cubitus interruptus to Smoothened in the Hedgehog signalosome complex. Nat. Commun. 2014, 5, 5034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Wen, X.; Ratti, N.; Loktev, A.; Rangell, L.; Scales, S.J.; Jackson, P.K. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 2013, 152, 210–223. [Google Scholar] [CrossRef] [Green Version]
- Pal, K.; Hwang, S.H.; Somatilaka, B.; Badgandi, H.; Jackson, P.K.; DeFea, K.; Mukhopadhyay, S. Smoothened determines beta-arrestin-mediated removal of the G protein-coupled receptor Gpr161 from the primary cilium. J. Cell Biol. 2016, 212, 861–875. [Google Scholar] [CrossRef] [Green Version]
- Ohlmeyer, J.T.; Kalderon, D. Hedgehog stimulates maturation of Cubitus interruptus into a labile transcriptional activator. Nature 1998, 396, 749–753. [Google Scholar] [CrossRef]
- Han, Y.; Shi, Q.; Jiang, J. Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc. Natl. Acad. Sci. USA 2015, 112, 6383–6388. [Google Scholar] [CrossRef] [Green Version]
- Ou, C.Y.; Lin, Y.F.; Chen, Y.J.; Chien, C.T. Distinct protein degradation mechanisms mediated by Cul1 and Cul3 controlling Ci stability in Drosophila eye development. Genes Dev. 2002, 16, 2403–2414. [Google Scholar] [CrossRef] [Green Version]
- Ou, C.Y.; Wang, C.H.; Jiang, J.; Chien, C.T. Suppression of Hedgehog signaling by Cul3 ligases in proliferation control of retinal precursors. Dev. Biol. 2007, 308, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, J.C.; Garcia-Garcia, E.; Sul, A.; Kalderon, D. Drosophila hedgehog can act as a morphogen in the absence of regulated Ci processing. eLife 2020, 9, e61083. [Google Scholar] [CrossRef]
- Zhang, Q.; Shi, Q.; Chen, Y.; Yue, T.; Li, S.; Wang, B.; Jiang, J. Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2009, 106, 21191–21196. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, M.; Calabrese, M.F.; Liu, J.; Waddell, M.B.; Nourse, A.; Hammel, M.; Miller, D.J.; Walden, H.; Duda, D.M.; Seyedin, S.N.; et al. Structures of SPOP-substrate complexes: Insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol. Cell 2009, 36, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Gan, W.; Dai, X.; Lunardi, A.; Li, Z.; Inuzuka, H.; Liu, P.; Varmeh, S.; Zhang, J.; Cheng, L.; Sun, Y.; et al. SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol. Cell 2015, 59, 917–930. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Gan, W.; Li, X.; Wang, S.; Zhang, W.; Huang, L.; Liu, S.; Zhong, Q.; Guo, J.; Zhang, J.; et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 2017, 23, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Marzahn, M.R.; Marada, S.; Lee, J.; Nourse, A.; Kenrick, S.; Zhao, H.; Ben-Nissan, G.; Kolaitis, R.M.; Peters, J.L.; Pounds, S.; et al. Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J. 2016, 35, 1254–1275. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, L.; Qi, X.; Zhang, Z.; Xia, Y.; Jia, J.; Jiang, J.; Zhao, Y.; Wu, G. Structural insight into the mutual recognition and regulation between Suppressor of Fused and Gli/Ci. Nat. Commun. 2013, 4, 2608. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhou, Z.; Yao, X.; Chen, P.; Sun, M.; Su, M.; Chang, C.; Yan, J.; Jiang, J.; Zhang, Q. Hedgehog signaling downregulates suppressor of fused through the HIB/SPOP-Crn axis in Drosophila. Cell Res. 2014, 24, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Li, S.; Li, S.; Jiang, A.; Chen, Y.; Jiang, J. Hedgehog-induced phosphorylation by CK1 sustains the activity of Ci/Gli activator. Proc. Natl. Acad. Sci. USA 2014, 111, E5651–E5660. [Google Scholar] [CrossRef] [Green Version]
- Ostertag, M.S.; Messias, A.C.; Sattler, M.; Popowicz, G.M. The Structure of the SPOP-Pdx1 Interface Reveals Insights into the Phosphorylation-Dependent Binding Regulation. Structure 2019, 27, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Vanderbilt, D.B.; Lin, C.C.; Martin, K.H.; Brundage, K.M.; Ruppert, J.M. SOX9 inhibits beta-TrCP-mediated protein degradation to promote nuclear GLI1 expression and cancer stem cell properties. J. Cell Sci. 2015, 128, 1123–1138. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.H.; Luo, J.; Mosley, Y.Y.; Hedrick, V.E.; Paul, L.N.; Chang, J.; Zhang, G.; Wang, Y.K.; Banko, M.R.; Brunet, A.; et al. AMP-Activated Protein Kinase Directly Phosphorylates and Destabilizes Hedgehog Pathway Transcription Factor GLI1 in Medulloblastoma. Cell Rep. 2015, 12, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Huang, S.Y.; Ka-Wai Li, K.; Li, Y.H.; Hsu, W.H.; Zhang, G.J.; Chang, C.J.; Yang, J.Y. Dual degradation signals destruct GLI1: AMPK inhibits GLI1 through beta-TrCP-mediated proteasome degradation. Oncotarget 2017, 8, 49869–49881. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.R.; Na, Y.J.; Kim, J.L.; Jeong, Y.A.; Park, S.H.; Jo, M.J.; Jeong, S.; Kang, S.; Oh, S.C.; Lee, D.H. RUNX3 suppresses metastasis and stemness by inhibiting Hedgehog signaling in colorectal cancer. Cell Death Differ. 2020, 27, 676–694. [Google Scholar] [CrossRef]
- Park, S.H.; Jeong, S.; Kim, B.R.; Jeong, Y.A.; Kim, J.L.; Na, Y.J.; Jo, M.J.; Yun, H.K.; Kim, D.Y.; Kim, B.G.; et al. Activating CCT2 triggers Gli-1 activation during hypoxic condition in colorectal cancer. Oncogene 2020, 39, 136–150. [Google Scholar] [CrossRef]
- Xiaoyun, S.; Yuyuan, Z.; Jie, X.; Yingjie, N.; Qing, X.; Yuezhen, D.; Haiguang, X. PHF19 activates hedgehog signaling and promotes tumorigenesis in hepatocellular carcinoma. Exp. Cell Res. 2021, 406, 112690. [Google Scholar] [CrossRef]
- Di Marcotullio, L.; Greco, A.; Mazza, D.; Canettieri, G.; Pietrosanti, L.; Infante, P.; Coni, S.; Moretti, M.; De Smaele, E.; Ferretti, E.; et al. Numb activates the E3 ligase Itch to control Gli1 function through a novel degradation signal. Oncogene 2011, 30, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Fang, Z.; Wang, A.; Luo, C.; Cheng, X.; Lu, M. Lithium Suppresses Hedgehog Signaling via Promoting ITCH E3 Ligase Activity and Gli1-SUFU Interaction in PDA Cells. Front. Pharmacol. 2017, 8, 820. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Mao, F.; Lu, Y.; Wu, W.; Zhang, L.; Zhao, Y. Transduction of the Hedgehog signal through the dimerization of Fused and the nuclear translocation of Cubitus interruptus. Cell Res. 2011, 21, 1436–1451. [Google Scholar] [CrossRef] [Green Version]
- Cheung, H.O.; Zhang, X.; Ribeiro, A.; Mo, R.; Makino, S.; Puviindran, V.; Law, K.K.; Briscoe, J.; Hui, C.C. The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci. Signal. 2009, 2, ra29. [Google Scholar] [CrossRef]
- Endoh-Yamagami, S.; Evangelista, M.; Wilson, D.; Wen, X.; Theunissen, J.W.; Phamluong, K.; Davis, M.; Scales, S.J.; Solloway, M.J.; de Sauvage, F.J.; et al. The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr. Biol. 2009, 19, 1320–1326. [Google Scholar] [CrossRef] [Green Version]
- Liem, K.F., Jr.; He, M.; Ocbina, P.J.; Anderson, K.V. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 13377–13382. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Subramanian, R.; Bangs, F.; Omelchenko, T.; Liem, K.F., Jr.; Kapoor, T.M.; Anderson, K.V. The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat. Cell Biol. 2014, 16, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Xiong, Y.; Shi, X.; Wu, J.; Zhao, Y.; Jiang, J. Regulation of Gli ciliary localization and Hedgehog signaling by the PY-NLS/karyopherin-beta2 nuclear import system. PLoS Biol. 2017, 15, e2002063. [Google Scholar] [CrossRef] [Green Version]
- Ruel, L.; Rodriguez, R.; Gallet, A.; Lavenant-Staccini, L.; Therond, P.P. Stability and association of Smoothened, Costal2 and Fused with Cubitus interruptus are regulated by Hedgehog. Nat. Cell Biol. 2003, 5, 907–913. [Google Scholar] [CrossRef]
- Yue, S.; Chen, Y.; Cheng, S.Y. Hedgehog signaling promotes the degradation of tumor suppressor Sufu through the ubiquitin-proteasome pathway. Oncogene 2009, 28, 492–499. [Google Scholar] [CrossRef]
- Chen, Y.; Yue, S.; Xie, L.; Pu, X.H.; Jin, T.; Cheng, S.Y. Dual Phosphorylation of suppressor of fused (Sufu) by PKA and GSK3beta regulates its stability and localization in the primary cilium. J. Biol. Chem. 2011, 286, 13502–13511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Hsia, E.Y.; Brigui, A.; Plessis, A.; Beachy, P.A.; Zheng, X. The role of ciliary trafficking in Hedgehog receptor signaling. Sci. Signal. 2015, 8, ra55. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hh Pathway Components | E3s | DUBs |
|---|---|---|
| Ptc (Drosophila) | Nedd4 [33,34], Smurf [34,35], Su(dx) [34] | |
| Ptch1 (Mammal) | Smurf1&2 [36], Itch [36,37] | ATXN3 [38] |
| Smo (Drosophila) | Smurf [39], Nedd4 [39], Su(dx) [39], CRL4Gβ [40], Herc4 [41] | USP8 [42,43], UCHL5 [44] |
| Smo (Mammals) | Wwp1 [45], MGRN1 [45,46], HERC4 [47] | UCH37 [44] |
| Ci | SCFSlimb [14,15,48], HIB [49,50] | USP7 [51] |
| Gli | SCFβ-TRCP [52], SPOP [49,53,54,55,56,57,58,59], Itch [60], RNF220 [61], PCAF [62], WWP2 [63] | HAUSP [51] OTUB2 [64] USP48 [65] |
| Cos2/Kif7 | UBR3 [66] | |
| Sufu (Mammal) | Fbxl17 [67], Itch [68], ROC1 [69], LNX1 [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Jiang, J. Regulation of Hedgehog Signal Transduction by Ubiquitination and Deubiquitination. Int. J. Mol. Sci. 2021, 22, 13338. https://doi.org/10.3390/ijms222413338
Zhang Q, Jiang J. Regulation of Hedgehog Signal Transduction by Ubiquitination and Deubiquitination. International Journal of Molecular Sciences. 2021; 22(24):13338. https://doi.org/10.3390/ijms222413338
Chicago/Turabian StyleZhang, Qing, and Jin Jiang. 2021. "Regulation of Hedgehog Signal Transduction by Ubiquitination and Deubiquitination" International Journal of Molecular Sciences 22, no. 24: 13338. https://doi.org/10.3390/ijms222413338
APA StyleZhang, Q., & Jiang, J. (2021). Regulation of Hedgehog Signal Transduction by Ubiquitination and Deubiquitination. International Journal of Molecular Sciences, 22(24), 13338. https://doi.org/10.3390/ijms222413338