
The Eclectic Nature of Glioma-Infiltrating Macrophages and Microglia

 , , ,
, , , 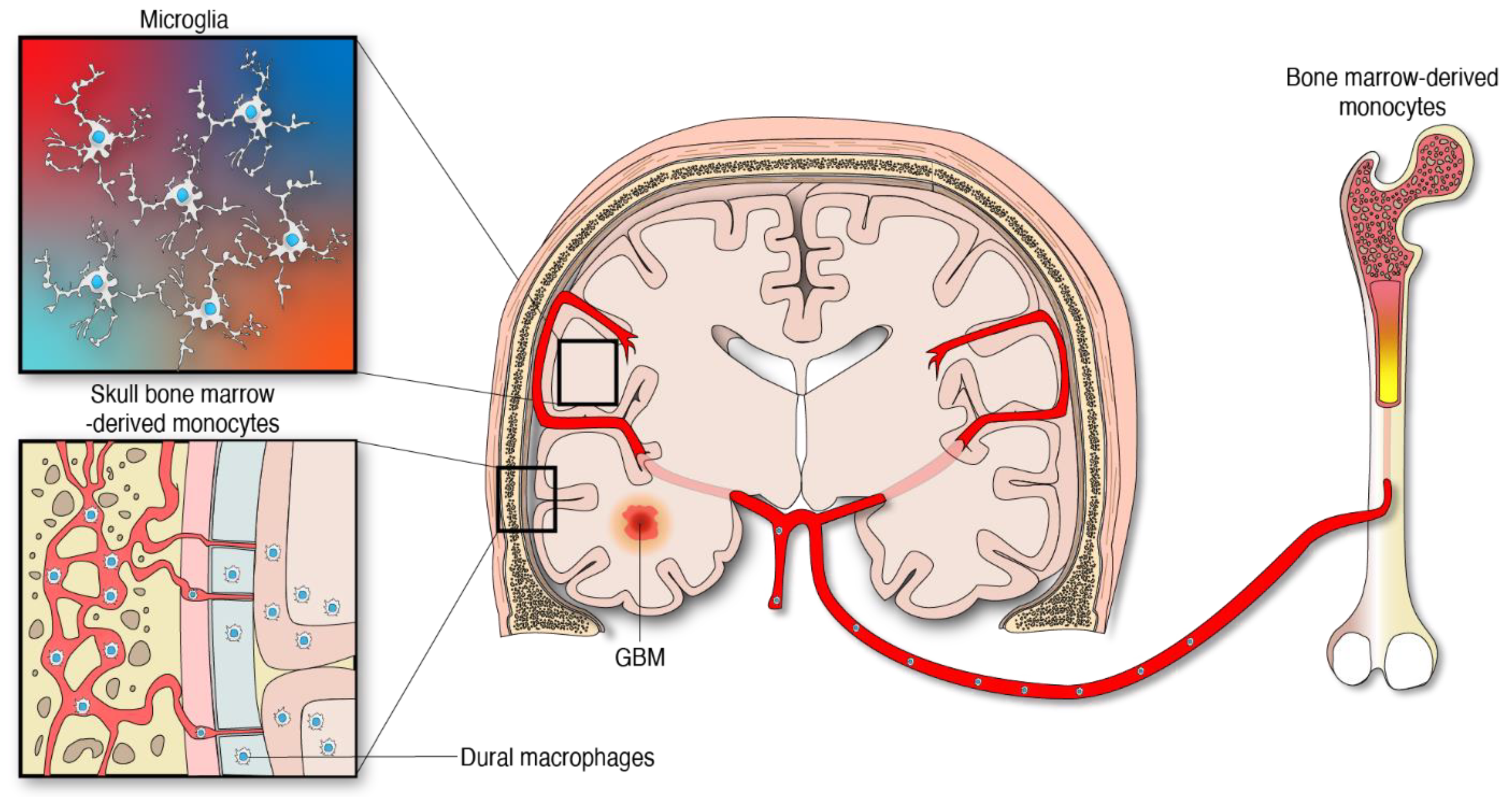{kind=link}
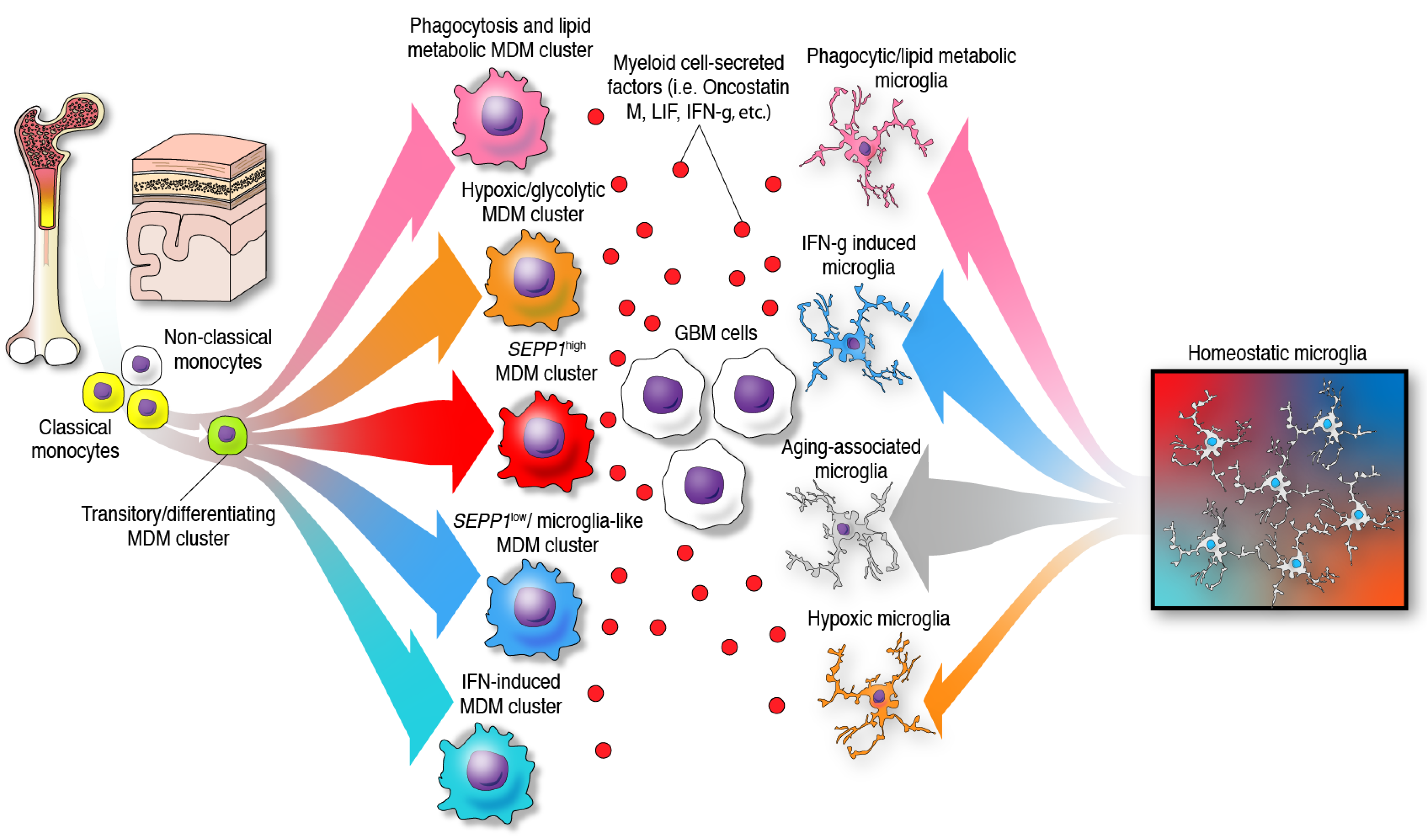{kind=link}
Abstract
:1. Introduction
2. Dissecting Myeloid Cells in Gliomas: Multiple Actors Come into Play
3. Differentiation Trajectories of Myeloid Cells under the Influence of Gliomas
4. Relationships and Interactions between the Tumor Genetics and Immune Landscape in Gliomas
5. Glioma-Infiltrating Myeloid Cells under Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 2018, 21, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Cugurra, A.; Mamuladze, T.; Rustenhoven, J.; Dykstra, T.; Beroshvili, G.; Greenberg, Z.J.; Baker, W.; Papadopoulos, Z.; Drieu, A.; Blackburn, S.; et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science 2021, 373, eabf7844. [Google Scholar] [CrossRef]
- Pulous, F.E.; Cruz-Hernández, J.C.; Yang, C.; Kaya, Z.; Wojtkiewicz, G.; Capen, D.; Brown, D.; Wu, J.W.; Vinegoni, C.; Yamazoe, M.; et al. Cerebrospinal fluid outflow through skull channels instructs cranial hematopoiesis. bioRxiv 2021, 52, 3692–3695. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Michell-Robinson, M.A.; Touil, H.; Healy, L.M.; Owen, D.R.; Durafourt, B.A.; Bar-Or, A.; Antel, J.P.; Moore, C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Chao, M.P.; Majeti, R.; Weissman, I.L. Macrophages as mediators of tumor immunosurveillance. Trends Immunol. 2010, 31, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Farhadian, S.F.; Mehta, S.S.; Zografou, C.; Robertson, K.; Price, R.W.; Pappalardo, J.; Chiarella, J.; Hafler, D.A.; Spudich, S.S. Single-cell RNA sequencing reveals microglia-like cells in cerebrospinal fluid during virologically suppressed HIV. JCI Insight 2018, 3, e121718. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Levitin, H.M.; Frattini, V.; Bush, E.C.; Boyett, D.M.; Samanamud, J.; Ceccarelli, M.; Dovas, A.; Zanazzi, G.; Canoll, P.; et al. Single-cell transcriptome analysis of lineage diversity in high-grade glioma. Genome Med. 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Gupta, S.; Liu, Y.; Fuhrman, K.; Gettinger, S.; Herbst, R.S.; Schalper, K.A.; Rimm, D.L. Biomarkers Associated with Beneficial PD-1 Checkpoint Blockade in Non-Small Cell Lung Cancer (NSCLC) Identified Using High-Plex Digital Spatial Profiling. Clin. Cancer Res. 2020, 26, 4360–4368. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Ng, A.H.C.; Chow, F.E.; Everson, R.G.; Helmink, B.A.; Tetzlaff, M.T.; Thakur, R.; Wargo, J.A.; Cloughesy, T.F.; Prins, R.M.; et al. Resolution of tissue signatures of therapy response in patients with recurrent GBM treated with neoadjuvant anti-PD1. Nat. Commun. 2021, 12, 4031. [Google Scholar] [CrossRef]
- Färkkilä, A.; Gulhan, D.C.; Casado, J.; Jacobson, C.A.; Nguyen, H.; Kochupurakkal, B.; Maliga, Z.; Yapp, C.; Chen, Y.A.; Schapiro, D.; et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat. Commun. 2020, 11, 1459. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Chanoch-Myers, R.; Mathewson, N.D.; Myskiw, C.; Atta, L.; Bussema, L.; Eichhorn, S.W.; Greenwald, A.C.; Kinker, G.S.; Rodman, C.; et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 2021, 39, 779–792.e711. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Füger, P.; Hefendehl, J.K.; Veeraraghavalu, K.; Wendeln, A.C.; Schlosser, C.; Obermüller, U.; Wegenast-Braun, B.M.; Neher, J.J.; Martus, P.; Kohsaka, S.; et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 2017, 20, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernández-Klett, F.; Lin, G.; Sagar; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e1617. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.X.; Gartrell, R.D.; Zhao, J.; Upadhyayula, P.S.; Zhao, W.; Yuan, J.; Minns, H.E.; Dovas, A.; Bruce, J.N.; Lasorella, A.; et al. Single-cell characterization of macrophages in glioblastoma reveals MARCO as a mesenchymal pro-tumor marker. Genome Med. 2021, 13, 88. [Google Scholar] [CrossRef]
- Kierdorf, K.; Masuda, T.; Jordão, M.J.C.; Prinz, M. Macrophages at CNS interfaces: Ontogeny and function in health and disease. Nat. Rev. Neurosci. 2019, 20, 547–562. [Google Scholar] [CrossRef]
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herisson, F.; Frodermann, V.; Courties, G.; Rohde, D.; Sun, Y.; Vandoorne, K.; Wojtkiewicz, G.R.; Masson, G.S.; Vinegoni, C.; Kim, J.; et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat. Neurosci. 2018, 21, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018, 3, e122264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Kohanbash, G.; Okada, H. Myeloid-derived suppressor cells (MDSCs) in gliomas and glioma-development. Immunol. Investig. 2012, 41, 658–679. [Google Scholar] [CrossRef]
- Fujita, M.; Kohanbash, G.; Fellows-Mayle, W.; Hamilton, R.L.; Komohara, Y.; Decker, S.A.; Ohlfest, J.R.; Okada, H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011, 71, 2664–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Rao, G.; Latha, K.; Ott, M.; Sabbagh, A.; Marisetty, A.; Ling, X.; Zamler, D.; Doucette, T.A.; Yang, Y.; Kong, L.Y.; et al. Anti-PD-1 Induces M1 Polarization in the Glioma Microenvironment and Exerts Therapeutic Efficacy in the Absence of CD8 Cytotoxic T Cells. Clin. Cancer Res. 2020, 26, 4699–4712. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, D.; Li, J.; Liang, X.; Chang, A.; Henry, V.K.; Lan, Z.; Spring, D.J.; Rao, G.; Wang, Y.A.; et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 2019, 35, 868–884.e866. [Google Scholar] [CrossRef]
- An, Z.; Knobbe-Thomsen, C.B.; Wan, X.; Fan, Q.W.; Reifenberger, G.; Weiss, W.A. EGFR Cooperates with EGFRvIII to Recruit Macrophages in Glioblastoma. Cancer Res. 2018, 78, 6785–6794. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, M.C.; Wolf, S.A.; Respondek, D.; Matyash, V.; Pohlmann, A.; Waiczies, S.; Waiczies, H.; Niendorf, T.; Synowitz, M.; Glass, R.; et al. GDNF mediates glioblastoma-induced microglia attraction but not astrogliosis. Acta Neuropathol. 2013, 125, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Sielska, M.; Przanowski, P.; Wylot, B.; Gabrusiewicz, K.; Maleszewska, M.; Kijewska, M.; Zawadzka, M.; Kucharska, J.; Vinnakota, K.; Kettenmann, H.; et al. Distinct roles of CSF family cytokines in macrophage infiltration and activation in glioma progression and injury response. J. Pathol. 2013, 230, 310–321. [Google Scholar] [CrossRef]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Hong, J.H.; Hsueh, C.; Chiang, C.S. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab. Investig. 2012, 92, 151–162. [Google Scholar] [CrossRef]
- Okada, M.; Saio, M.; Kito, Y.; Ohe, N.; Yano, H.; Yoshimura, S.; Iwama, T.; Takami, T. Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int. J. Oncol. 2009, 34, 1621–1627. [Google Scholar]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, B.C.; Maier, L.M.; D’Amico, R.; Mandigo, C.E.; Fontana, E.J.; Waziri, A.; Assanah, M.C.; Canoll, P.; Anderson, R.C.; Anderson, D.E.; et al. Dynamics of central and peripheral immunomodulation in a murine glioma model. BMC Immunol. 2009, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.; Brandenburg, S.; Turkowski, K.; Müller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e1620. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Burguillos, M.A.; Osman, A.M.; Frijhoff, J.; Carrillo-Jiménez, A.; Kanatani, S.; Augsten, M.; Saidi, D.; Rodhe, J.; Kavanagh, E.; et al. Glioma-induced inhibition of caspase-3 in microglia promotes a tumor-supportive phenotype. Nat. Immunol. 2016, 17, 1282–1290. [Google Scholar] [CrossRef]
- Wagner, S.; Czub, S.; Greif, M.; Vince, G.H.; Süss, N.; Kerkau, S.; Rieckmann, P.; Roggendorf, W.; Roosen, K.; Tonn, J.C. Microglial/macrophage expression of interleukin 10 in human glioblastomas. Int. J. Cancer 1999, 82, 12–16. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [Green Version]
- a Dzaye, O.D.; Hu, F.; Derkow, K.; Haage, V.; Euskirchen, P.; Harms, C.; Lehnardt, S.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Glioma Stem Cells but Not Bulk Glioma Cells Upregulate IL-6 Secretion in Microglia/Brain Macrophages via Toll-like Receptor 4 Signaling. J. Neuropathol. Exp. Neurol. 2016, 75, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Ping, Y.F.; Zhou, W.; He, Z.C.; Chen, C.; Bian, B.S.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.C.; et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-beta. J. Neurooncol. 2001, 53, 177–185. [Google Scholar] [CrossRef]
- Hu, F.; Ku, M.C.; Markovic, D.; a Dzaye, O.D.; Lehnardt, S.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Glioma-associated microglial MMP9 expression is upregulated by TLR2 signaling and sensitive to minocycline. Int. J. Cancer 2014, 135, 2569–2578. [Google Scholar] [CrossRef]
- Hu, F.; a Dzaye, O.D.; Hahn, A.; Yu, Y.; Scavetta, R.J.; Dittmar, G.; Kaczmarek, A.K.; Dunning, K.R.; Ricciardelli, C.; Rinnenthal, J.L.; et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro-Oncology 2015, 17, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro-Oncology 2012, 14, 958–978. [Google Scholar] [CrossRef] [Green Version]
- Sa, J.K.; Chang, N.; Lee, H.W.; Cho, H.J.; Ceccarelli, M.; Cerulo, L.; Yin, J.; Kim, S.S.; Caruso, F.P.; Lee, M.; et al. Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol. 2020, 21, 216. [Google Scholar] [CrossRef]
- Khan, A.B.; Lee, S.; Latha, K.; Harmanci, A.; Yang, Y.; Marisetty, A.; Patel, R.; Heimberger, A.B.; Fuller, G.N.; Deneen, B.; et al. Cxcr4 expression drives proneural-to-mesenchymal transition in glioblastoma. Submitted. Neuro-Oncology, Unpublished work.
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Tomaszowski, K.H.; Marisetty, A.; Kong, L.Y.; Wei, J.; Duna, M.; Blumberg, K.; Ji, X.; Jacobs, C.; Fuller, G.N.; et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight 2020, 5, e134386. [Google Scholar] [CrossRef] [PubMed]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro-Oncology 2016, 18, 1253–1264. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Rayman, P.; Ireland, J.; Ko, J.; Rini, B.; Borden, E.C.; Garcia, J.; Vogelbaum, M.A.; Finke, J. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro-Oncology 2011, 13, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Sippel, T.R.; White, J.; Nag, K.; Tsvankin, V.; Klaassen, M.; Kleinschmidt-DeMasters, B.K.; Waziri, A. Neutrophil degranulation and immunosuppression in patients with GBM: Restoration of cellular immune function by targeting arginase I. Clin. Cancer Res. 2011, 17, 6992–7002. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Zea, A.H.; Culotta, K.S.; Zabaleta, J.; Ochoa, J.B.; Ochoa, A.C. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J. Biol. Chem. 2002, 277, 21123–21129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohanbash, G.; McKaveney, K.; Sakaki, M.; Ueda, R.; Mintz, A.H.; Amankulor, N.; Fujita, M.; Ohlfest, J.R.; Okada, H. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-α. Cancer Res. 2013, 73, 6413–6423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [Green Version]
- Chabas, D.; Baranzini, S.E.; Mitchell, D.; Bernard, C.C.; Rittling, S.R.; Denhardt, D.T.; Sobel, R.A.; Lock, C.; Karpuj, M.; Pedotti, R.; et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 2001, 294, 1731–1735. [Google Scholar] [CrossRef]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef]
- Unruh, D.; Zewde, M.; Buss, A.; Drumm, M.R.; Tran, A.N.; Scholtens, D.M.; Horbinski, C. Methylation and transcription patterns are distinct in IDH mutant gliomas compared to other IDH mutant cancers. Sci. Rep. 2019, 9, 8946. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Wilhelm, D.; Rajky, O.; Kurscheid, S.; Kresl, P.; Wöhrer, A.; Marosi, C.; Hegi, M.E.; et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro-Oncology 2017. [Google Scholar] [CrossRef] [Green Version]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Kaffes, I.; Szulzewsky, F.; Chen, Z.; Herting, C.J.; Gabanic, B.; Velázquez Vega, J.E.; Shelton, J.; Switchenko, J.M.; Ross, J.L.; McSwain, L.F.; et al. Human Mesenchymal glioblastomas are characterized by an increased immune cell presence compared to Proneural and Classical tumors. Oncoimmunology 2019, 8, e1655360. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Company, C.; Dramaretska, Y.; Barozzi, I.; Göhrig, A.; Kertalli, S.; Großmann, M.; Naumann, H.; Sanchez-Bailon, M.P.; Hulsman, D.; et al. Phenotypic Mapping of Pathologic Cross-Talk between Glioblastoma and Innate Immune Cells by Synthetic Genetic Tracing. Cancer Discov. 2021, 11, 754–777. [Google Scholar] [CrossRef] [PubMed]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Güç, E.; Kapourani, C.A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021, 184, 2454–2470.e2426. [Google Scholar] [CrossRef]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Pitter, K.L.; Tamagno, I.; Alikhanyan, K.; Hosni-Ahmed, A.; Pattwell, S.S.; Donnola, S.; Dai, C.; Ozawa, T.; Chang, M.; Chan, T.A.; et al. Corticosteroids compromise survival in glioblastoma. Brain 2016, 139, 1458–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herting, C.J.; Chen, Z.; Maximov, V.; Duffy, A.; Szulzewsky, F.; Shayakhmetov, D.M.; Hambardzumyan, D. Tumour-associated macrophage-derived interleukin-1 mediates glioblastoma-associated cerebral oedema. Brain 2019, 142, 3834–3851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat Commun 2021, 12, 6938. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- de Groot, J.; Penas-Prado, M.; Alfaro-Munoz, K.; Hunter, K.; Pei, B.L.; O’Brien, B.; Weathers, S.P.; Loghin, M.; Kamiya Matsouka, C.; Yung, W.K.A.; et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro-Oncology 2020, 22, 539–549. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020. [Google Scholar] [CrossRef]
- Arrieta, V.A.; Chen, A.X.; Kane, J.R.; Kang, S.J.; Kassab, C.; Dmello, C.; Zhao, J.; Burdett, K.B.; Upadhyayula, P.S.; Lee-Chang, C.; et al. ERK1/2 phosphorylation predicts survival following anti-PD-1 immunotherapy in recurrent glioblastoma. Nat. Cancer 2021. [Google Scholar] [CrossRef]
- Ochocka, N.; Segit, P.; Walentynowicz, K.A.; Wojnicki, K.; Cyranowski, S.; Swatler, J.; Mieczkowski, J.; Kaminska, B. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021, 12, 1151. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Brown, M.C.; Zhang, G.; Lin, X.; Chen, Y.; Wei, Z.; Beaubier, N.; Yan, H.; He, Y.; Desjardins, A.; et al. Very low mutation burden is a feature of inflamed recurrent glioblastomas responsive to cancer immunotherapy. Nat. Commun. 2021, 12, 352. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Stupp, R. Withholding temozolomide in glioblastoma patients with unmethylated MGMT promoter—Still a dilemma? Neuro-Oncology 2015, 17, 1425–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrieta, V.A.; Najem, H.; Petrosyan, E.; Lee-Chang, C.; Chen, P.; Sonabend, A.M.; Heimberger, A.B. The Eclectic Nature of Glioma-Infiltrating Macrophages and Microglia. Int. J. Mol. Sci. 2021, 22, 13382. https://doi.org/10.3390/ijms222413382
Arrieta VA, Najem H, Petrosyan E, Lee-Chang C, Chen P, Sonabend AM, Heimberger AB. The Eclectic Nature of Glioma-Infiltrating Macrophages and Microglia. International Journal of Molecular Sciences. 2021; 22(24):13382. https://doi.org/10.3390/ijms222413382
Chicago/Turabian StyleArrieta, Víctor A., Hinda Najem, Edgar Petrosyan, Catalina Lee-Chang, Peiwen Chen, Adam M. Sonabend, and Amy B. Heimberger. 2021. "The Eclectic Nature of Glioma-Infiltrating Macrophages and Microglia" International Journal of Molecular Sciences 22, no. 24: 13382. https://doi.org/10.3390/ijms222413382
APA StyleArrieta, V. A., Najem, H., Petrosyan, E., Lee-Chang, C., Chen, P., Sonabend, A. M., & Heimberger, A. B. (2021). The Eclectic Nature of Glioma-Infiltrating Macrophages and Microglia. International Journal of Molecular Sciences, 22(24), 13382. https://doi.org/10.3390/ijms222413382