
Metabolic Effects of CCN5/WISP2 Gene Deficiency and Transgenic Overexpression in Mice


Abstract
:1. Introduction
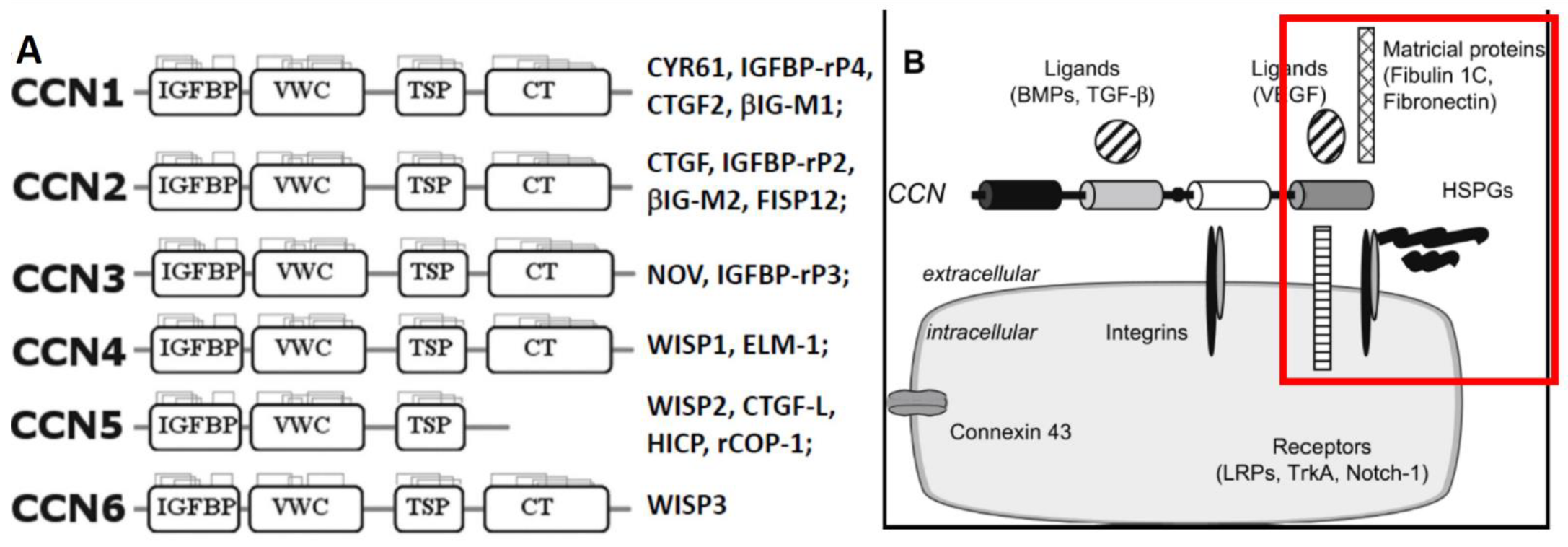
2. General Overview of CCN/WISP Family of Matricellular Proteins
3. The Effects of a Systemic CCN5/WISP2 Gene Deficiency
3.1. Adipocyte Hypertrophy, Increased Adipogenesis, and Mild Obesity
3.2. Increased Lipid Accumulation and Fibrosis in the Heart
3.3. Mild Hyperglycemia, Hyperinsulinemia and Cardiac Dysfunction
4. Differential Roles Played by CCN2, CCN3, and CCN4 in Obesity, Fibrosis, and Pancreatic Islets
5. Metabolic Effects of Adipocyte-Specific and Systemic Overexpression of CCN5
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chowdhury, S.; Wang, X.; Srikant, C.B.; Li, Q.; Fu, M.; Gong, Y.J.; Ning, G.; Liu, J.L. IGF-I stimulates CCN5/WISP2 gene expression in pancreatic beta-cells, which promotes cell proliferation and survival against streptozotocin. Endocrinology 2014, 155, 1629–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaddour, N.; Zhang, D.; Gao, Z.H.; Liu, J.L. Recombinant protein CCN5/WISP2 promotes islet cell proliferation and survival in vitro. Growth Factors 2019, 37, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Kaddour, N.; Chowdhury, S.; Li, Q.; Gao, Z.H. Role of CCN5 (WNT1 inducible signaling pathway protein 2) in pancreatic islets. J. Diabetes 2017, 9, 462–474. [Google Scholar] [CrossRef]
- Kim, J.; Joo, S.; Eom, G.H.; Lee, S.H.; Lee, M.A.; Lee, M.; Kim, K.W.; Kim, D.H.; Kook, H.; Kwak, T.H.; et al. CCN5 knockout mice exhibit lipotoxic cardiomyopathy with mild obesity and diabetes. PLoS ONE 2018, 13, e0207228. [Google Scholar] [CrossRef]
- Grunberg, J.R.; Hoffmann, J.M.; Hedjazifar, S.; Nerstedt, A.; Jenndahl, L.; Elvin, J.; Castellot, J.; Wei, L.; Moverare-Skrtic, S.; Ohlsson, C.; et al. Overexpressing the novel autocrine/endocrine adipokine WISP2 induces hyperplasia of the heart, white and brown adipose tissues and prevents insulin resistance. Sci. Rep. 2017, 7, 43515. [Google Scholar] [CrossRef] [Green Version]
- Grotendorst, G.R.; Lau, L.F.; Perbal, B. CCN Proteins Are Distinct from and Should Not Be Considered Members of the Insulin-Like Growth Factor-Binding Protein Superfamily. Endocrinology 2000, 141, 2254–2256. [Google Scholar] [CrossRef] [PubMed]
- Holbourn, K.P.; Malfois, M.; Acharya, K.R. First Structural Glimpse of CCN3 and CCN5 Multifunctional Signaling Regulators Elucidated by Small Angle X-ray Scattering. J. Biol. Chem. 2011, 286, 22243–22249. [Google Scholar] [CrossRef] [Green Version]
- Brigstock, D.R.; Goldschmeding, R.; Katsube, K.-I.; Lam, S.C.-T.; Lau, L.F.; Lyons, K.; Naus, C.; Perbal, B.; Riser, B.; Takigawa, M.; et al. Proposal for a unified CCN nomenclature. Mol. Pathol. 2003, 56, 127–128. [Google Scholar] [CrossRef]
- Rachfal, A.W.; Brigstock, D.R. Structural and functional properties of CCN proteins. Vitam. Horm. 2005, 70, 69–103. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. All in the CCN family: Essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 2006, 119, 4803–4810. [Google Scholar] [CrossRef] [Green Version]
- Perbal, B. The concept of the CCN protein family revisited: A centralized coordination network. J. Cell Commun. Signal. 2018, 12, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-C.; Lau, L.F. Functions and mechanisms of action of CCN matricellular proteins. Intern. J Biochem. Cell Biol. 2009, 41, 771–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segarini, P.R.; Nesbitt, J.E.; Li, D.; Hays, L.G.; Yates, J.R., 3rd; Carmichael, D.F. The low density lipoprotein receptor-related protein/alpha2-macroglobulin receptor is a receptor for connective tissue growth factor. J. Biol. Chem. 2001, 276, 40659–40667. [Google Scholar] [CrossRef] [Green Version]
- Rayego-Mateos, S.; Rodrigues-Díez, R.; Morgado-Pascual, J.L.; Rodrigues Díez, R.R.; Mas, S.; Lavoz, C.; Alique, M.; Pato, J.; Keri, G.; Ortiz, A.; et al. Connective tissue growth factor is a new ligand of epidermal growth factor receptor. J. Mol. Cell Biol. 2013, 5, 323–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.-I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [Green Version]
- Kular, L.; Pakradouni, J.; Kitabgi, P.; Laurent, M.; Martinerie, C. The CCN family: A new class of inflammation modulators? Biochimie 2011, 93, 377–388. [Google Scholar] [CrossRef]
- Gray, M.R.; Malmquist, J.A.; Sullivan, M.; Blea, M.; Castellot, J.J., Jr. CCN5 Expression in mammals. II. Adult rodent tissues. J. Cell Commun. Signal. 2007, 1, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.A.; Gray, M.R.; Oliveira, B.E.; Koch, M.; Castellot, J.J., Jr. CCN5 expression in mammals: I. Embryonic and fetal tissues of mouse and human. J. Cell Commun. Signal. 2007, 1, 127–143. [Google Scholar] [CrossRef] [Green Version]
- Dahlman, I.; Elsen, M.; Tennagels, N.; Korn, M.; Brockmann, B.; Sell, H.; Eckel, J.; Arner, P. Functional annotation of the human fat cell secretome. Arch. Physiol. Biochem. 2012, 118, 84–91. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Hedjazifar, S.; Jenndahl, L.; Gogg, S.; Grünberg, J.; Gustafson, B.; Klimcakova, E.; Stich, V.; Langin, D.; Laakso, M.; et al. WISP2 regulates preadipocyte commitment and PPARγ activation by BMP4. Proc. Natl. Acad. Sci. USA 2013, 110, 2563–2568. [Google Scholar] [CrossRef] [Green Version]
- Mason, H.R.; Grove-Strawser, D.; Rubin, B.S.; Nowak, R.A.; Castellot, J.J., Jr. Estrogen induces CCN5 expression in the rat uterus in vivo. Endocrinology 2004, 145, 976–982. [Google Scholar] [CrossRef] [Green Version]
- Grunberg, J.R.; Elvin, J.; Paul, A.; Hedjazifar, S.; Hammarstedt, A.; Smith, U. CCN5/WISP2 and metabolic diseases. J. Cell Commun. Signal. 2018, 12, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Twigg, S.M. Regulation and bioactivity of the CCN family of genes and proteins in obesity and diabetes. J. Cell Commun. Signal. 2018, 12, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Zhao, G.; Lyons, K.M. Characterization of bone morphology in CCN5/WISP2 knockout mice. J. Cell Commun. Signal. 2018, 12, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.K.; Chong, H.C.; Tan, E.H.P.; Tan, N.S. Getting ‘Smad’ about obesity and diabetes. Nutr. Diabetes 2012, 2, e29. [Google Scholar] [CrossRef] [Green Version]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from Obesity and Diabetes by Blockade of TGF-β/Smad3 Signaling. Cell Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.-Q.; Zhang, J.-W.; Daniel Lane, M. Sequential gene promoter interactions of C/EBPβ, C/EBPα, and PPARγ during adipogenesis. Biochem. Biophys. Res. Commun. 2004, 319, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Li, H.; Zeng, C.; Chen, W.; Wei, L.; Liu, Y.; Qi, X. Endogenous CCN5 Participates in Angiotensin II/TGF-β1 Networking of Cardiac Fibrosis in High Angiotensin II-Induced Hypertensive Heart Failure. Front. Pharmacol. 2020, 11, 1235. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Lee, M.A.; Li, Y.; Yang, D.K.; Kho, C.; Oh, J.G.; Hong, G.; Lee, A.; Song, M.H.; LaRocca, T.J.; et al. Matricellular Protein CCN5 Reverses Established Cardiac Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Cavalera, M.; Wang, J.; Frangogiannis, N.G. Obesity, metabolic dysfunction, and cardiac fibrosis: Pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl. Res. 2014, 164, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, R.; Lau, D.H.; Sanders, P. Impact of obesity on cardiac metabolism, fibrosis, and function. Trends Cardiovasc. Med. 2015, 25, 119–126. [Google Scholar] [CrossRef]
- Martinerie, C.; Garcia, M.; Do, T.T.H.; Antoine, B.; Moldes, M.; Dorothee, G.; Kazazian, C.; Auclair, M.; Buyse, M.; Ledent, T.; et al. NOV/CCN3: A New Adipocytokine Involved in Obesity-Associated Insulin Resistance. Diabetes 2016, 65, 2502–2515. [Google Scholar] [CrossRef] [Green Version]
- Paradis, R.; Lazar, N.; Antinozzi, P.; Perbal, B.; Buteau, J. Nov/Ccn3, a novel transcriptional target of FoxO1, impairs pancreatic beta-cell function. PLoS ONE 2013, 8, e64957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Ruiz, R.; García-Alamán, A.; Esteban, Y.; Mir-Coll, J.; Serra-Navarro, B.; Fontcuberta-PiSunyer, M.; Broca, C.; Armanet, M.; Wojtusciszyn, A.; Kram, V.; et al. Wisp1 is a circulating factor that stimulates proliferation of adult mouse and human beta cells. Nat. Commun. 2020, 11, 5982. [Google Scholar] [CrossRef] [PubMed]
- Charrier, A.; Brigstock, D.R. Regulation of pancreatic function by connective tissue growth factor (CTGF, CCN2). Cytokine Growth Factor Rev. 2013, 24, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henley, K.D.; Gooding, K.A.; Economides, A.N.; Gannon, M. Inactivation of the dual Bmp/Wnt inhibitor Sostdc1 enhances pancreatic islet function. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E752–E761. [Google Scholar] [CrossRef] [Green Version]
- Guney, M.A.; Petersen, C.P.; Boustani, A.; Duncan, M.R.; Gunasekaran, U.; Menon, R.; Warfield, C.; Grotendorst, G.R.; Means, A.L.; Economides, A.N.; et al. Connective tissue growth factor acts within both endothelial cells and beta cells to promote proliferation of developing beta cells. Proc. Natl. Acad. Sci. USA 2011, 108, 15242–15247. [Google Scholar] [CrossRef] [Green Version]
- Crawford, L.A.; Guney, M.A.; Oh, Y.A.; Deyoung, R.A.; Valenzuela, D.M.; Murphy, A.J.; Yancopoulos, G.D.; Lyons, K.M.; Brigstock, D.R.; Economides, A.; et al. Connective tissue growth factor (CTGF) inactivation leads to defects in islet cell lineage allocation and b-cell proliferation during embryogenesis. Mol. Endocrinol. 2009, 23, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Zhang, H.; Liu, X. Emerging role of CCN family proteins in fibrosis. J. Cell. Physiol. 2020, 236, 4195–4206. [Google Scholar] [CrossRef]
- Liu, S.; Thompson, K.; Leask, A. CCN2 expression by fibroblasts is not required for cutaneous tissue repair. Wound Repair Regen. 2014, 22, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Dorn, L.E.; Petrosino, J.M.; Wright, P.; Accornero, F. CTGF/CCN2 is an autocrine regulator of cardiac fibrosis. J. Mol. Cell. Cardiol. 2018, 121, 205–211. [Google Scholar] [CrossRef]
- Yoon, P.O.; Lee, M.A.; Cha, H.; Jeong, M.H.; Kim, J.; Jang, S.P.; Choi, B.Y.; Jeong, D.; Yang, D.K.; Hajjar, R.J.; et al. The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. J. Mol. Cell Cardiol. 2010, 49, 294–303. [Google Scholar] [CrossRef]
- Chakrabarti, P. Promoting Adipose Specificity: The Adiponectin Promoter. Endocrinology 2010, 151, 2408–2410. [Google Scholar] [CrossRef] [PubMed]
- Brejchova, K.; Balas, L.; Paluchova, V.; Brezinova, M.; Durand, T.; Kuda, O. Understanding FAHFAs: From structure to metabolic regulation. Prog. Lipid Res. 2020, 79, 101053. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| CCN5-Knockout | aP2-CCN5 | |
|---|---|---|
| When fed normal chow diet (NCD or LFD) | ||
| Body weight | Slight increase | Normal |
| Adipocyte | Hypertrophy | Normal |
| Fat mass (obesity) | Increased | Normal |
| Lean mass | Increased | |
| Insulin sensitivity | Decreased | Increased |
| Glucose tolerance | Decreased | Increased |
| Glycemia | Elevated | Normal |
| Insulinemia | Normal/Elevated | Normal |
| Water intake | Decreased | |
| Food intake | Normal | Normal |
| Energy expenditure | Normal | |
| Cardiomyocyte | Hypertrophy | Hyperplasia |
| Cardiac function | Systolic + Diastolic deficiency | Not tested |
| Lipid accumulation | Increased in heart | |
| Fibrosis | Heart, fat | Not tested |
| Adipogenic genes | 4 Increased (SREBP, C/EBP, PPARγ, aP2) | |
| Adiponectin level | Decreased | |
| When fed high-fat diet (HFD) | ||
| Body weight | Increased | Slight increase |
| Adipocyte | Further hypertrophy | sWAT: hyperplasia, hypotrophy |
| Fat mass (obesity) | Increased | Decreased |
| Lean mass | Increased | |
| Insulin sensitivity | Decreased | Increased |
| Glucose tolerance | Decreased/Intolerance | Increased |
| Glycemia | Higher, Mild diabetes | Decreased |
| Insulinemia | Elevated/Normal | Decreased |
| Water intake | Increased | |
| Food intake | Increased | Increased |
| Energy expenditure | Increased (per body weight) | |
| Cardiomyocyte | Hypertrophy | Hyperplasia |
| Cardiac function | Deficient | Not tested |
| Lipid accumulation | Further increased in heart | |
| Fibrosis | Heart (more), fat | Not tested |
| Adipogenic genes | 2 Increased (PPARγ, aP2) | 13 Increased including SREBP |
| Adiponectin level | Decreased | Increased |
| CCN2 | CCN3 | CCN4 | CCN5 | |
|---|---|---|---|---|
| Adipocyte and adiposity | ||||
| Proliferation | Promote | Promote | ||
| Hypertrophy | Increase | Limit | ||
| Adipocity | Increase | Decrease | ||
| Energy expenditure | Decrease | |||
| Pancreatic β-cell and insulin secretion | ||||
| β-cell proliferation | Promote, early | Decrease | Promote | Promote |
| Islet vascularity | Increase | |||
| β-cell maturition | Promote | |||
| Insulin secretion | Impair | |||
| Fibrosis, heart | Pro- | Anti- | Pro- | Anti- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alami, T.; Liu, J.-L. Metabolic Effects of CCN5/WISP2 Gene Deficiency and Transgenic Overexpression in Mice. Int. J. Mol. Sci. 2021, 22, 13418. https://doi.org/10.3390/ijms222413418
Alami T, Liu J-L. Metabolic Effects of CCN5/WISP2 Gene Deficiency and Transgenic Overexpression in Mice. International Journal of Molecular Sciences. 2021; 22(24):13418. https://doi.org/10.3390/ijms222413418
Chicago/Turabian StyleAlami, Tara, and Jun-Li Liu. 2021. "Metabolic Effects of CCN5/WISP2 Gene Deficiency and Transgenic Overexpression in Mice" International Journal of Molecular Sciences 22, no. 24: 13418. https://doi.org/10.3390/ijms222413418
APA StyleAlami, T., & Liu, J. -L. (2021). Metabolic Effects of CCN5/WISP2 Gene Deficiency and Transgenic Overexpression in Mice. International Journal of Molecular Sciences, 22(24), 13418. https://doi.org/10.3390/ijms222413418