
An Insight on Novel Molecular Pathways in Metastatic Prostate Cancer: A Focus on DDR, MSI and AKT

 ,
,  , and
, and 
Abstract
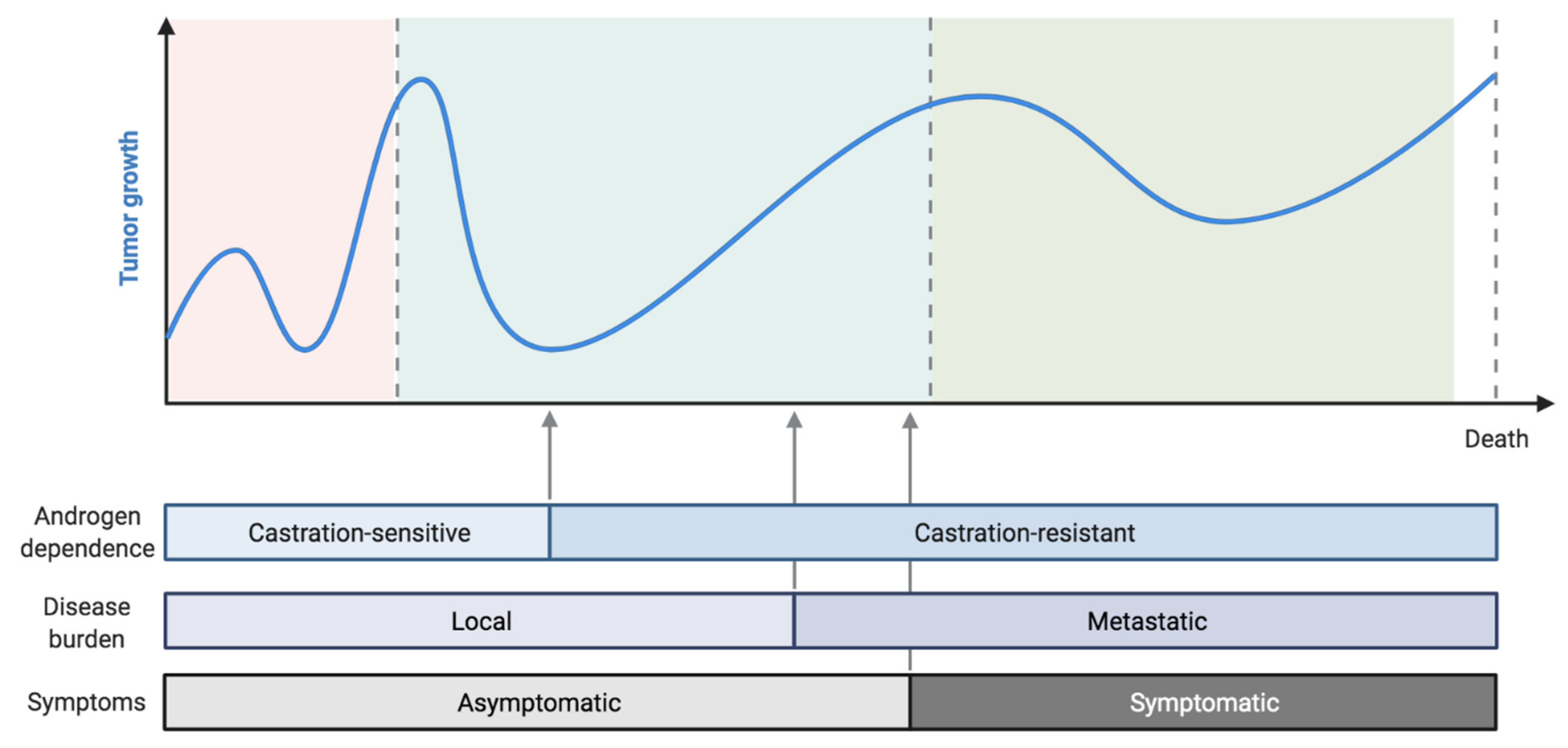
:1. Introduction
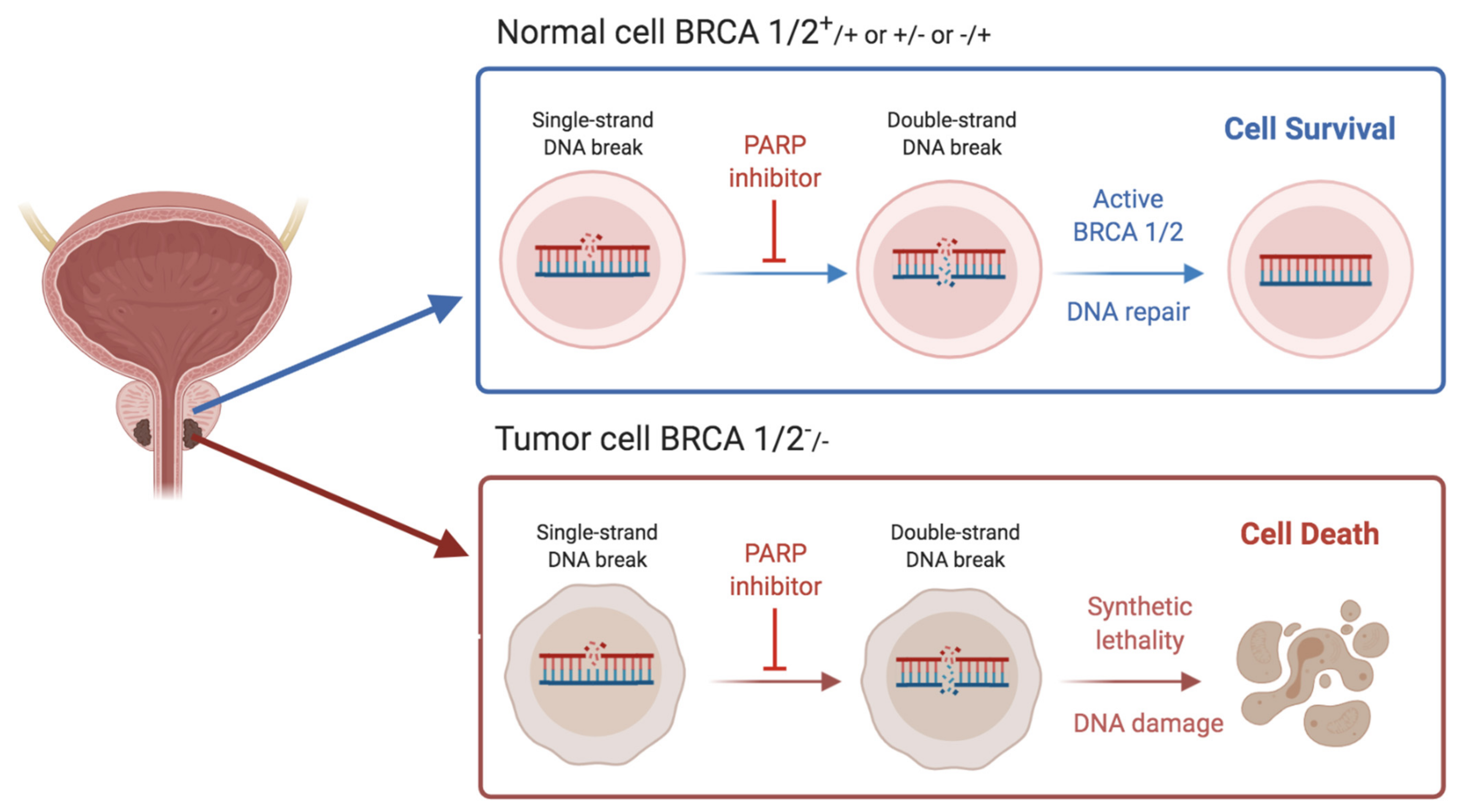
2. DDR Alterations, PARP Inhibitors and Platinum-Based Chemotherapy
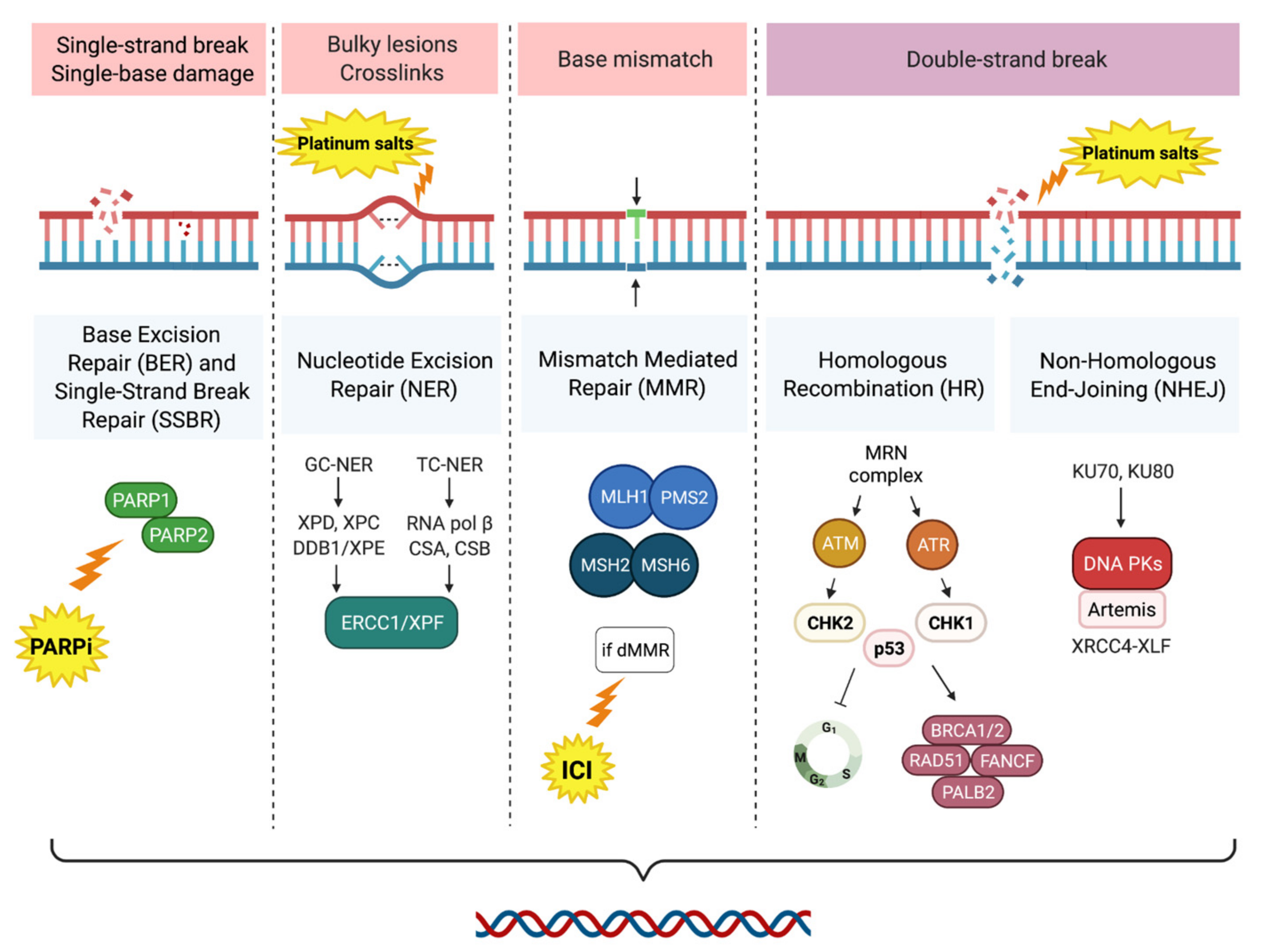
2.1. DNA Damage Response Systems
2.2. Main Trials on PARPi in Metastatic Prostate Cancer and New Approaches under Investigation
2.3. Role of Platinum-Based Chemotherapy
2.4. PARPi and Platinum Salts: Current and Future Directions
3. Microsatellite Instability and Immunotherapy
3.1. Microsatellite Instability as a Predictor for Immunotherapy
3.2. Microsatellite Instability and Prostate Cancer
3.3. Immunotherapy in mCRPC: Vaccines
3.4. Immunotherapy in mCRPC: Anti-CTLA-4 and Anti-PD-1/PD-L1 Agents
3.5. Immunotherapy in mCRPC: Combining ICIs with ARTAs
3.6. Immunotherapy in mCRPC: Combining ICIs with Chemotherapy
3.7. Immunotherapy in mCRPC: Other Immuno-Combinations
3.8. Immunotherapy in mCRPC: Where Are We Running?
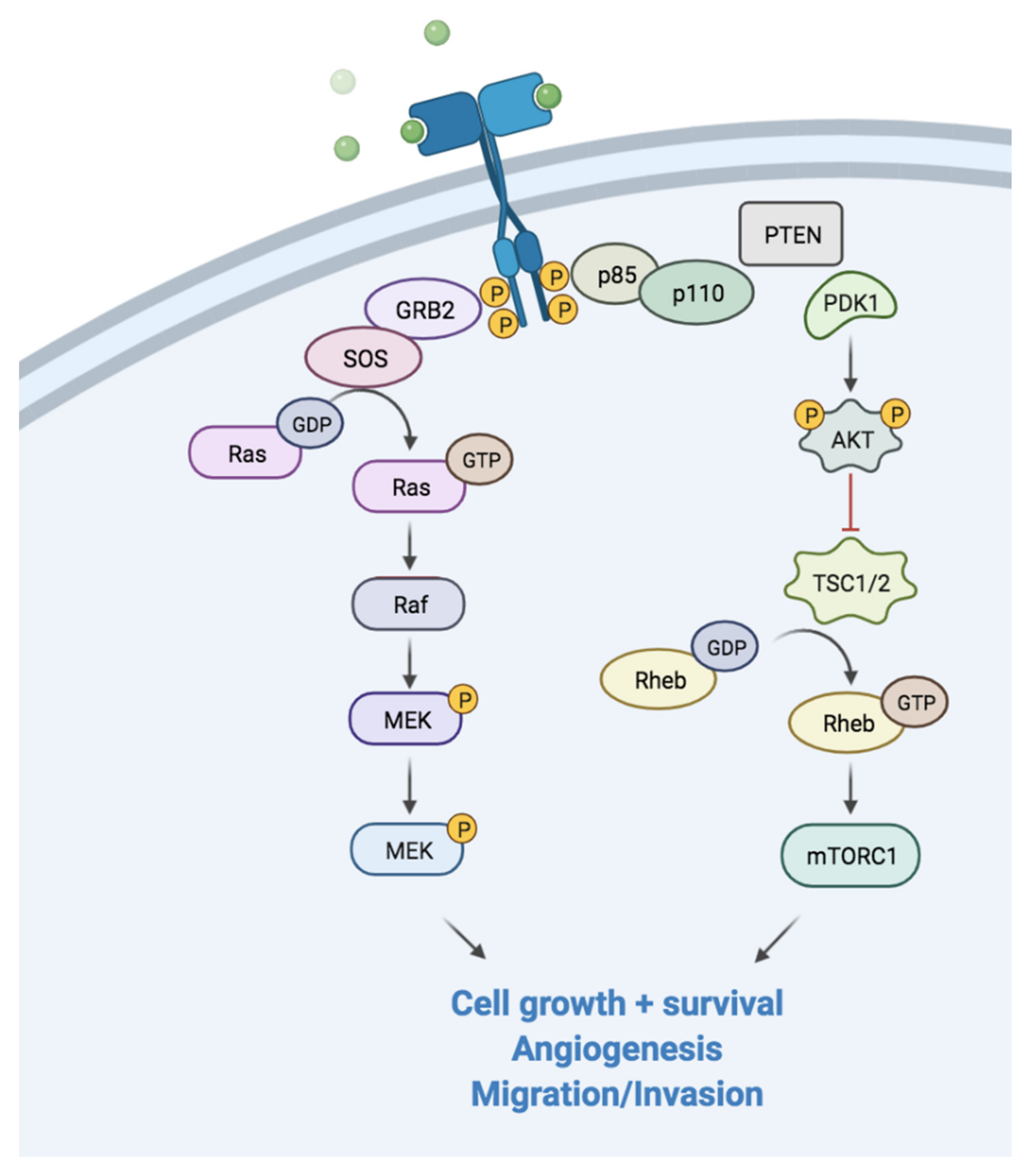
4. PI3K and Akt Pathway
4.1. Functioning and Role in Carcinogenesis Process
4.2. Development of PI3K/AKT Targeted Therapy in Prostate Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Samson, D.J.; Seidenfeld, J.; Schmitt, B.; Hasselblad, V.; Albertsen, P.C.; Bennett, C.L.; Wilt, T.J.; Aronson, N. Systematic review and meta-analysis of monotherapy compared with combined androgen blockade for patients with advanced prostate carcinoma. Cancer 2002, 95, 361–376. [Google Scholar] [CrossRef]
- Tannock, I.F.; De Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; De Bono, J.S.; Molina, A.; Logothetis, C.J.; De Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; De Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- De Wit, R.; De Bono, J.; Sternberg, C.N.; Fizazi, K.; Tombal, B.; Wülfing, C.; Kramer, G.; Eymard, J.C.; Bamias, A.; Carles, J.; et al. Cabazitaxel versus Abiraterone or Enzalutamide in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 2506–2518. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Mollica, V.; Di Nunno, V.; Cimadamore, A.; Lopez-Beltran, A.; Cheng, L.; Santoni, M.; Scarpelli, M.; Montironi, R.; Massari, F. Molecular Mechanisms Related to Hormone Inhibition Resistance in Prostate Cancer. Cells 2019, 8, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; McKay, R.; Abida, W.; Aggarwal, R.; Alumkal, J.; Alva, A.; Feng, F.; Gao, X.; Graff, J.; Hussain, M.; et al. Accelerating precision medicine in metastatic prostate cancer. Nat. Cancer 2020, 1, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer: I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Boysen, G.; Barbieri, C.E.; Bryant, H.E.; Castro, E.; Nelson, P.S.; Olmos, D.; Pritchard, C.C.; Rubin, M.A.; De Bono, J.S. DNA Repair in Prostate Cancer: Biology and Clinical Implications. Eur. Urol. 2017, 71, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; De Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- De Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Smith, M.R.; Sandhu, S.K.; Kelly, W.K.; Scher, H.I.; Efstathiou, E.; Lara, P.N.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; et al. Pre-specified interim analysis of GALAHAD: A phase II study of niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD). Ann. Oncol. 2019, 30, v884–v885. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Carducci, M.A.; Slovin, S.; Cetnar, J.; Qian, J.; McKeegan, E.M.; Refici-Buhr, M.; Chyla, B.; Shepherd, S.P.; Giranda, V.L.; et al. Targeting DNA repair with combination veliparib (ABT-888) and temozolomide in patients with metastatic castration-resistant prostate cancer. Investig. New Drugs 2014, 32, 904–912. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Daignault-Newton, S.; Twardowski, P.W.; Albany, C.; Stein, M.N.; Kunju, L.P.; Siddiqui, J.; Wu, Y.M.; Robinson, D.; Lonigro, R.J.; et al. Targeting Androgen Receptor and DNA Repair in Metastatic Castration-Resistant Prostate Cancer: Results From NCI 9012. J. Clin. Oncol. 2018, 36, 991–999. [Google Scholar] [CrossRef]
- Chao, O.S.; Goodman, O.B., Jr. Synergistic loss of prostate cancer cell viability by coinhibition of HDAC and PARP. Mol. Cancer Res. 2014, 12, 1755–1766. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Li, Y.C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, N.; Cai, D.; Kennedy, R.D.; Pathania, S.; Arora, M.; Li, Y.C.; D’Andrea, A.D.; Parvin, J.D.; Shapiro, G.I. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol. Cell. 2006, 35, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, C.N.; Petrylak, D.P.; Sartor, O.; Witjes, J.A.; Demkow, T.; Ferrero, J.M.; Eymard, J.C.; Falcon, S.; Calabrò, F.; James, N.; et al. Multinational, double-blind, phase III study of prednisone and either satraplatin or placebo in patients with castrate-refractory prostate cancer progressing after prior chemotherapy: The SPARC trial. J. Clin. Oncol. 2009, 27, 5431–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, J.M.; Barnett, E.; Nauseef, J.T.; Nguyen, B.; Stopsack, K.H.; Wibmer, A.; Flynn, J.R.; Heller, G.; Danila, D.C.; Rathkopf, D.; et al. Platinum-Based Chemotherapy in Metastatic Prostate Cancer With DNA Repair Gene Alterations. JCO Precis. Oncol. 2020, 4, 355–366. [Google Scholar] [CrossRef]
- Toyoda, M.; Minami, H. Clinical development of PARP inhibitors. Gan Kagaku. Ryoho. 2012, 39, 519–524. [Google Scholar]
- Ku, S.Y.; Gleave, M.E.; Beltran, H. Towards precision oncology in advanced prostate cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J.; et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef] [Green Version]
- Page, E.C.; Bancroft, E.K.; Brook, M.N.; Assel, M.; Hassan Al Battat, M.; Thomas, S.; Taylor, N.; Chamberlain, A.; Pope, J.; Raghallaigh, H.N.; et al. Interim Results from the IMPACT Study: Evidence for Prostate-specific Antigen Screening in BRCA2 Mutation Carriers. Eur. Urol. 2019, 76, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Massari, F.; Mollica, V. Re: Platinum-based Chemotherapy in Metastatic Prostate Cancer with DNA Repair Gene Alterations. Eur. Urol. 2020, 78, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. MSI-H: A truly agnostic biomarker? Nat. Rev. Clin. Oncol. 2020, 17, 68. [Google Scholar] [CrossRef] [PubMed]
- Vitkin, N.; Nersesian, S.; Siemens, D.R.; Koti, M. The Tumor Immune Contexture of Prostate Cancer. Front. Immunol. 2019, 10, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, D.N.; Rescigno, P.; Liu, D.; Yuan, W.; Carreira, S.; Lambros, M.B.; Seed, G.; Mateo, J.; Riisnaes, R.; Mullane, S.; et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J. Clin. Investig. 2018, 128, 5185. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Morrissey, C.; Kumar, A.; Zhang, X.; Smith, C.; Coleman, I.; Salipante, S.J.; Milbank, J.; Yu, M.; Grady, W.M.; et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat. Commun. 2014, 5, 4988. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Cheng, H.H.; Tretiakova, M.S.; Vakar-Lopez, F.; Klemfuss, N.; Konnick, E.Q.; Mostaghel, E.A.; Nelson, P.S.; Yu, E.Y.; Montgomery, B.; et al. Mismatch repair deficiency may be common in ductal adenocarcinoma of the prostate. Oncotarget 2016, 7, 82504–82510. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.; Mollica, V.; Cimadamore, A.; Santoni, M.; Scarpelli, M.; Giunchi, F.; Cheng, L.; Lopez-Beltran, A.; Fiorentino, M.; Montironi, R.; et al. Is There a Role for Immunotherapy in Prostate Cancer? Cells 2020, 9, 2051. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Bansal, D.; Reimers, M.A.; Knoche, E.M.; Pachynski, R.K. Immunotherapy and Immunotherapy Combinations in Metastatic Castration-Resistant Prostate Cancer. Cancers 2021, 13, 334. [Google Scholar] [CrossRef]
- Schellhammer, P.F.; Chodak, G.; Whitmore, J.B.; Sims, R.; Frohlich, M.W.; Kantoff, P.W. Lower baseline prostate-specific antigen is associated with a greater overall survival benefit from sipuleucel-T in the Immunotherapy for Prostate Adenocarcinoma Treatment (IMPACT) trial. Urology 2013, 81, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.R.; Massard, C.; Ott, P.A.; Haas, N.B.; Lopez, J.S.; Ejadi, S.; Wallmark, J.M.; Keam, B.; Delord, J.P.; Aggarwal, R.; et al. Pembrolizumab for advanced prostate adenocarcinoma: Findings of the KEYNOTE-028 study. Ann. Oncol. 2018, 29, 1807–1813. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Small, E.J.; Tchekmedyian, N.S.; Rini, B.I.; Fong, L.; Lowy, I.; Allison, J.P. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin. Cancer Res. 2007, 13, 1810–1815. [Google Scholar] [CrossRef] [Green Version]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; Van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients With Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Loriot, Y.; Shaffer, D.R.; Braiteh, F.; Powderly, J.; Harshman, L.C.; Conkling, P.; Delord, J.P.; Gordon, M.; Kim, J.W.; et al. Safety and Clinical Activity of Atezolizumab in Patients with Metastatic Castration-Resistant Prostate Cancer: A Phase I Study. Clin. Cancer Res. 2021, 27, 3360–3369. [Google Scholar] [CrossRef]
- Fakhrejahani, F.; Madan, R.A.; Dahut, W.L.; Karzai, F.; Cordes, L.M.; Schlom, J.; Gulley, J.L. Avelumab in metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2017, 35 (Suppl. S6), 159. [Google Scholar] [CrossRef]
- Bishop, J.L.; Sio, A.; Angeles, A.; Roberts, M.E.; Azad, A.A.; Chi, K.N.; Zoubeidi, A. PD-L1 is highly expressed in Enzalutamide resistant prostate cancer. Oncotarget 2015, 6, 234–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graff, J.N.; Antonarakis, E.S.; Hoimes, C.J.; Tagawa, S.T.; Hwang, C.; Kilari, D.; Tije, A.J.T.; Omlin, A.G.; McDermott, R.S.; Vaishampayan, U.N.; et al. Pembrolizumab (pembro) plus enzalutamide (enza) for enza-resistant metastatic castration-resistant prostate cancer (mCRPC): KEYNOTE-199 cohorts 4–5. J. Clin. Oncol. 2020, 38 (Suppl. 6), 15. [Google Scholar] [CrossRef]
- Yu, E.Y.; Fong, P.; Piulats, J.M.; Appleman, L.; Conter, H.; Feyerabend, S.; Shore, N.; Gravis, G.; Laguerre, B.; Gurney, H.; et al. Pembrolizumab plus enzalutamide in abiraterone-pretreated patients with metastatic castration-resistant prostate cancer: Updated results from KEYNOTE-365 Cohort C. J. Urol. 2020, 203 (Suppl. S4), e368. [Google Scholar]
- Sweeney, C.J.; Gillessen, S.; Rathkopf, D.; Matsubara, N.; Drake, C.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; Buchschacher, J.L., Jr.; Doss, J.; et al. CT014—Imbassador250: A phase III Trial Comparing Atezolizumab with Enzalutamide vs Enzalutamide Alone in Patients with Metastatic Castration-Resistant Prostate Cancer (mCRPC). AACR Annu. Meeting 2020, Session VCTPL01—Opening Clinical Plenary. Available online: https://www.abstractsonline.com/pp8/#!/9045/presentation/10596 (accessed on 26 July 2020).
- Wong, R.L.; Yu, E.Y. Refining Immuno-Oncology Approaches in Metastatic Prostate Cancer: Transcending Current Limitations. Curr. Treat. Options Oncol. 2021, 22, 13. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Mella, P.G.; Castellano, D.; Minatta, J.N.; Rezazadeh, A.; Shaffer, D.R.; Vazquez Limon, J.C.; Sánchez López, H.M.; Armstrong, A.J.; Horvath, L.; et al. CheckMate 9KD Arm B final analysis: Efficacy and safety of nivolumab plus docetaxel for chemotherapy-naïve metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2021, 39 (Suppl. 12), 12. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, A.; Rosellini, M.; Rizzo, A.; Mollica, V.; Battelli, N.; Massari, F.; Santoni, M. An up-to-date evaluation of cabozantinib for the treatment of renal cell carcinoma. Expert. Opin. Pharmacother. 2021, 18, 1–14. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunother. Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Agarwal, N.; Loriot, Y.; McGregor, B.A.; Dreicer, R.; Dorff, T.B.; Maughan, B.L.; Kelly, W.K.; Pagliaro, L.C.; Srinivas, S.; Squillante, C.M.; et al. Cabozantinib in combination with atezolizumab (A) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): Results of Cohort 6 of the COSMIC-021 Study. J. Clin. Oncol. 2020, 38 (Suppl. 6), 13. [Google Scholar] [CrossRef]
- Boudadi, K.; Suzman, D.L.; Anagnostou, V.; Fu, W.; Luber, B.; Wang, H.; Niknafs, N.; White, J.R.; Silberstein, J.L.; Sullivan, R.; et al. Ipilimumab plus nivolumab and DNA-repair defects in AR-V7-expressing metastatic prostate cancer. Oncotarget 2018, 9, 28561–28571. [Google Scholar] [CrossRef] [Green Version]
- Cimadamore, A.; Cheng, L.; Massari, F.; Santoni, M.; Pepi, L.; Franzese, C.; Scarpelli, M.; Lopez-Beltran, A.; Galosi, A.B.; Montironi, R.; et al. Circulating Tumor DNA Testing for Homology Recombination Repair Genes in Prostate Cancer: From the Lab to the Clinic. Int. J. Mol. Sci. 2021, 22, 5522. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell. Bio. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhu, M.; Pan, R.; Fang, T.; Cao, Y.Y.; Chen, S.; Zhao, X.; Lei, C.Q.; Guo, L.; Chen, Y.; et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat. Immunol. 2016, 17, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; De Bono, J.S. Metastatic Prostate Cancer. N. Engl. J. Med. 2018, 378, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Correia, N.C.; Gírio, A.; Antunes, I.; Martins, L.R.; Barata, J.T. The multiple layers of non-genetic regulation of PTEN tumour suppressor activity. Eur. J. Cancer 2014, 50, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.H.; Attard, G.; Ambroisine, L.; Fisher, G.; Kovacs, G.; Brewer, D.; Clark, J.; Flohr, P.; Edwards, S.; Berney, D.M.; et al. Molecular characterisation of ERG, ETV1 and PTEN gene loci identifies patients at low and high risk of death from prostate cancer. Br. J. Cancer 2010, 102, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Reid, A.H.; Attard, G.; Brewer, D.; Miranda, S.; Riisnaes, R.; Clark, J.; Hylands, L.; Merson, S.; Vergis, R.; Jameson, C.; et al. Novel, gross chromosomal alterations involving PTEN cooperate with allelic loss in prostate cancer. Mod. Pathol. 2012, 25, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Ferraldeschi, R.; Nava Rodrigues, D.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D.; et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhu, J.; Efferson, C.L.; Ware, C.; Tammam, J.; Angagaw, M.; Laskey, J.; Bettano, K.A.; Kasibhatla, S.; Reilly, J.F.; et al. Inhibition of tumor growth progression by antiandrogens and mTOR inhibitor in a Pten-deficient mouse model of prostate cancer. Cancer Res. 2009, 69, 7466–7472. [Google Scholar] [CrossRef] [Green Version]
- Sarker, D.; Reid, A.H.; Yap, T.A.; De Bono, J.S. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [Green Version]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Li, Y.; Buttyan, R.; Dong, X. Implications of PI3K/AKT inhibition on REST protein stability and neuroendocrine phenotype acquisition in prostate cancer cells. Oncotarget 2017, 8, 84863–84876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blake, J.F.; Xu, R.; Bencsik, J.R.; Xiao, D.; Kallan, N.C.; Schlachter, S.; Mitchell, I.S.; Spencer, K.L.; Banka, A.L.; Wallace, E.M.; et al. Discovery and preclinical pharmacology of a selective ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human tumors. J. Med. Chem. 2012, 55, 8110–8127. [Google Scholar] [CrossRef]
- Lin, J.; Sampath, D.; Nannini, M.A.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, C.; Roda, D.; Roselló, S.; Oliveira, M.; Macarulla, T.; Pérez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-García, J.M.; Musib, L.; Budha, N.; et al. A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov. 2017, 7, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Isakoff, S.J.; Tabernero, J.; Molife, L.R.; Soria, J.C.; Cervantes, A.; Vogelzang, N.J.; Patel, M.R.; Hussain, M.; Baron, A.; Argilés, G.; et al. Antitumor activity of ipatasertib combined with chemotherapy: Results from a phase Ib study in solid tumors. Ann. Oncol. 2020, 31, 626–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, T.; Fujiwara, Y.; Matsubara, N.; Tomomatsu, J.; Iwasa, S.; Tanaka, A.; Endo-Tsukude, C.; Nakagawa, S.; Takahashi, S. Phase I study of ipatasertib as a single agent and in combination with abiraterone plus prednisolone in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2019, 84, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): A multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: Pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Kolinsky, M.P.; Rescigno, P.; Bianchini, D.; Zafeiriou, Z.; Mehra, N.; Mateo, J.; Michalarea, V.; Riisnaes, R.; Crespo, M.; Figueiredo, I.; et al. A phase I dose-escalation study of enzalutamide in combination with the AKT inhibitor AZD5363 (capivasertib) in patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2020, 31, 619–625. [Google Scholar] [CrossRef] [Green Version]
- Crabb, S.J.; Griffiths, G.; Marwood, E.; Dunkley, D.; Downs, N.; Martin, K.; Light, M.; Northey, J.; Wilding, S.; Whitehead, A.; et al. Pan-AKT Inhibitor Capivasertib with Docetaxel and Prednisolone in Metastatic Castration-Resistant Prostate Cancer: A Randomized, Placebo-Controlled Phase II Trial (ProCAID). J. Clin. Oncol. 2021, 39, 190–201. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT (Acronym) | Phase | Number of Patients | Experimental Arm | Control Arm | Setting | Status |
|---|---|---|---|---|---|---|
| TOPARP-A (NCT01682772) [18] | II | 16 | Olaparib | - | mCRPC (previously treated with chemotherapy and novel anti-androgens) | Active, not recruiting |
| TOPARP-B (NCT01682772) [19] | II | 711 (161 with DDR gene alterations) | Olaparib (400 or 300 mg) | - | mCRPC (at least one previous taxane-based line of therapy) | Active, not recruiting |
| PROfound (NCT02987543) [9] | III | 245 in cohort A, 142 in cohort B | Olaparib | Abirateroneor Enzalutamide | mCRPC (previously treated with Abiraterone or Enzalutamide, taxane-based chemotherapy was permitted) | Active, not recruiting |
| TALAPRO-1 (NCT03148795) [21] | II | 128 | Talazoparib | - | mCRPC (previously treated with Abiraterone or Enzalutamide and at least one taxane-based chemotherapy) | Active, not recruiting |
| TALAPRO-2 (NCT03395197) | III | 1150 (estimated) | Talazoparib + Enzalutamide | Placebo + Enzalutamide | mCRPC | Recruiting |
| TALAPRO-3 (NCT04821622) | III | 550 (estimated) | Talazoparib + Enzalutamide | Placebo + Enzalutamide | mHSPC | Recruiting |
| GALAHAD (NCT02854436) [22] | II | 165 | Niraparib | - | mCRPC (androgen receptor-target treatment and taxanes) | Ongoing, not recruiting |
| MAGNITUDE (NCT03748641) | III | 765 | Niraparib + Abiraterone | Placebo + Abiraterone | mCRPC | Ongoing, not recruiting |
| TRITON2 (NCT02952534) [23] | II | 277 | Rucaparib | - | mCRPC (one or two previous novel anti-androgens and one taxane-based chemotherapy) | Completed |
| NCT (Acronym) | Phase | Number of Patients | Experimental Arm | Control Arm | Setting | Pharmaco- Dynamic | Status |
|---|---|---|---|---|---|---|---|
| NCT04382898 (PRO-MERIT) | I/II | 80 | W_pro1/W_pro1 + goserelin/W_pro1 + cemiplimab + goserelin | - | mCRPC | mRNA liposome complex of five Ags, NSAA and anti-PD-1 | Recruiting |
| NCT02933255 | I/II | 29 | Nivolumab + PROST-VAC | - | mCRPC | Anti-PD-1 and virus-based vaccine targeting PSA | Recruiting |
| NCT03493945 | I/II | 113 | BN-Brachyury + M7824/ BN-Brachyury + ALT-803/ BN-Brachyury + ALT-803 + epacadostat | - | mCRPC | MVA cancer vaccine, anti-PD-1/TGF-beta (M7824), IL-14 agonist (ALT-803) and IDO-1 inhibitor (epacadostat) | Recruiting |
| NCT02985957 | II | 497 | Nivolumab/ ipilimumab or ipilimumab | Cabazitaxel | mCRPC | Anti-PD-1 and anti-CTLA-4 | Recruiting |
| NCT03570619 (IMPACT) | II | 40 | Nivolumab + ipilimumab | - | mCRPC (CDK12 mutations) | Anti-PD-1 and anti-CTLA-4 | Recruiting |
| NCT04104893 (CHOMP) | II | 30 | Pembrolizumab | - | mCRPC (CDK12, MLH1, MSH2, MLH3, PMS1, MSH6, PMS2 mutations or MSI-H) | Anti-PD-1 | Recruiting |
| NCT03040791 (ImmunoProst) | II | 29 | Nivolumab | - | mCRPC (BRCA1/2, ATM, PTEN, CHEK2, RAD51C, RAD51D, PALb12, MLH1, MSH2, MSH6, PMS2 mutations) | Anti-PD-1 | Recruiting |
| NCT03570619 | II | 40 | Nivolumab + ipilimumab | - | Advanced solid tumors with biallelic CDK12 loss | Anti-PD-1 + anti-CTLA-4 | Recruiting |
| NCT00583024 (APP22) | II | 66 | AdPSA | - | mCRPC | PSA AdV vaccine | Active, not recruiting |
| NCT (Acronym) | Phase | Number of Patients | Experimental Arm | Control Arm | Setting | Pharmaco- Dynamic | Status |
|---|---|---|---|---|---|---|---|
| NCT03170960 (COSMIC-021) | I/II | 1732 | Atezolizumab + cabozantinib | - | mCRPC | Anti-PD-L1 and anti-VEGF and MET TKI | Recruiting |
| NCT02861573 (KEYNOTE- 365) | I/II | 1000 | Pembrolizumab + olaparib (cohort A)/docetaxel (cohort B)/enzalutamide (cohort C)/abiraterone (cohort D)/lenvatinib (cohorts E-F)/vibostolimab (cohort G)/CBDCA + etoposide (cohort H) | CBDCA + etoposide (only in cohort H’s arm 2) | mCRPC | Anti-PD-1 in combination with: PARPi, taxane, ARSIs, TKI. | Recruiting |
| NCT03673787 | I/II | 51 | Atezolizumab + ipatasertib | - | mCRPC (PTEN loss) | Anti-PD-L1 and inhibitor of the serine/threonine protein kinase Akt | Recruiting |
| NCT03330405 (JAVELIN PARP Medley) | I/II | 216 | Avelumab + talazoparib | - | Locally advanced or metastatic solid tumors (including CRPC) | Anti-PD-L1 and PARPi | Active, not recruiting |
| NCT03658447 (PRINCE) | I/II | 37 | 177Lu- PSMA + pembrolizumab | - | mCRPC | Conjugate of a PSMA ligand and a beta-emitting radioisotope Lu177 and anti-PD-1 | Active, not recruiting |
| NCT04109729 (Rad2Nivo) | I/II | 36 | Radium-223 + nivolumab | - | mCRPC | Radio-isotope Rad223 and anti-PD-1 | Recruiting |
| NCT01688492 | I/II | 57 | Ipilimumab + abiraterone | - | mCRPC | Anti-CTLA-4 and ARSI | Active, not recruiting |
| NCT03409458 | I/II | 52 | Avelumab + PT-112 | - | Advanced solid tumors | Anti-PD-L1 and a platinum agent complexed to a pyrophosphatase ligand (PT-112) | Recruiting |
| NCT02740985 | I | 307 | Durvalumab + AZD4635 | - | Advanced solid tumors | Anti-PD-L1 and adenosine A2A receptor antagonist | Active, not recruiting |
| NCT03805594 | I | 30 | 177Lu-PSMA + pembrolizumab | - | mCRPC | Conjugate of a PSMA ligand and a beta-emitting radio-isotope Lu177 and anti-PD-1isotope Lu177 and anti-PD-1 | Recruiting |
| NCT03549000 | I | 344 | NZV930 alone or + PDR001/ NIR178/ both | - | mCRPC | Anti-CD73, anti-PD-1 (PDR001) and A2AR antagonist (NIR178) | Recruiting |
| NCT04159896 | II | 49 | CEP-11981 + nivolumab | - | mCRPC | Pan-TKI with selectivity for VEGF-R/TIE2 and anti-PD-1 | Recruiting |
| NCT03338790 (CheckMate 9KD) | II | 330 | Nivolumab + rucaparib/ docetaxel/ enzalutamide | - | mCRPC | Anti-PD-1 with PARPi or taxane or ARSI | Active, not recruiting |
| NCT01867333 | II | 57 | PROST-VAC + enzalutamide | Enzalutamide | mCRPC | Virus-based vaccine and ARSI | Active, not recruiting |
| NCT04446117 (CONTACT- 02) | III | 580 | Atezolizumab + cabozantinib | Enzalutamide or abiraterone acetate | mCRPC | Anti-PD-L1 and anti-VEGF and MET TKI | Recruiting |
| NCT03834493 (KEYNOTE- 641) | III | 1200 | Pembrolizumab + enzalutamide | Placebo + enzalutamide | mCRPC | Anti-PD-1 and ARSI | Recruiting |
| NCT04191096 (KEYNOTE- 991) | III | 1232 | Pembrolizumab + enzalutamide | Placebo + enzalutamide | mHSPC | Anti-PD-1 and ARSI | Active, not recruiting |
| NCT03834506 (KEYNOTE- 921) | III | 1000 | Pembrolizumab + docetaxel | Placebo + docetaxel | mCRPC | Anti-PD-1 and taxane | Active, not recruiting |
| NCT03834519 (KEYLYNK- 010) | III | 780 | Pembrolizumab + olaparib | Abiraterone or enzalutamide | mCRPC | Anti-PD-1 and PARPi | Active, not recruiting |
| NCT04100018 (CheckMate 7DX) | III | 984 | Nivolumab + docetaxel | Placebo + docetaxel | mCRPC | Anti-PD-1 and taxane | Recruiting |
| NCT03879122 (PROSTRA- TEGY) | II/III | 135 | Docetaxel + nivolumab (arm 1)/docetaxel + ipilimumab → nivolumab (arm 2) | Docetaxel (arm 3) | mHSPC | Taxane, anti-PD-1 and anti-CTLA-4 | Active, not recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollica, V.; Marchetti, A.; Rosellini, M.; Nuvola, G.; Rizzo, A.; Santoni, M.; Cimadamore, A.; Montironi, R.; Massari, F. An Insight on Novel Molecular Pathways in Metastatic Prostate Cancer: A Focus on DDR, MSI and AKT. Int. J. Mol. Sci. 2021, 22, 13519. https://doi.org/10.3390/ijms222413519
Mollica V, Marchetti A, Rosellini M, Nuvola G, Rizzo A, Santoni M, Cimadamore A, Montironi R, Massari F. An Insight on Novel Molecular Pathways in Metastatic Prostate Cancer: A Focus on DDR, MSI and AKT. International Journal of Molecular Sciences. 2021; 22(24):13519. https://doi.org/10.3390/ijms222413519
Chicago/Turabian StyleMollica, Veronica, Andrea Marchetti, Matteo Rosellini, Giacomo Nuvola, Alessandro Rizzo, Matteo Santoni, Alessia Cimadamore, Rodolfo Montironi, and Francesco Massari. 2021. "An Insight on Novel Molecular Pathways in Metastatic Prostate Cancer: A Focus on DDR, MSI and AKT" International Journal of Molecular Sciences 22, no. 24: 13519. https://doi.org/10.3390/ijms222413519
APA StyleMollica, V., Marchetti, A., Rosellini, M., Nuvola, G., Rizzo, A., Santoni, M., Cimadamore, A., Montironi, R., & Massari, F. (2021). An Insight on Novel Molecular Pathways in Metastatic Prostate Cancer: A Focus on DDR, MSI and AKT. International Journal of Molecular Sciences, 22(24), 13519. https://doi.org/10.3390/ijms222413519