
SARS-CoV-2 Exacerbates Beta-Amyloid Neurotoxicity, Inflammation and Oxidative Stress in Alzheimer’s Disease Patients


Abstract
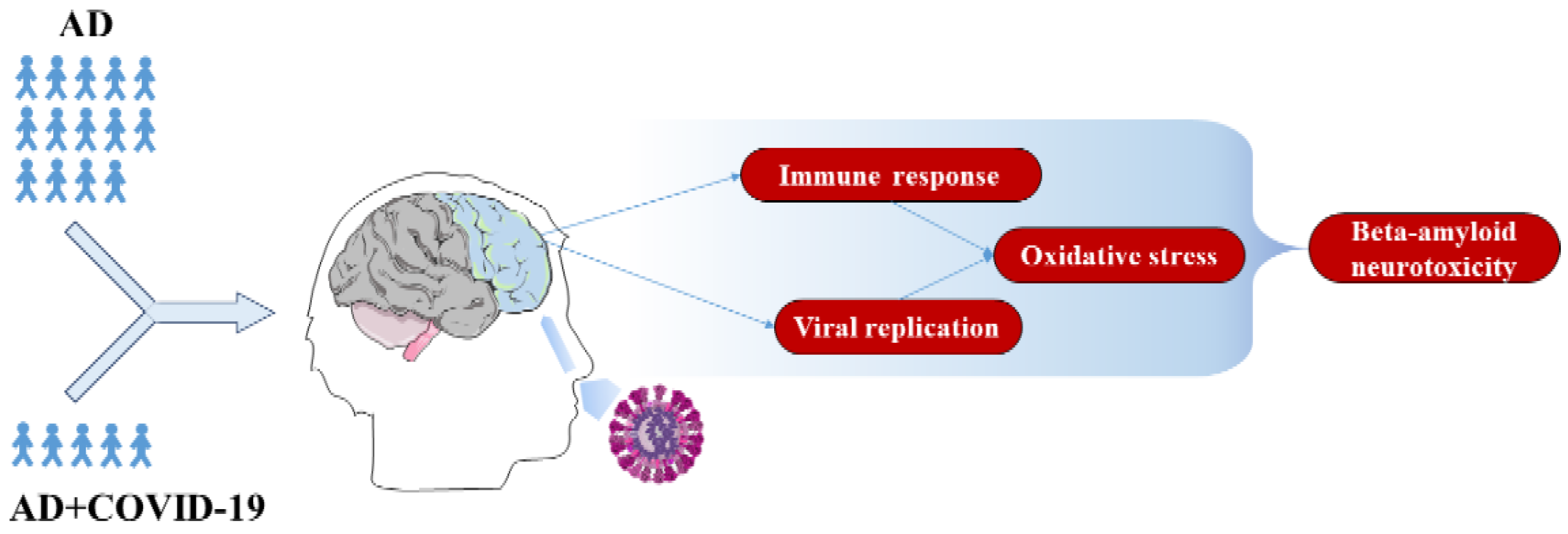
:1. Introduction
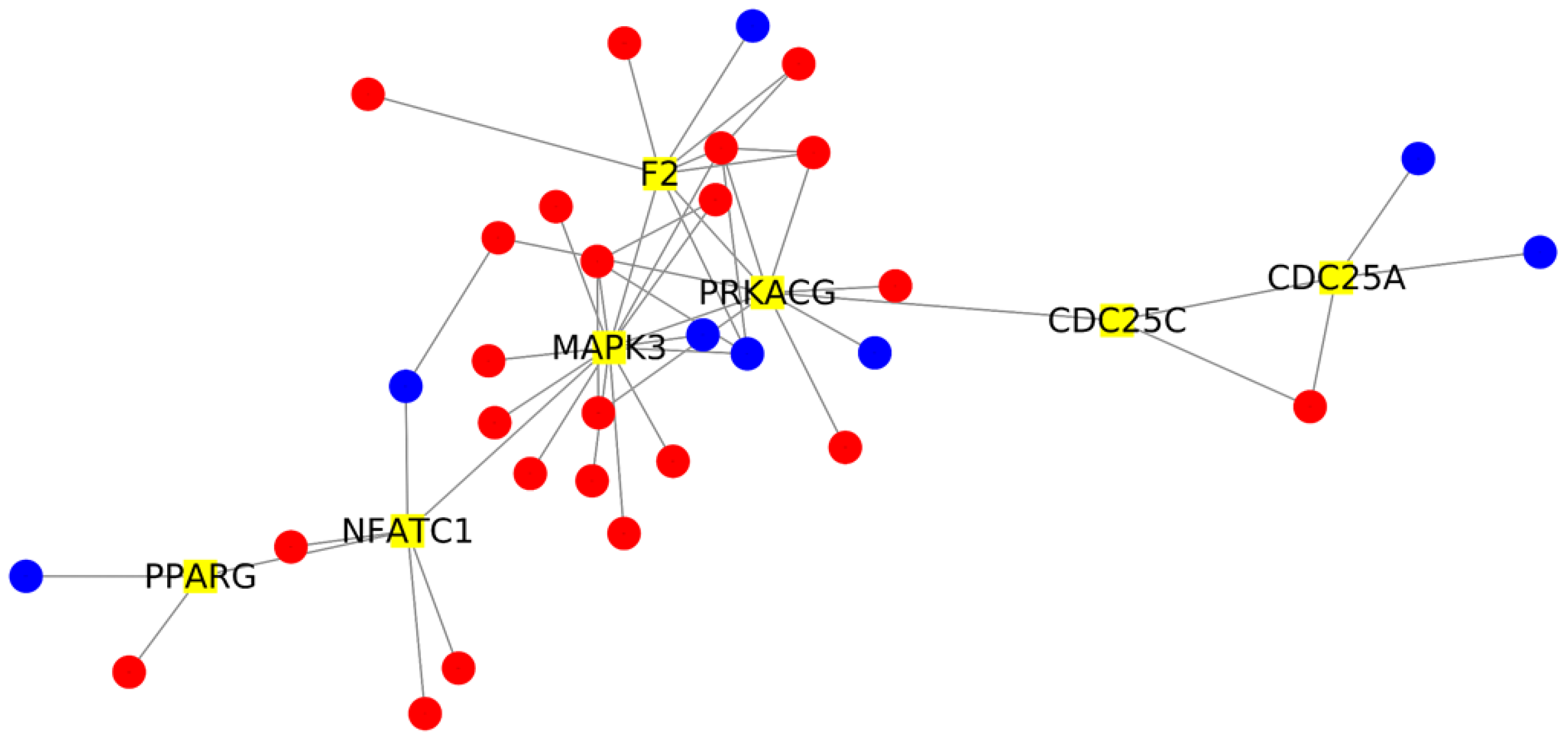
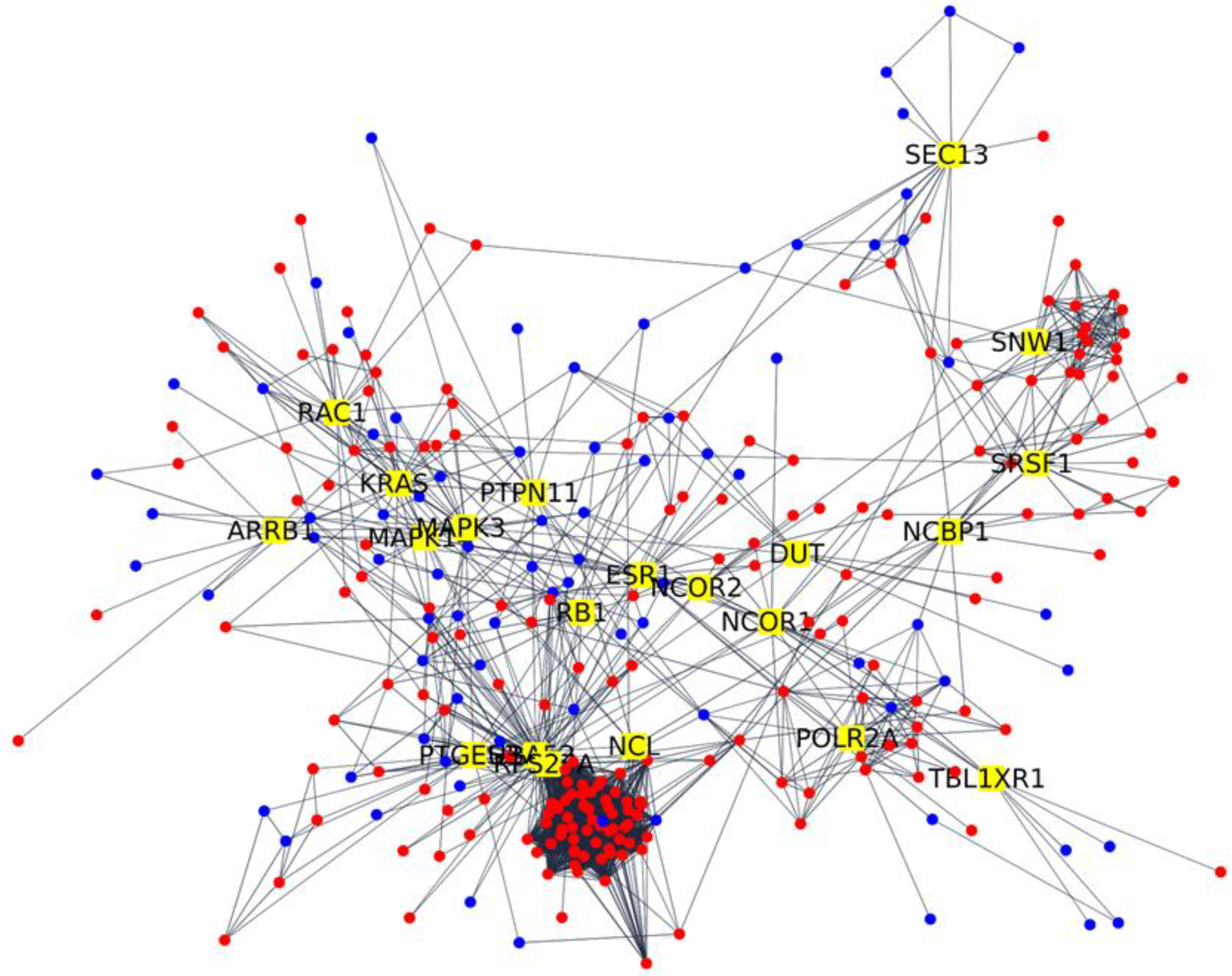
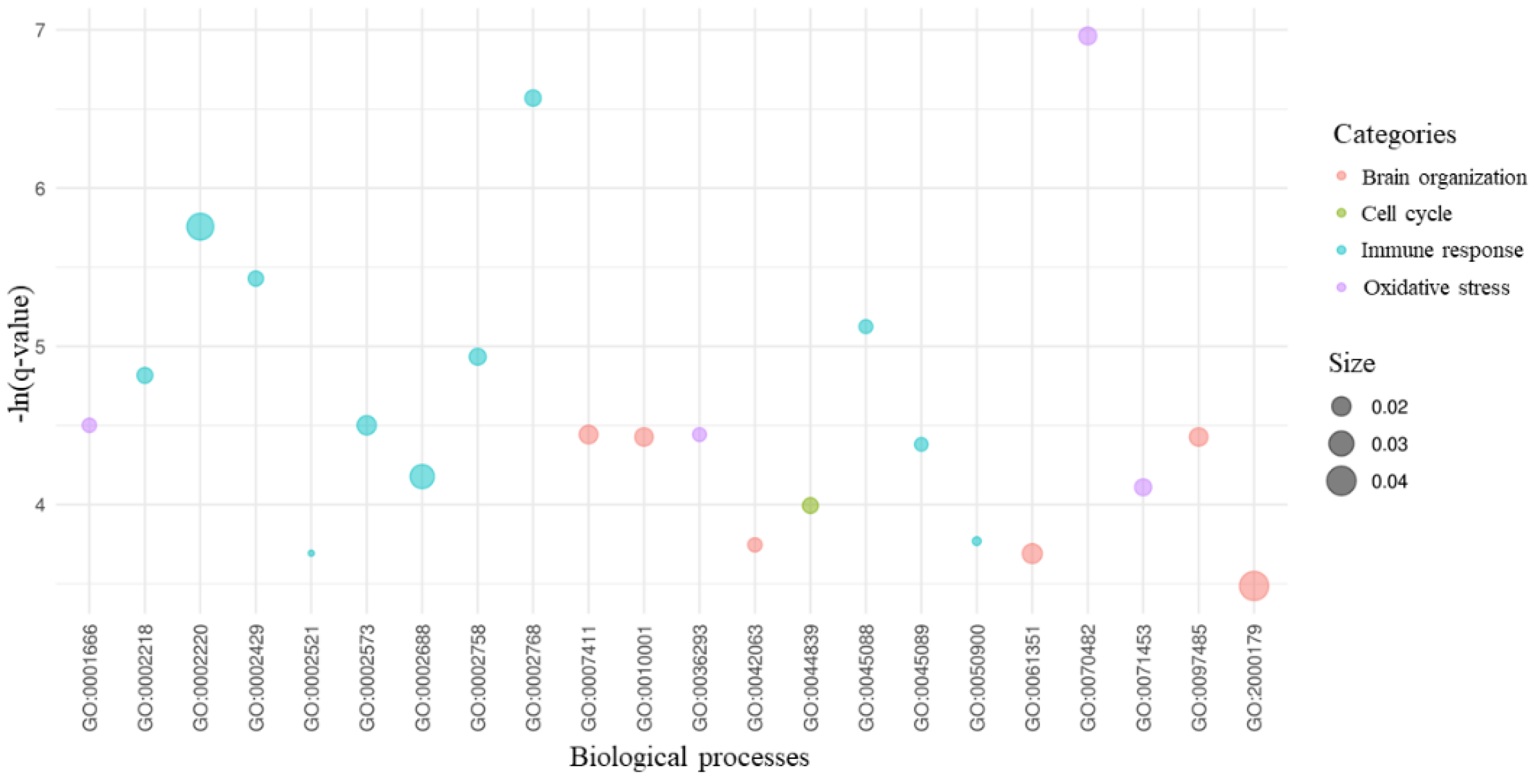
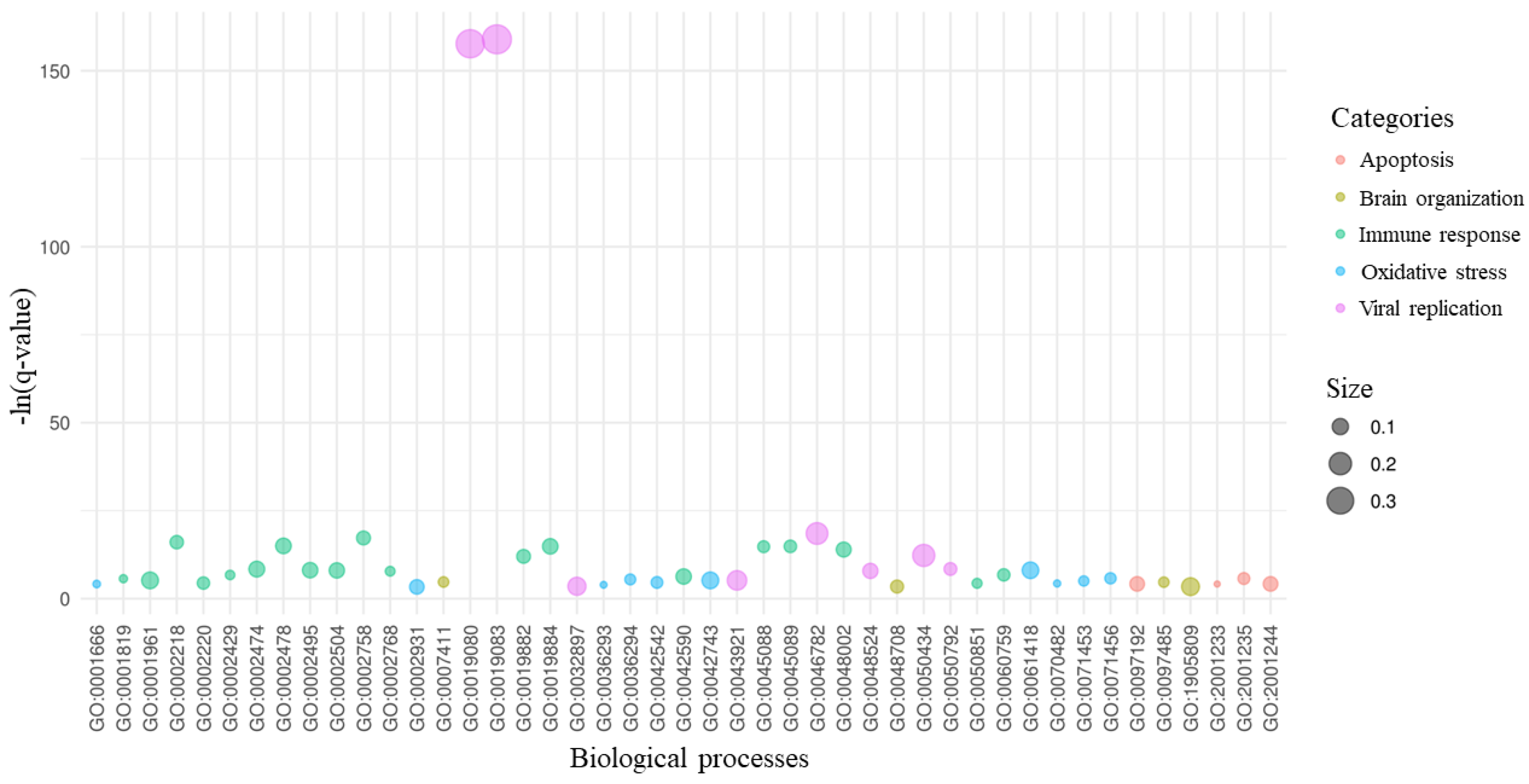
2. Results
3. Discussion
4. Materials and Methods
4.1. Cohort Selection
4.2. Bioinformatics Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Available online: https://covid19.who.int/ (accessed on 26 November 2021).
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 long-term effects of COVID-19: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 16144. [Google Scholar] [CrossRef]
- Thakur, B.; Dubey, P.; Benitez, J.; Torres, J.P.; Reddy, S.; Shokar, N.; Aung, K.; Mukherjee, D.; Dwivedi, A.K. A systematic review and meta-analysis of geographic differences in comorbidities and associated severity and mortality among individuals with COVID-19. Sci. Rep. 2021, 11, 8562. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 26 November 2021).
- Jeremic, D.; Jimenez-Diaz, L.; Navarro-Lopez, J.D. Past, present and future of therapeutic strategies against amyloid-beta peptides in Alzheimer’s disease a systematic review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef] [PubMed]
- Ferini-Strambi, L.; Salsone, M. COVID-19 and neurological disorders: Are neurodegenerative or neuroimmunological diseases more vulnerable? J. Neurol. 2021, 268, 409–419. [Google Scholar] [CrossRef]
- Rahman, M.A.; Islam, K.; Rahman, S.; Alamin, M. Neurobiochemical Cross-talk Between COVID-19 and Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Shults, N.V.; Gychka, S.G.; Harris, B.T.; Suzuki, Y.J. Protein Expression of Angiotensin-Converting Enzyme 2 (ACE2) is Upregulated in Brains with Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1687. [Google Scholar] [CrossRef] [PubMed]
- Politi, L.S.; Salsano, E.; Grimaldi, M. Magnetic Resonance Imaging Alteration of the Brain in a Patient With Coronavirus Disease 2019 (COVID-19) and Anosmia. JAMA Neurol. 2020, 77, 1028–1029. [Google Scholar] [CrossRef]
- O’Reilly, R.C. The What and How of prefrontal cortical organization. Trends Neurosci. 2010, 33, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Bianchetti, A.; Rozzini, R.; Guerini, F.; Boffelli, S.; Ranieri, P.; Minelli, G.; Bianchetti, L.; Trabucchi, M. Clinical Presentation of COVID-19 in Dementia Patients. J. Nutr. Health Aging 2020, 24, 560–562. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020, 87, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.K.; Hsieh, S.T.; Chang, Y.C. Neurological manifestations in severe acute respiratory syndrome. Acta Neurol. Taiwanica 2005, 14, 113–119. [Google Scholar]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Heo, J.H.; Kim, H.O.; Song, S.H.; Park, S.S.; Park, T.H.; Ahn, J.Y.; Kim, M.K.; Choi, J.P. Neurological Complications during Treatment of Middle East Respiratory Syndrome. J. Clin. Neurol. 2017, 13, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.; Omrani, A.S.; Baig, K.; Bahloul, A.; Elzein, F.; Matin, M.A.; Selim, M.A.; Al Mutairi, M.; Al Nakhli, D.; Al Aidaroos, A.Y.; et al. Clinical aspects and outcomes of 70 patients with Middle East respiratory syndrome coronavirus infection: A single-center experience in Saudi Arabia. Int. J. Infect. Dis. 2014, 29, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brunink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 24, 168–175. [Google Scholar] [CrossRef]
- Kipshidze, N.; Dangas, G.; White, C.J.; Kipshidze, N.; Siddiqui, F.; Lattimer, C.R.; Carter, C.A.; Fareed, J. Viral Coagulopathy in Patients With COVID-19: Treatment and Care. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620936776. [Google Scholar] [CrossRef]
- Perwitasari, O.; Torrecilhas, A.C.; Yan, X.; Johnson, S.; White, C.; Tompkins, S.M.; Tripp, R.A. Targeting cell division cycle 25 homolog B to regulate influenza virus replication. J. Virol. 2013, 87, 13775–13784. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Li, Z.; Guan, J.; Hu, S.; Zhang, J.; Lan, Y.; Zhao, K.; Lu, H.; Song, D.; He, H.; et al. Porcine Hemagglutinating Encephalomyelitis Virus Activation of the Integrin alpha5beta1-FAK-Cofilin Pathway Causes Cytoskeletal Rearrangement To Promote Its Invasion of N2a Cells. J. Virol. 2019, 93, 93. [Google Scholar] [CrossRef] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Usera, F.; Enjuanes, L. Coronavirus virulence genes with main focus on SARS-CoV envelope gene. Virus Res. 2014, 194, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Tian, Y.; Nguyen, V.; Zhang, Y.; Gao, C.; Yin, R.; Carver, W.; Fan, D.; Albrecht, H.; Cui, T.; et al. Spike protein of SARS-CoV-2 activates macrophages and contributes to induction of acute lung inflammation in male mice. FASEB J. 2021, 35, e21801. [Google Scholar] [CrossRef]
- Aslam, M.; Ladilov, Y. Targeting the sAC-Dependent cAMP Pool to Prevent SARS-Cov-2 Infection. Cells 2020, 9, 1962. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Eychene, A. The Raf/MEK/ERK pathway: New concepts of activation. Biol. Cell 2001, 93, 53–62. [Google Scholar] [CrossRef]
- Ghasemnejad-Berenji, M.; Pashapour, S. SARS-CoV-2 and the Possible Role of Raf/MEK/ERK Pathway in Viral Survival: Is This a Potential Therapeutic Strategy for COVID-19? Pharmacology 2021, 106, 119–122. [Google Scholar] [CrossRef]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Tanaka, K. Regulatory mechanisms involved in the control of ubiquitin homeostasis. J. Biochem. 2010, 147, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Limanaqi, F.; Biagioni, F.; Gaglione, A.; Busceti, C.L.; Fornai, F. A Sentinel in the Crosstalk Between the Nervous and Immune System: The (Immuno)-Proteasome. Front. Immunol. 2019, 10, 628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.J.; Xu, J.; Lahousse, S.A.; Caggiano, N.L.; de la Monte, S.M. Transient hypoxia causes Alzheimer-type molecular and biochemical abnormalities in cortical neurons: Potential strategies for neuroprotection. JAD 2003, 5, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Perissi, V.; Aggarwal, A.; Glass, C.K.; Rose, D.W.; Rosenfeld, M.G. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell 2004, 116, 511–526. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.; Lozach, J.; Jepsen, K.; Sawka-Verhelle, D.; Perissi, V.; Sasik, R.; Rose, D.W.; Johnson, R.S.; Rosenfeld, M.G.; Glass, C.K. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc. Natl. Acad. Sci. USA 2004, 101, 14461–14466. [Google Scholar] [CrossRef] [Green Version]
- Moreira, T.G.; Zhang, L.; Shaulov, L.; Harel, A.; Kuss, S.K.; Williams, J.; Shelton, J.; Somatilaka, B.; Seemann, J.; Yang, J.; et al. Sec13 Regulates Expression of Specific Immune Factors Involved in Inflammation In Vivo. Sci. Rep. 2015, 5, 17655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, A.L. The coat protein complex II, COPII, protein Sec13 directly interacts with presenilin-1. Biochem. Biophys. Res. Commun. 2009, 388, 571–575. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, X.; Zeng, X.; Bossers, K.; Swaab, D.F.; Zhao, J.; Pei, G. beta-arrestin1 regulates gamma-secretase complex assembly and modulates amyloid-beta pathology. Cell Res. 2013, 23, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Pike, C.J.; Carroll, J.C.; Rosario, E.R.; Barron, A.M. Protective actions of sex steroid hormones in Alzheimer’s disease. Front. Neuroendocrinol. 2009, 30, 239–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, J.F.; Bienias, J.L.; Shah, A.; Meeke, K.A.; Schneider, J.A.; Soriano, E.; Bennett, D.A. Levels of estrogen receptors alpha and beta in frontal cortex of patients with Alzheimer’s disease: Relationship to Mini-Mental State Examination scores. Curr. Alzheimer Res. 2008, 5, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.P.; Wang, P. Integrated communications between cyclooxygenase-2 and Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 13–33. [Google Scholar] [CrossRef]
- Lubkowska, A.; Pluta, W.; Stronska, A.; Lalko, A. Role of Heat Shock Proteins (HSP70 and HSP90) in Viral Infection. Int. J. Mol. Sci. 2021, 22, 9366. [Google Scholar] [CrossRef]
- Bello-Perez, M.; Sola, I.; Novoa, B.; Klionsky, D.J.; Falco, A. Canonical and Noncanonical Autophagy as Potential Targets for COVID-19. Cells 2020, 9, 1619. [Google Scholar] [CrossRef]
- Schoneborn, H.; Raudzus, F.; Coppey, M.; Neumann, S.; Heumann, R. Perspectives of RAS and RHEB GTPase Signaling Pathways in Regenerating Brain Neurons. Int. J. Mol. Sci. 2018, 19, 4052. [Google Scholar] [CrossRef] [Green Version]
- Borin, M.; Saraceno, C.; Catania, M.; Lorenzetto, E.; Pontelli, V.; Paterlini, A.; Fostinelli, S.; Avesani, A.; Di Fede, G.; Zanusso, G.; et al. Rac1 activation links tau hyperphosphorylation and Abeta dysmetabolism in Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Liu, G.; Leugers, C.J.; Mueller, J.D.; Francis, M.B.; Hefti, M.M.; Schneider, J.A.; Lee, G. Tau interacts with SHP2 in neuronal systems and in Alzheimer’s disease brains. J. Cell Sci. 2019, 132, 132. [Google Scholar] [CrossRef]
- MacPherson, D.; Sage, J.; Crowley, D.; Trumpp, A.; Bronson, R.T.; Jacks, T. Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol. Cell. Biol. 2003, 23, 1044–1053. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.; Oh, H.; Nam, D.; Seol, W.; Seo, M.K.; Park, S.W.; Kim, H.G.; Seo, H.; Son, I.; Ho, D.H. Increase in anti-apoptotic molecules, nucleolin, and heat shock protein 70, against upregulated LRRK2 kinase activity. Anim. Cells Syst. 2018, 22, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Husseman, J.W.; Hallows, J.L.; Bregman, D.B.; Leverenz, J.B.; Nochlin, D.; Jin, L.W.; Vincent, I. Hyperphosphorylation of RNA polymerase II and reduced neuronal RNA levels precede neurofibrillary tangles in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 1219–1232. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, A.; Habjan, M.; Benda, C.; Meiler, A.; Haas, D.A.; Hein, M.Y.; Mann, A.; Mann, M.; Habermann, B.; Pichlmair, A. mRNA export through an additional cap-binding complex consisting of NCBP1 and NCBP3. Nat. Commun. 2015, 6, 8192. [Google Scholar] [CrossRef] [Green Version]
- Jakubauskiene, E.; Vilys, L.; Peciuliene, I.; Kanopka, A. The role of hypoxia on Alzheimer’s disease-related APP and Tau mRNA formation. Gene 2021, 766, 145146. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M. Viral subversion of apoptotic enzymes: Escape from death row. Annu. Rev. Microbiol. 2008, 62, 171–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alwazeer, D.; Liu, F.F.; Wu, X.Y.; LeBaron, T.W. Combating Oxidative Stress and Inflammation in COVID-19 by Molecular Hydrogen Therapy: Mechanisms and Perspectives. Oxidative Med. Cell. Longev. 2021, 2021, 5513868. [Google Scholar] [CrossRef]
- Gagliardi, S.; Poloni, E.T.; Pandini, C.; Garofalo, M.; Dragoni, F.; Medici, V.; Davin, A.; Visona, S.D.; Moretti, M.; Sproviero, D.; et al. Detection of SARS-CoV-2 genome and whole transcriptome sequencing in frontal cortex of COVID-19 patients. Brain Behav. Immun. 2021, 97, 13–21. [Google Scholar] [CrossRef]
- Scheckel, C.; Drapeau, E.; Frias, M.A.; Park, C.Y.; Fak, J.; Zucker-Scharff, I.; Kou, Y.; Haroutunian, V.; Ma’ayan, A.; Buxbaum, J.D.; et al. Regulatory consequences of neuronal ELAV-like protein binding to coding and non-coding RNAs in human brain. eLife 2016, 5, e10421. [Google Scholar] [CrossRef]
- Fastq-dump. Available online: https://github.Com/Ncbi/Sra-Tools (accessed on 4 October 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- STRING. Available online: https://string-db.org/ (accessed on 18 October 2021).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Control | COVID-19 | Fold Change | Betweenness Centrality | Degree |
|---|---|---|---|---|---|
| MAPK3 | 365.56 | 695.22 | 0.93 | 0.51 | 15 |
| PRKACG | 0.06 | 20.49 | 6.69 | 0.44 | 10 |
| CDC25C | 2.79 | 43.05 | 3.80 | 0.34 | 3 |
| NFATC1 | 33.21 | 166.23 | 2.32 | 0.29 | 6 |
| CDC25A | 7.03 | 61.62 | 3.16 | 0.25 | 4 |
| F2 | 1.07 | 33.31 | 5.13 | 0.19 | 9 |
| PPARG | 42.25 | 128.56 | 1.60 | 0.19 | 3 |
| Gene | AD | AD+COVID-19 | Fold Change | Betweenness Centrality | Degree |
|---|---|---|---|---|---|
| RPS27A | 911.74 | 1780.96 | 0.97 | 0.20 | 103 |
| UBA52 | 677.56 | 1204.86 | 0.82 | 0.15 | 95 |
| NCOR1 | 2542.38 | 3646.46 | 0.52 | 0.13 | 18 |
| MAPK3 | 444.68 | 763.02 | 0.78 | 0.13 | 26 |
| ESR1 | 74.27 | 19.81 | −1.84 | 0.10 | 23 |
| KRAS | 673.02 | 411.00 | −0.72 | 0.09 | 26 |
| PTGES3 | 1154.09 | 676.38 | −0.75 | 0.08 | 16 |
| NCOR2 | 1127.04 | 2296.71 | 1.03 | 0.08 | 14 |
| RB1 | 600.81 | 340.79 | −0.81 | 0.07 | 16 |
| SNW1 | 269.40 | 404.53 | 0.56 | 0.06 | 17 |
| SEC13 | 228.79 | 131.39 | −0.69 | 0.06 | 16 |
| MAPK14 | 485.07 | 267.52 | −0.84 | 0.06 | 18 |
| TBL1XR1 | 2220.09 | 1191.37 | −0.90 | 0.06 | 8 |
| RAC1 | 948.07 | 1678.89 | 0.83 | 0.05 | 22 |
| NCL | 1784.81 | 6608.74 | 1.89 | 0.05 | 17 |
| SRSF1 | 506.54 | 284.79 | −0.78 | 0.05 | 19 |
| PTPN11 | 2778.66 | 5429.21 | 0.97 | 0.04 | 16 |
| DUT | 237.24 | 152.47 | −0.67 | 0.04 | 9 |
| NCBP1 | 600.36 | 409.11 | −0.58 | 0.04 | 14 |
| POLR2A | 1164.20 | 1595.16 | 0.45 | 0.04 | 22 |
| ARRB1 | 1183.63 | 1826.55 | 0.62 | 0.04 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiricosta, L.; Gugliandolo, A.; Mazzon, E. SARS-CoV-2 Exacerbates Beta-Amyloid Neurotoxicity, Inflammation and Oxidative Stress in Alzheimer’s Disease Patients. Int. J. Mol. Sci. 2021, 22, 13603. https://doi.org/10.3390/ijms222413603
Chiricosta L, Gugliandolo A, Mazzon E. SARS-CoV-2 Exacerbates Beta-Amyloid Neurotoxicity, Inflammation and Oxidative Stress in Alzheimer’s Disease Patients. International Journal of Molecular Sciences. 2021; 22(24):13603. https://doi.org/10.3390/ijms222413603
Chicago/Turabian StyleChiricosta, Luigi, Agnese Gugliandolo, and Emanuela Mazzon. 2021. "SARS-CoV-2 Exacerbates Beta-Amyloid Neurotoxicity, Inflammation and Oxidative Stress in Alzheimer’s Disease Patients" International Journal of Molecular Sciences 22, no. 24: 13603. https://doi.org/10.3390/ijms222413603
APA StyleChiricosta, L., Gugliandolo, A., & Mazzon, E. (2021). SARS-CoV-2 Exacerbates Beta-Amyloid Neurotoxicity, Inflammation and Oxidative Stress in Alzheimer’s Disease Patients. International Journal of Molecular Sciences, 22(24), 13603. https://doi.org/10.3390/ijms222413603