
Pseudopeptide Amyloid Aggregation Inhibitors: In Silico, Single Molecule and Cell Viability Studies

 ,
,  and
and
Abstract
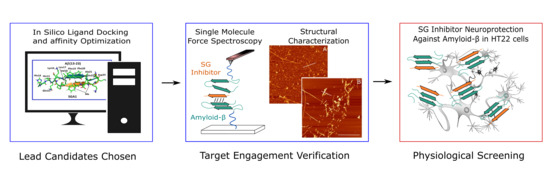
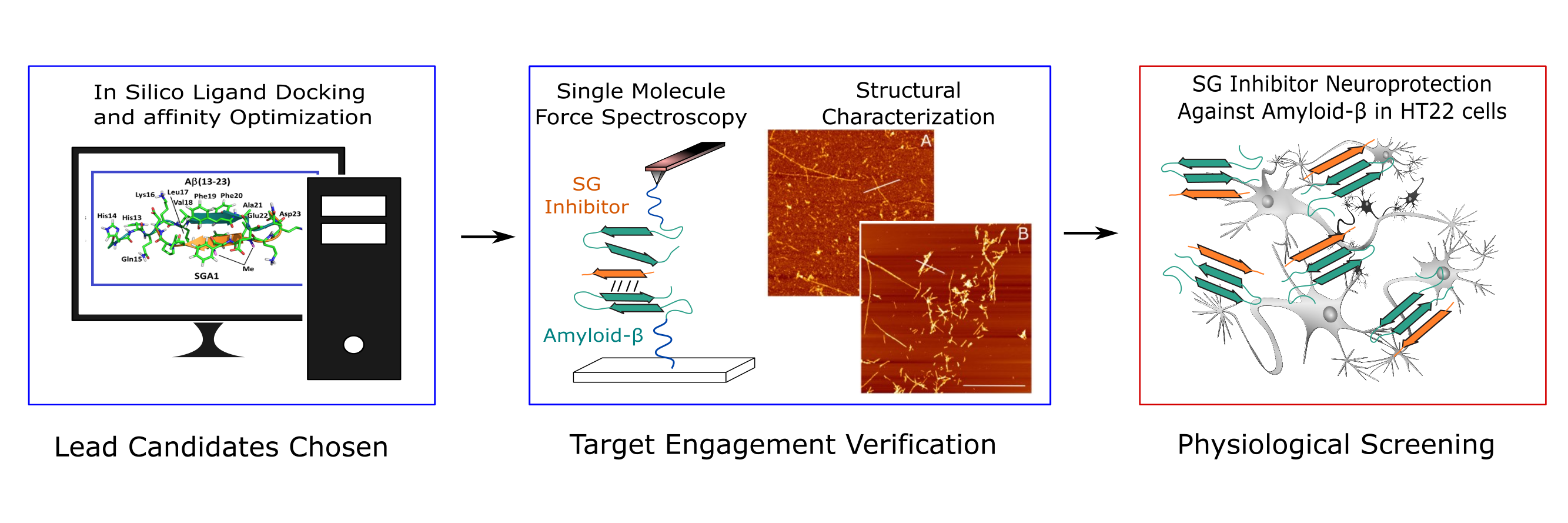
:
1. Introduction
1.1. Amyloid-β Cascade as A Target for Alzheimer’s Disease Treatment
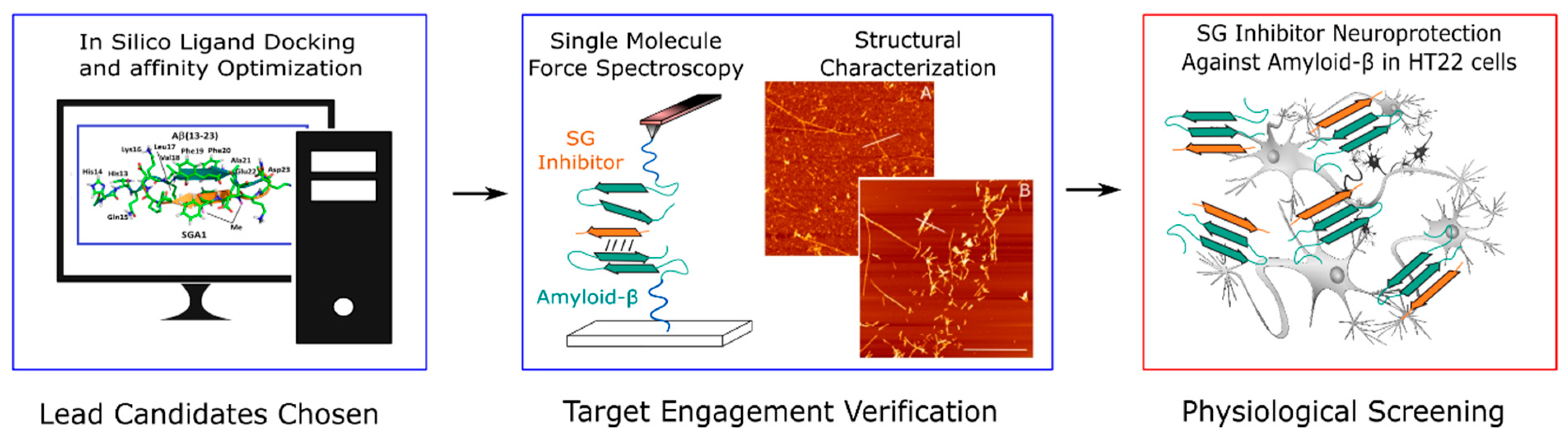
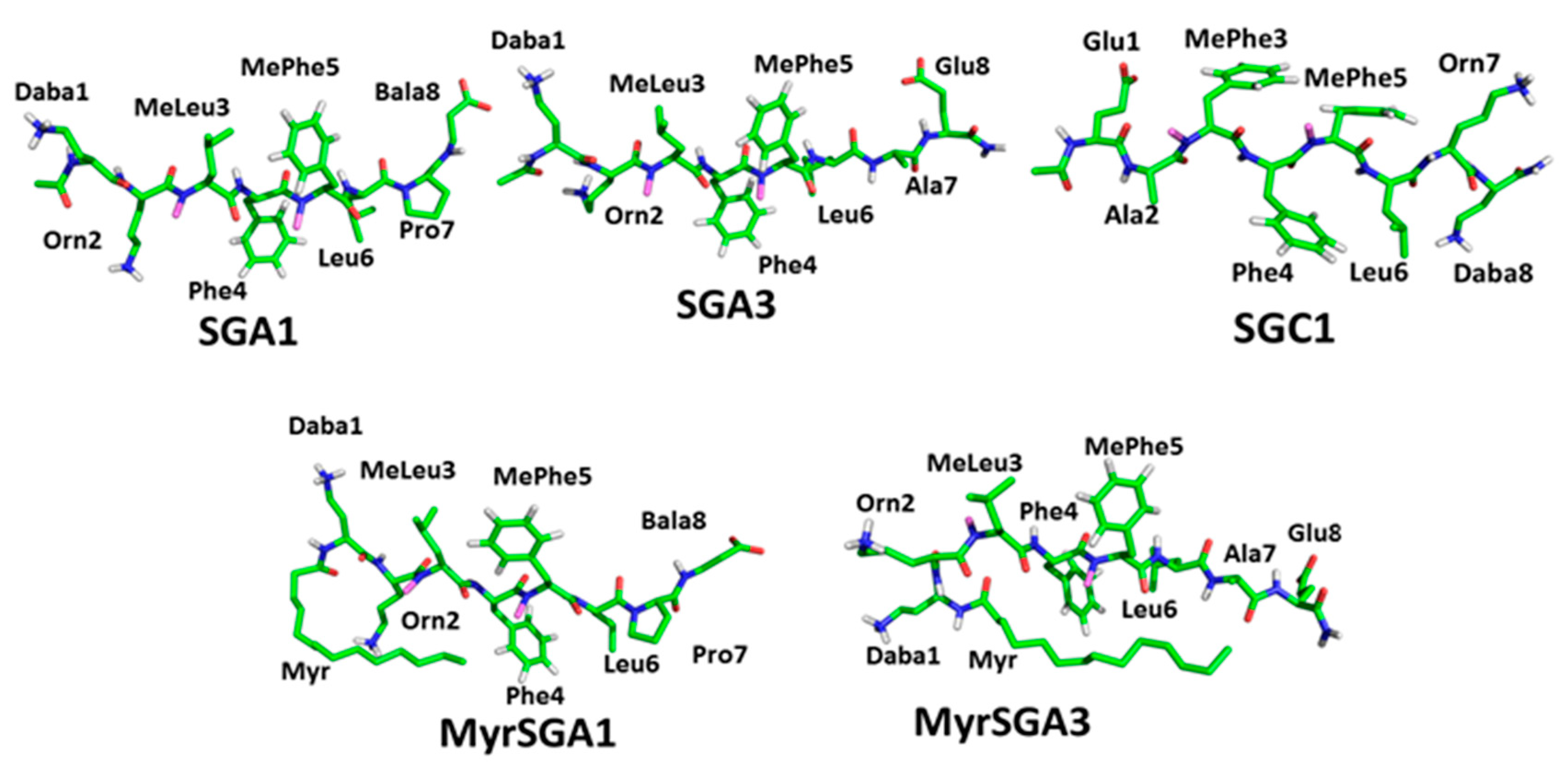
1.2. SG Inhibitor Design Rationale
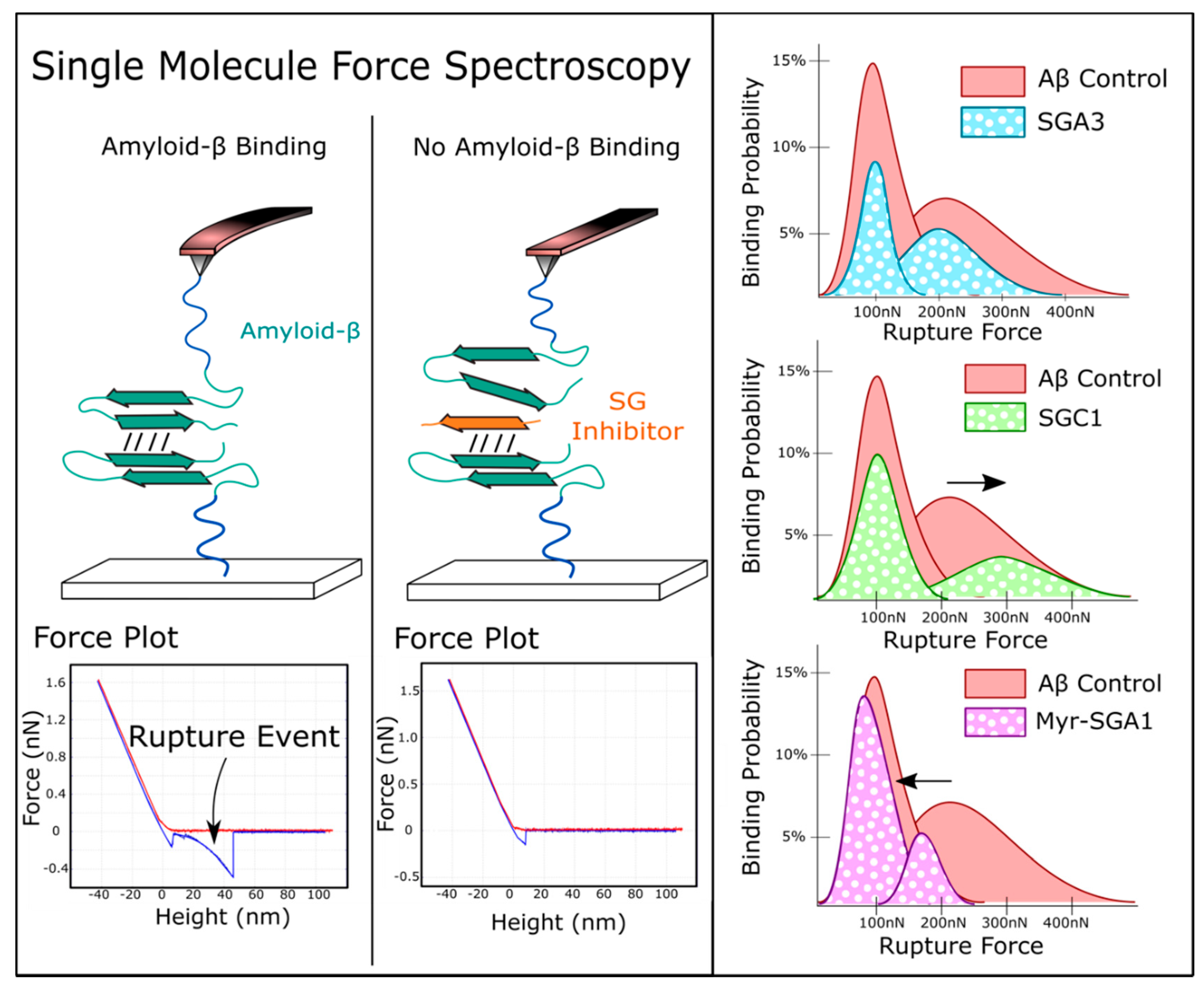
1.3. Target Verification by Single Molecule Force Spectroscopy
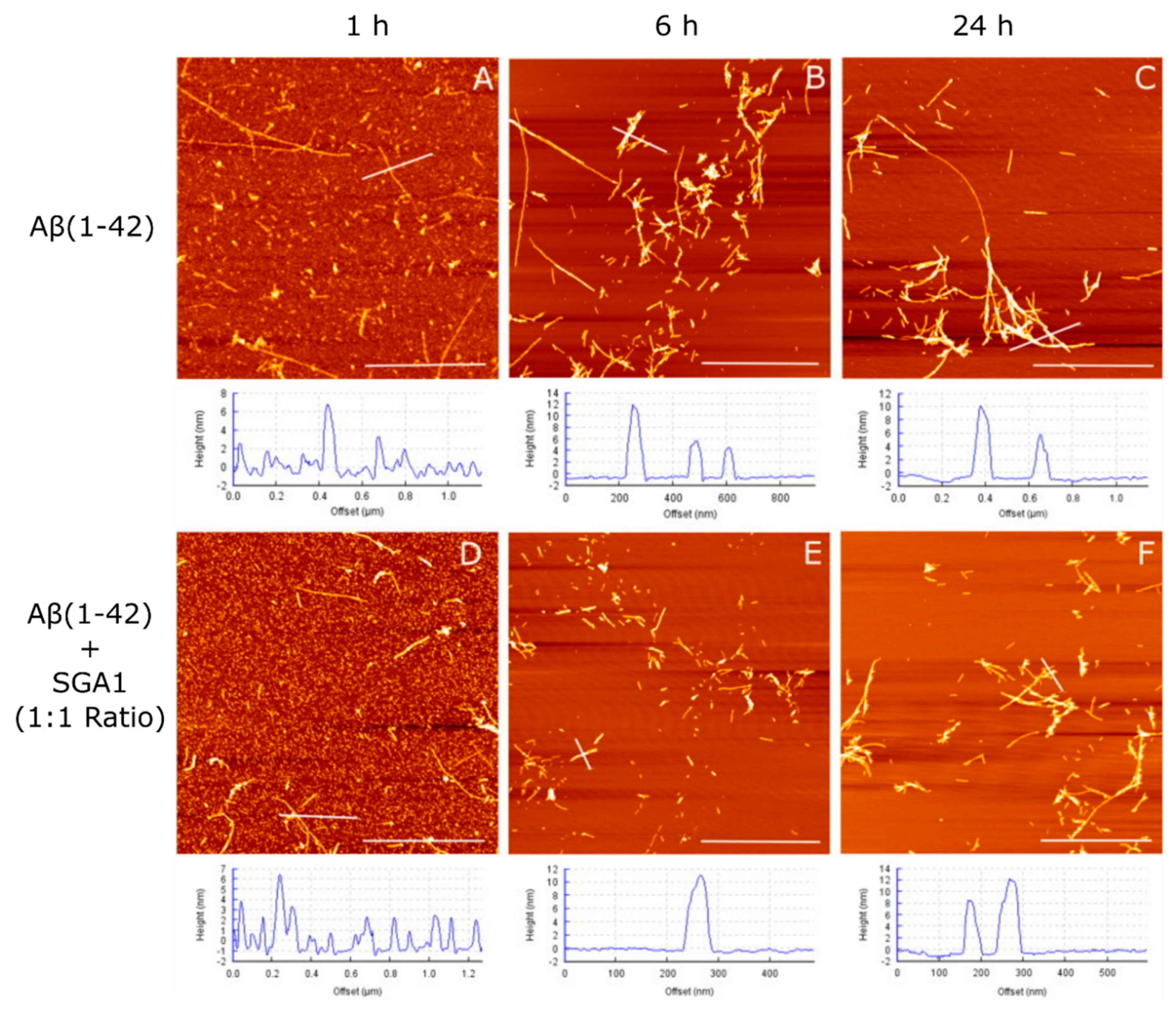
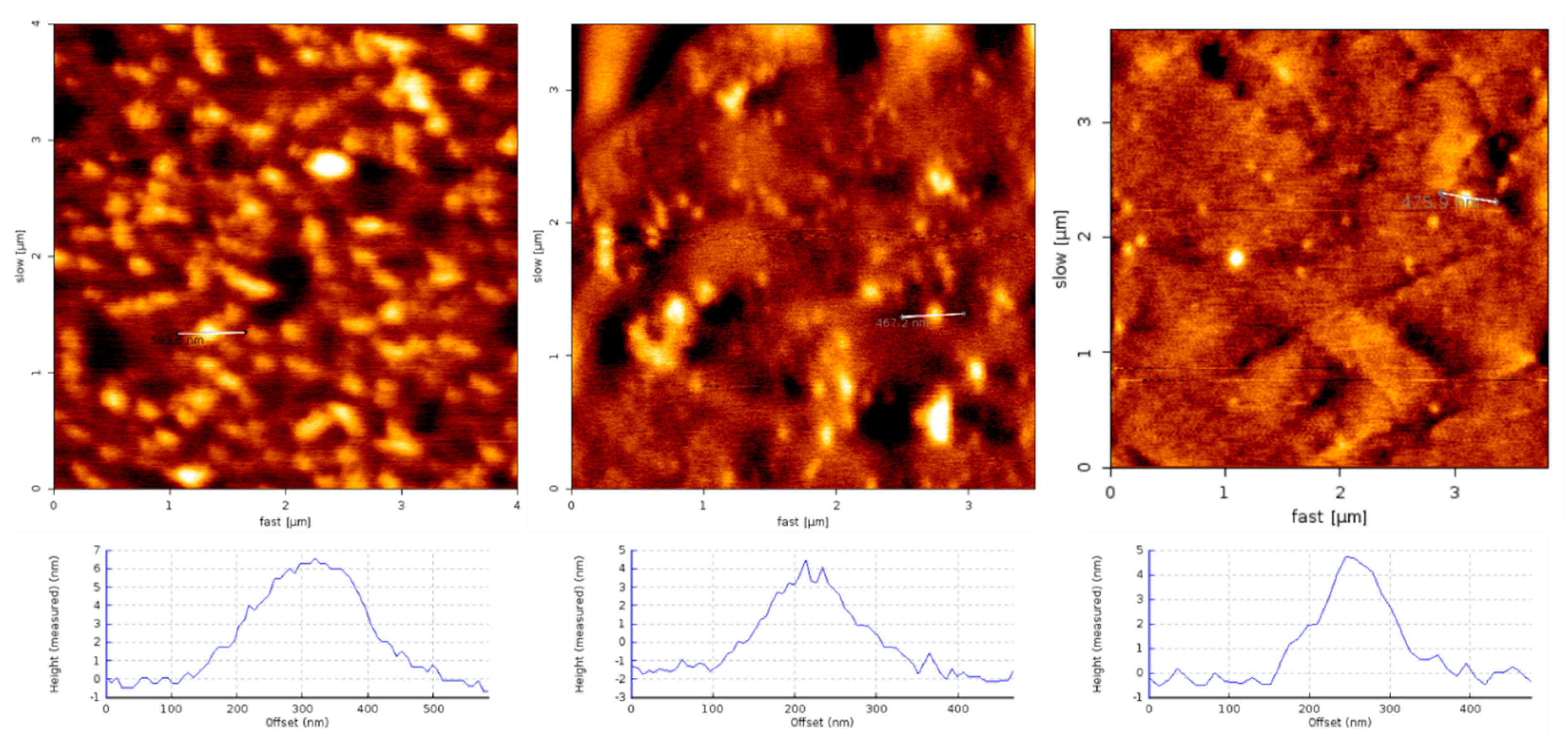
1.4. Structural Characterization of SG-Aβ Aggregation
1.5. Cell Viability Studies.
2. Results
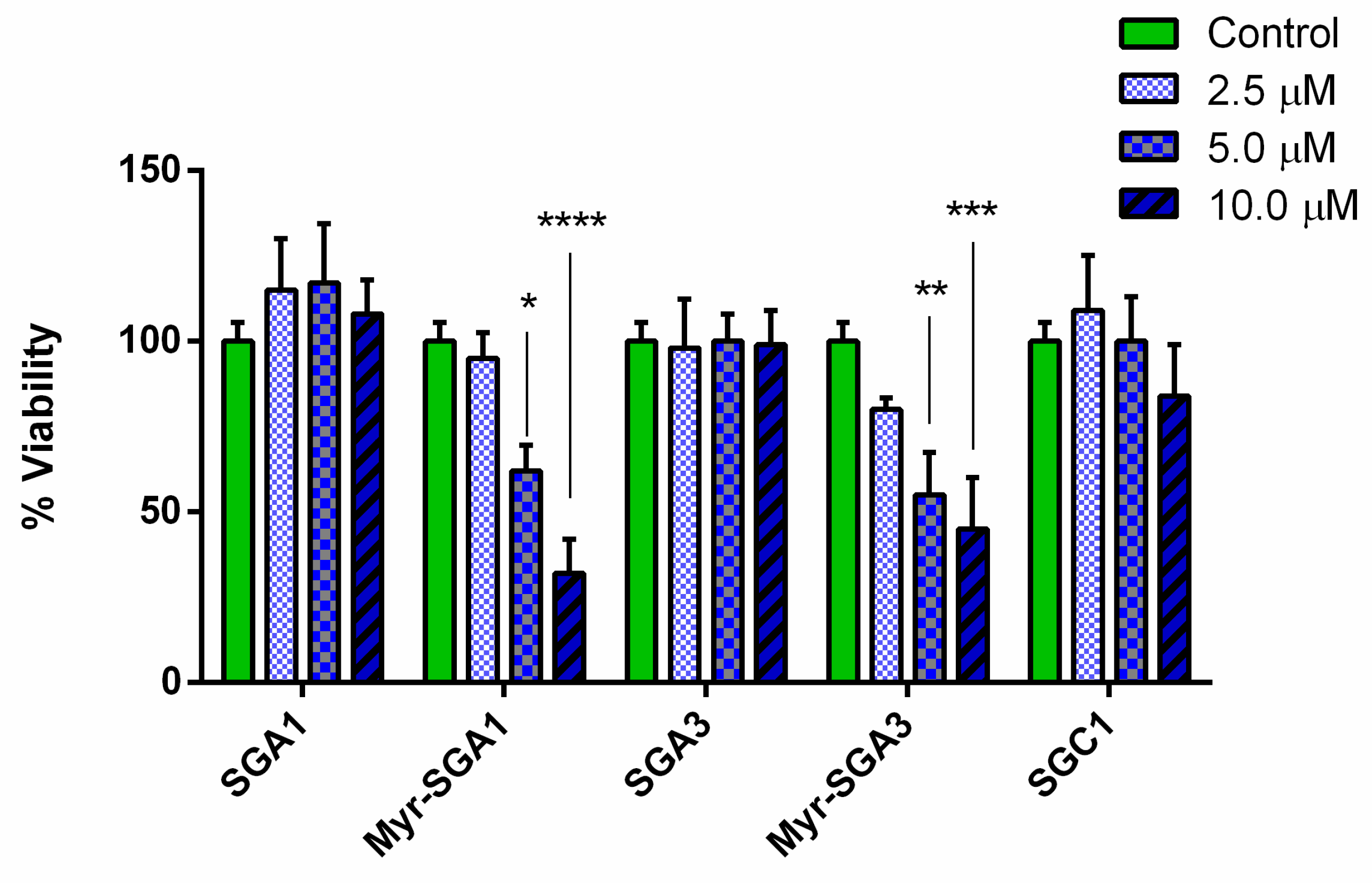
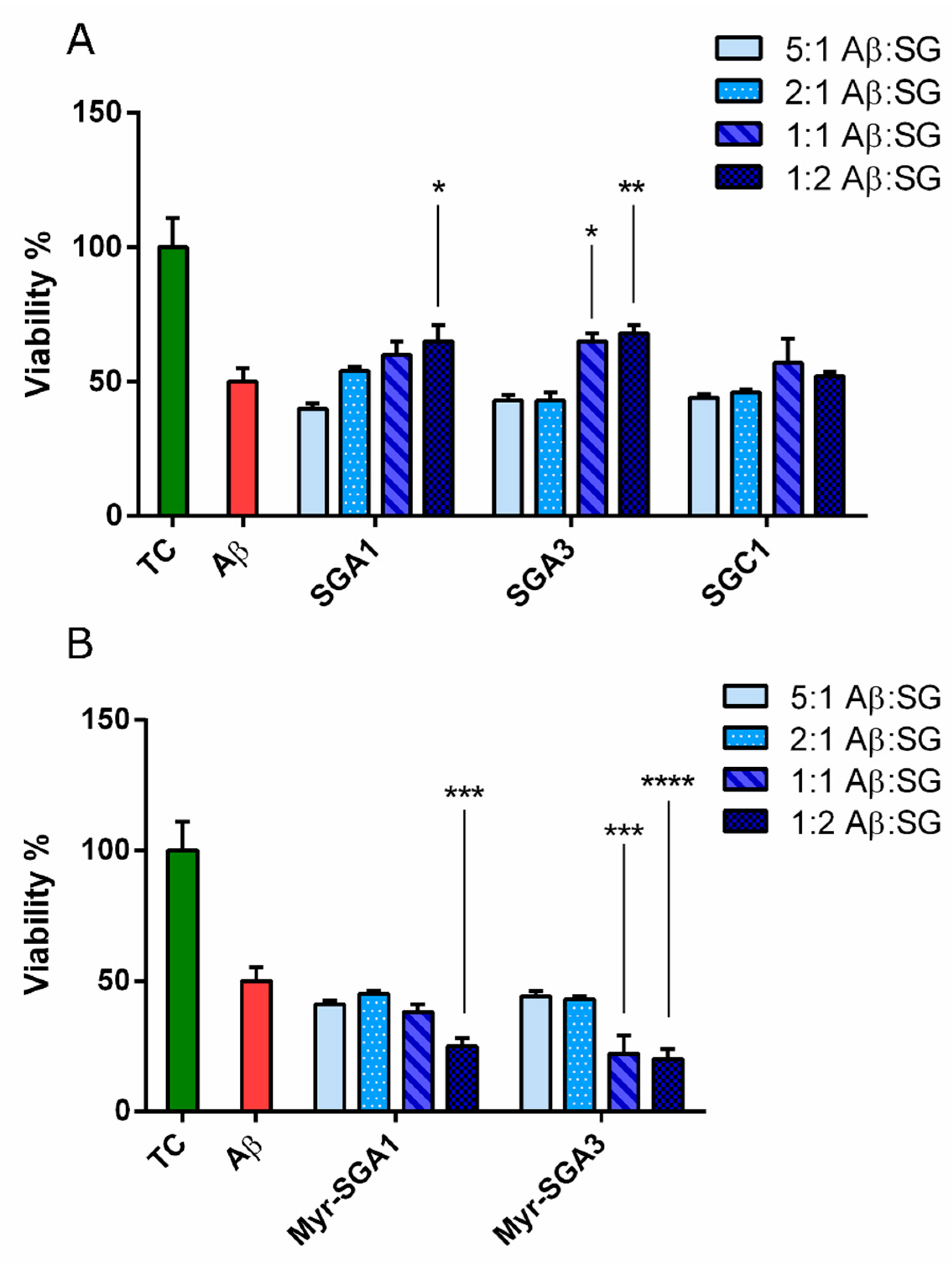
2.1. SG Inhibitor Toxicity
2.2. The Effects of SG Inhibitors on Aβ Oligomer Toxicity
3. Discussion
3.1. In vitro HT22 Cell Models for Testing Anti-Aβ Aggregation Drugs
3.2. Myristic Acid to Improve BBB Delivery of Peptides
3.3. Comparing SG Inhibitors In Silico, Single Molecule Force Spectroscopy and Cell Viability Assays
4. Materials and Methods
4.1. Reagents and SG Inhibitors
4.2. HT22 Cells and MTT Assay
4.3. Aβ and Aβ-SG Inhibitor Treatment Preparation and AFM Characterization
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β |
| AD | Alzheimer’s disease |
| ANOVA | analysis of variance |
| BBB | blood-brain barrier |
| CNS | central nervous system |
| DMSO | dimethylsulfoxide |
| HFIP | hexafluoroisopropanol |
| MD | molecular dynamics |
| MOE | Molecular Environment |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide |
| mAb | monoclonal antibody |
| SMFS | single-molecule force spectroscopy |
References
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar] [CrossRef] [Green Version]
- Cecchi, C.; Stefani, M. The amyloid-cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity. Biophys. Chem. 2013, 182, 30–43. [Google Scholar] [CrossRef]
- Williams, T.L.; Johnson, B.R.G.; Urbanc, B.; Jenkins, A.T.A.; Connell, S.D.A.; Serpell, L.C. Aβ42 oligomers, but not fibrils, simultaneously bind to and cause damage to ganglioside-containing lipid membranes. Biochem. J. 2011, 439, 67–77. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Laske, C. Phase 3 Trials of Solanezumab and Bapineuzumab for Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 1459–1460. [Google Scholar] [CrossRef]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef]
- Panza, F.; Solfrizzi, V.; Imbimbo, B.P.; Giannini, M.; Santamato, A.; Seripa, D.; Logroscino, G. Efficacy and safety studies of gantenerumab in patients with Alzheimer’s disease. Expert Rev. Neurother. 2014, 14, 973–986. [Google Scholar] [CrossRef]
- Siemers, E.R.; Sundell, K.L.; Carlson, C.; Case, M.; Sethuraman, G.; Liu-Seifert, H.; Dowsett, S.A.; Pontecorvo, M.J.; Dean, R.A.; DeMattos, R. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimer’s Dement. 2016, 12, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu-Seifert, H.; Siemers, E.; Holdridge, K.C.; Andersen, S.W.; Lipkovich, I.; Carlson, C.; Sethuraman, G.; Hoog, S.; Hayduk, R.; Doody, R.; et al. Delayed-start analysis: Mild Alzheimer’s disease patients in solanezumab trials, 3.5 years. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2015, 1, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Ghanta, J.; Shen, C.L.; Kiessling, L.L.; Murphy, R.M. A strategy for designing inhibitors of β-amyloid toxicity. J. Biol. Chem. 1996, 271, 29525–29528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjernberg, L.O.; Näslundt, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Tereniust, L.; Nordstedt, C. Arrest of β-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [Green Version]
- Tjernberg, L.O.; Lilliehöök, C.; Callaway, D.J.E.; Näslund, J.; Hahne, S.; Thyberg, J.; Terenius, L.; Nordstedt, C. Controlling amyloid β-peptide fibril formation with protease-stable ligand. J. Biol. Chem. 1997, 272, 12601–12605. [Google Scholar] [CrossRef] [Green Version]
- Austen, B.M.; Paleologou, K.E.; Ali, S.A.E.; Qureshi, M.M.; Allsop, D.; El-Agnaf, O.M.A. Designing peptide inhibitors for oligomerization and toxicity of Alzheimer’s β-amyloid peptide. Biochemistry 2008, 47, 1984–1992. [Google Scholar] [CrossRef]
- Lowe, T.L.; Strzelec, A.; Kiessling, L.L.; Murphy, R.M. Structure—Function relationships for inhibitors ofβ-Amyloid toxicity containing the recognition sequence KLVFF. Biochemistry 2001, 40, 7882–7889. [Google Scholar] [CrossRef]
- Grillo-Bosch, D.; Carulla, N.; Cruz, M.; Sánchez, L.; Pujol-Pina, R.; Madurga, S.; Rabanal, F.; Giralt, E. Retro-enantio N-methylated peptides as β-amyloid aggregation inhibitors. ChemMedChem 2009, 4, 1488–1494. [Google Scholar] [CrossRef]
- Soto, C.; Kindy, M.S.; Baumann, M.; Frangione, B. Inhibition of Alzheimer’s Amyloidosis by Peptides That Prevent β-Sheet Conformation. Biochem. Biophys. Res. Commun. 1996, 226, 672–680. [Google Scholar] [CrossRef]
- Hane, F.T.; Lee, B.Y.; Petoyan, A.; Rauk, A.; Leonenko, Z. Testing synthetic amyloid-β aggregation inhibitor using single molecule atomic force spectroscopy. Biosens. Bioelectron. 2014, 54, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Sigurdsson, E.M. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat. Med. 1998, 4, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Chacón, M.A.; Barría, M.I.; Soto, C.; Inestrosa, N.C. β-sheet breaker peptide prevents Aβ-induced spatial memory impairments with partial reduction of amyloid deposits. Mol. Psychiatry 2004, 9, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parthsarathy, V.; McClean, P.L.; Hölscher, C.; Taylor, M.; Tinker, C.; Jones, G.; Kolosov, O.; Salvati, E.; Gregori, M.; Masserini, M.; et al. A Novel Retro-Inverso Peptide Inhibitor Reduces Amyloid Deposition, Oxidation and Inflammation and Stimulates Neurogenesis in the APPswe/PS1ΔE9 Mouse Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e54769. [Google Scholar] [CrossRef]
- Taylor, M.; Moore, S.; Mayes, J.; Parkin, E.; Beeg, M.; Canovi, M.; Gobbi, M.; Mann, D.M.A.; Allsop, D. Development of a proteolytically stable retro-inverso peptide inhibitor of β-amyloid oligomerization as a potential novel treatment for Alzheimers Disease. Biochemistry 2010, 49, 3261–3272. [Google Scholar] [CrossRef]
- Pallitto, M.M.; Ghanta, J.; Heinzelman, P.; Kiessling, L.L.; Murphy, R.M. Recognition sequence design for peptidyl modulators of β-amyloid aggregation and toxicity. Biochemistry 1999, 38, 3570–3578. [Google Scholar] [CrossRef]
- Hane, F.T.; Robinson, M.; Lee, B.Y.; Bai, O.; Leonenko, Z.; Albert, M.S. Recent Progress in Alzheimer’s Disease Research, Part 3: Diagnosis and Treatment. J. Alzheimer’s Dis. 2017, 57, 645–665. [Google Scholar] [CrossRef] [Green Version]
- Opare, S.K.A.; Petoyan, A.; Mehrazma, B.; Rauk, A. Molecular dynamics study of the monomers and dimers of N-AcAβ (13–23) NH2: On the effect of pH on the aggregation of the amyloid beta peptide of Alzheimer’s disease. Can. J. Chem. 2015, 94, 273–281. [Google Scholar] [CrossRef]
- Mehrazma, B.; Opare, S.; Petoyan, A.; Rauk, A. D-amino acid pseudopeptides as potential amyloid-beta aggregation inhibitors. Molecules 2018, 23, 2387. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.; Mehrazma, B.; Opare, S.K.A.; Petoyan, A.; Lou, J.; Hane, F.T.; Rauk, A.; Leonenko, Z. Pseudo-peptide Amyloid-β Blocking Inhibitors: Molecular Dynamics and Single Molecule Force Spectroscopy Study. BBA Proteins Proteom. 2017, 1865, 1707–1718. [Google Scholar]
- Mehrazma, B.; Petoyan, A.; Opare, S.K.A.; Rauk, A. Interaction of the N -AcAβ (13–23) NH 2 segment of the beta amyloid peptide with beta-sheet-blocking peptides: Site and edge specificity. Can. J. Chem. 2016, 10, 1–10. [Google Scholar] [CrossRef]
- Gordon, D.J.; Tappe, R.; Meredith, S.C. Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits Abeta1–40 fibrillogenesis. J. Pept. Res. 2002, 60, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Roy, S. Designing Novel Peptidic Inhibitors of Beta Amyloid Oligomerization. Ph.D. Thesis, University of Calgary, Calgary, AB, Canada, 2010. [Google Scholar]
- Cruz, M.; Tusell, J.M.; Grillo-Bosch, D.; Albericio, F.; Serratosa, J.; Rabanal, F.; Giralt, E. Inhibition of β-amyloid toxicity by short peptides containing N-methyl amino acids. J. Pept. Res. 2004, 63, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Poduslo, J.F.; Curran, G.L.; Kumar, A.; Frangione, B.; Soto, C. β-Sheet breaker peptide inhibitor of Alzheimer’s amyloidogenesis with increased blood-brain barrier permeability and resistance to proteolytic degradation in plasma. J. Neurobiol. 1999, 39, 371–382. [Google Scholar] [CrossRef]
- Kokkoni, N.; Stott, K.; Amijee, H.; Mason, J.M.; Doig, A.J. N-methylated peptide inhibitors of β-amyloid aggregation and toxicity. Optimization of the inhibitor structure. Biochemistry 2006, 45, 9906–9918. [Google Scholar] [CrossRef]
- Robinson, M.; Yasie Lee, B.; Leonenko, Z. Drugs and drug delivery systems targeting amyloid-β in Alzheimer’s disease. AIMS Mol. Sci. 2015, 2, 332–358. [Google Scholar] [CrossRef]
- Buser, C.A.; Sigal, C.T.; Resh, M.D.; McLaughlin, S. Membrane binding of myristylated peptides corresponding to the NH2 terminus of Src. Biochemistry 1994, 33, 13093–13101. [Google Scholar] [CrossRef]
- Nelson, A.R.; Borland, L.; Allbritton, N.L.; Sims, C.E. Myristoyl-based transport of peptides into living cells. Biochemistry 2007, 46, 14771–14781. [Google Scholar] [CrossRef] [Green Version]
- Hane, F.T.; Attwood, S.J.; Leonenko, Z. Comparison of three competing dynamic force spectroscopy models to study binding forces of amyloid-β (1–42). Soft Matter 2014, 10, 1924–1930. [Google Scholar] [CrossRef]
- Hane, F.; Leonenko, Z. Effect of Metals on Kinetic Pathways of Amyloid-β Aggregation. Biomolecules 2014, 4, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Jia, J.; Lei, C.; Ji, L.; Chen, X.; Sang, H.; Xiong, L. Cannabinoid Receptor CB1 Is Involved in Nicotine-Induced Protection Against Aβ 1–42 Neurotoxicity in HT22 Cells. J. Mol. Neurosci. 2014, 55, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Saffi, G.T.; Vasefi, M.S.; Choi, Y.; Kruk, J.S.; Ahmed, N.; Gondora, N.; Mielke, J.; Leonenko, Z.; Beazely, M.A. Amyloid-β Inhibits PDGFβ Receptor Activation and Prevents PDGF-BB-induced Neuroprotection. Curr. Alzheimer Res. 2018, 15, 618–627. [Google Scholar] [CrossRef]
- Stine, W.B.; Jungbauer, L.; Yu, C.; LaDu, M.J. Preparing synthetic Aβ in different aggregation states. Methods Mol. Biol. 2011, 670, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.B.; Maher, P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994, 652, 169–173. [Google Scholar] [CrossRef]
- Liu, J.; Li, L.; Suo, W.Z. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009, 84, 267–271. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Gursoy, E.; Cardounel, A.; Kalimi, M. Pregnenolone Protects Mouse Hippocampal (HT-22) Cells against Glutamate and Amyloid Beta Protein Toxicity. Neurochem. Res. 2001, 26, 15–21. [Google Scholar] [CrossRef]
- Charron, G. Protein Lipidation and Lipid Trafficking. Acc. Chem. Res. 2011, 44, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Aicart-Ramos, C.; Valero, R.A.; Rodriguez-Crespo, I. Protein palmitoylation and subcellular trafficking. Biochim. Biophys. Acta Biomembr. 2011, 1808, 2981–2994. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, F.J.; Parodi, J.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS ONE 2010, 5, e11820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drolle, E.; Negoda, A.; Hammond, K.; Pavlov, E.; Leonenko, Z. Changes in lipid membranes may trigger amyloid toxicity in Alzheimer’s disease. PLoS ONE 2017, 12, e0182194. [Google Scholar] [CrossRef] [PubMed]
- Ulloth, J.E.; Casiano, C.A.; De Leon, M. Palmitic and stearic fatty acids induce caspase-dependent and-independent cell death in nerve growth factor differentiated PC12 cells. J. Neurochem. 2003, 84, 655–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, S.; Chan, C. Palmitic and stearic fatty acids induce Alzheimer-like hyperphosphorylation of tau in primary rat cortical neurons. Neurosci. Lett. 2005, 384, 288–293. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mothana, B.; Roy, S.; Rauk, A. Molecular dynamics study of the interaction of Aβ (13–23) with β-sheet inhibitors. Arkivoc 2009, 116–134. [Google Scholar] [CrossRef] [Green Version]
- Porat, Y.; Mazor, Y.; Efrat, S.; Gazit, E. Inhibition of islet amyloid polypeptide fibril formation: A potential role for heteroaromatic interactions. Biochemistry 2004, 43, 14454–14462. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Frydman-Marom, A.; Rechter, M.; Gazit, E. Inhibition of amyloid fibril formation and cytotoxicity by hydroxyindole derivatives. Biochemistry 2006, 45, 4727–4735. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SG Inhibitor Classes | Anti-Parallel | Parallel |
| L-enantiomer | SGA | SGC |
| D-enantiomer | SGB | SGD |
| SG Inhibitor Binding | ΔGdimer (kJ/mol) | Average ΔGeff (kJ/mol) |
| SGA1 | 21 | 3 |
| SGA3 | 46 | -4 |
| SGC1 | 26 | -24 |
| Myr-SGA1 | 62 | 6 |
| SG Inhibitor | Inhibitor Sequence |
|---|---|
| SGA1 | Daba-Orn-(Me)Leu-Phe-(Me)Phe-Leu-Pro-Bala |
| MyrSGA1 | Myr-Daba-Orn-(Me)Leu-Phe-(Me)Phe-Leu-Pro-Bala |
| SGA3 | Daba-Orn-(Me)Leu-Phe-(Me)Phe-Leu-Ala-Glu |
| MyrSGA3 | Myr-Daba-Orn-(Me)Leu-Phe-(Me)Phe-Leu-Ala-Glu |
| SGC1 | Glu-Ala-(Me)Phe-Phe-(Me)Phe-Leu-Orn-Daba |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, M.; Lou, J.; Mehrazma, B.; Rauk, A.; Beazely, M.; Leonenko, Z. Pseudopeptide Amyloid Aggregation Inhibitors: In Silico, Single Molecule and Cell Viability Studies. Int. J. Mol. Sci. 2021, 22, 1051. https://doi.org/10.3390/ijms22031051
Robinson M, Lou J, Mehrazma B, Rauk A, Beazely M, Leonenko Z. Pseudopeptide Amyloid Aggregation Inhibitors: In Silico, Single Molecule and Cell Viability Studies. International Journal of Molecular Sciences. 2021; 22(3):1051. https://doi.org/10.3390/ijms22031051
Chicago/Turabian StyleRobinson, Morgan, Jennifer Lou, Banafsheh Mehrazma, Arvi Rauk, Michael Beazely, and Zoya Leonenko. 2021. "Pseudopeptide Amyloid Aggregation Inhibitors: In Silico, Single Molecule and Cell Viability Studies" International Journal of Molecular Sciences 22, no. 3: 1051. https://doi.org/10.3390/ijms22031051
APA StyleRobinson, M., Lou, J., Mehrazma, B., Rauk, A., Beazely, M., & Leonenko, Z. (2021). Pseudopeptide Amyloid Aggregation Inhibitors: In Silico, Single Molecule and Cell Viability Studies. International Journal of Molecular Sciences, 22(3), 1051. https://doi.org/10.3390/ijms22031051