
Differential Role of Threonine and Tyrosine Phosphorylation in the Activation and Activity of the Yeast MAPK Slt2



Abstract
:1. Introduction
2. Results
2.1. Detection of Slt2 Monophosphorylation at the Activation Loop with Commercial Antibodies
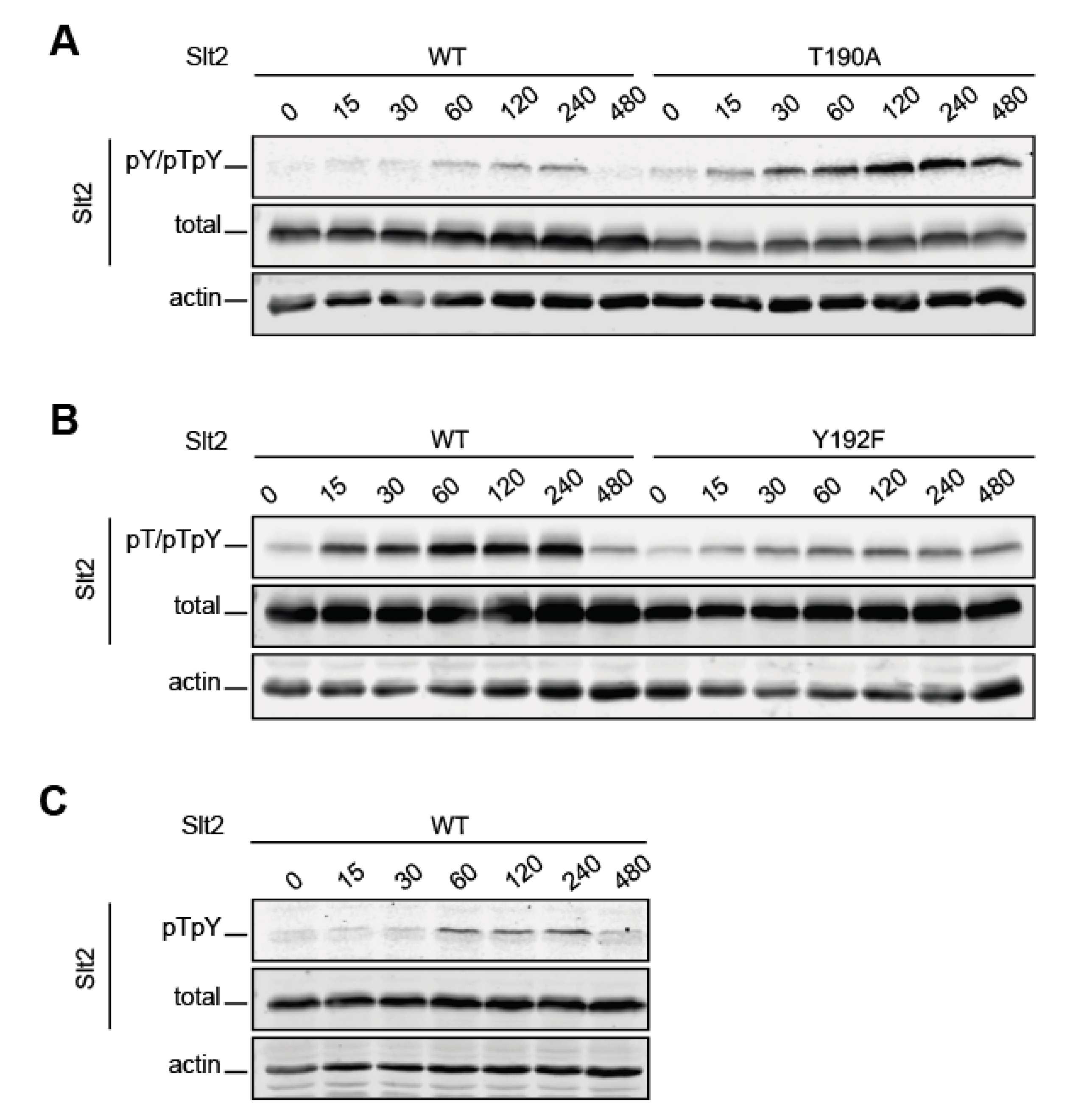
2.2. The Lack of T190 Phosphorylation Results in Increased Phosphorylation of Y192 in Slt2
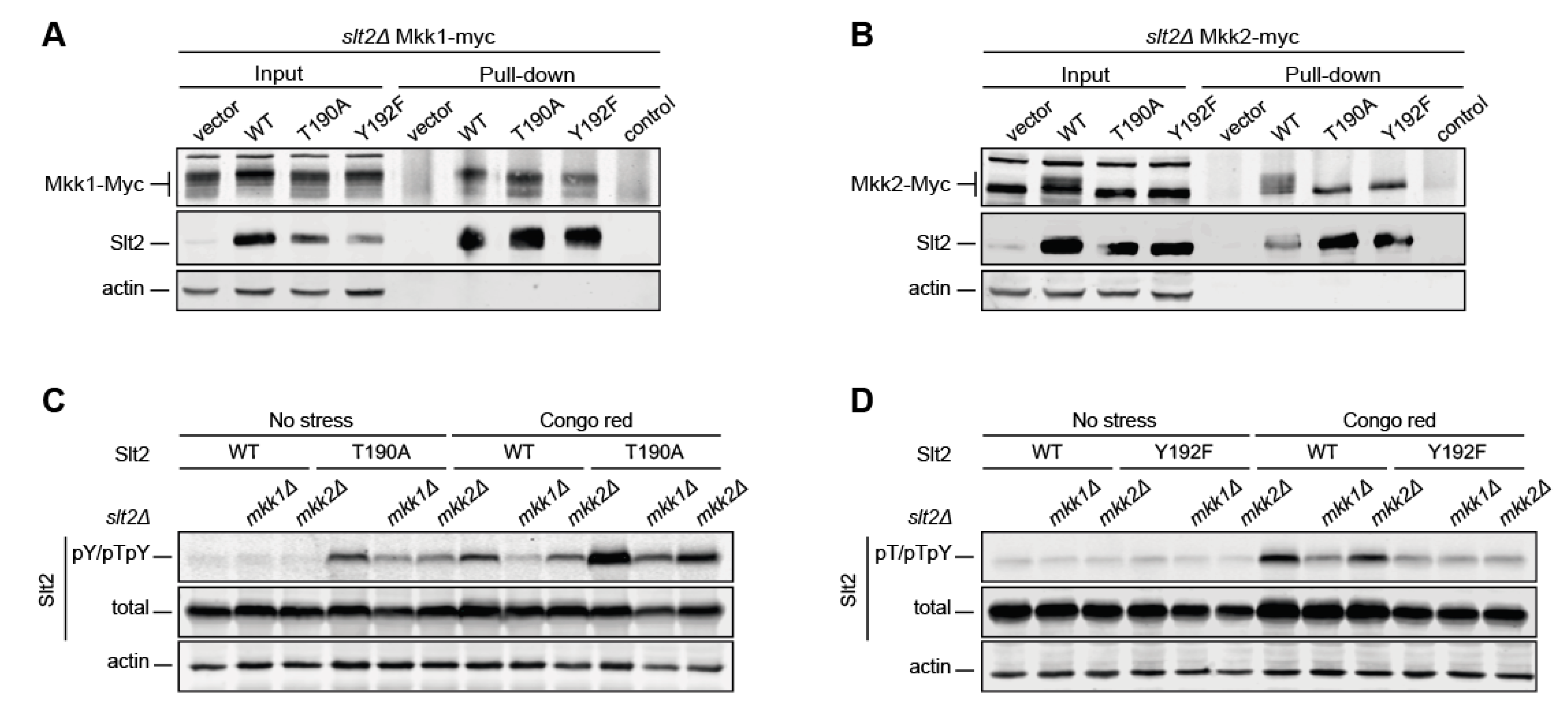
2.3. Y192 Phosphorylation by Mkk1 and Mkk2 Is a Stepping-Stone to the Dual Phosphorylation of Slt2
2.4. Dephosphorylation of T190 on Slt2 by Msg5 Depends on the Previous Dephosphorylation of Y192
2.5. T190 Is Essential for the Catalytic Activity and Biological Role of Slt2
2.6. T195 Plays a Key Role in Slt2 Activity
2.7. T190 and T195 Are Essential for Maintaining a Low Level of Y192 Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Plasmids and DNA Manipulation
4.2. Yeast Strains and Culture Conditions
4.3. Preparation of Yeast Extracts and Immunoblotting Analysis
4.4. Yeast Drop Dilution Growth Assays
4.5. Co-Purification Experiment with DynabeadsTM Protein G
4.6. β-galactosidase Activity Assay
4.7. Microscopy Techniques
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Caf | Caffeine |
| CD | Common docking |
| CFW | Calcofluor White |
| CR | Congo Red |
| CWI | Cell wall integrity |
| DSP | Dual-specificity phosphatases |
| EDTA | Ethylenediaminetetraacetic acid |
| ERK | Extracellular signal-regulated kinase |
| GFP | Green fluorescent protein |
| GST | Glutathione S-transferase |
| IB | Immunoblotting |
| IP | Immunoprecipitation |
| JNK | c-Jun N-terminal kinase |
| KD | Kinase dead |
| MAPK | Mitogen-activated protein kinase |
| MAPKKK/MAP3K | MAPK kinase kinase |
| MAPKK/MAP2K/MEK | MAPK kinase |
| MKP | MAPK phosphatase |
| mC | Monoclonal |
| OD | Optical density |
| ONPG | o-nitrophenyl-β-D-galactopyranoside |
| PAGE | Polyacrylamide gel electrophoresis |
| PBS | Phosphate-buffered saline |
| pC | Policlonal |
| PMSF | Phenylmethylsulfonyl fluoride |
| Pro | Proline |
| PSA | Ammonium peroxysulfate |
| PTMs | Post-translational modifications |
| SD | Synthetic dextrose |
| SDS | Sodium dodecyl sulfate |
| Ser | Serine |
| TEMED | tetramethylethylenediamine |
| Thr | Threonine |
| Tuni | Tunicamycin |
| Tyr | Tyrosine |
| WB | Western blotting |
| WT | Wild type |
| Zym | Zymolyase |
References
- Marshall, C.J. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr. Opin. Genet. Dev. 1994, 4, 82–89. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.E.; Thorner, J. Function and regulation in MAPK signaling pathways: Lessons learned from the yeast Saccharomyces cerevisiae. Biochim. Et Biophys. Acta 2007, 1773, 1311–1340. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.G.; Maller, J.L.; Tonks, N.K.; Sturgill, T.W. Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature 1990, 343, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Ahn, N.G.; Seger, R.; Bratlien, R.L.; Diltz, C.D.; Tonks, N.K.; Krebs, E.G. Multiple components in an epidermal growth factor-stimulated protein kinase cascade. In vitro activation of a myelin basic protein/microtubule-associated protein 2 kinase. J. Biol. Chem. 1991, 266, 4220–4227. [Google Scholar] [CrossRef]
- Ferrell, J.E., Jr.; Bhatt, R.R. Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J. Biol. Chem. 1997, 272, 19008–19016. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Yamada, M.; Kunida, K.; Yasuda, S.; Matsuda, M. Processive phosphorylation of ERK MAP kinase in mammalian cells. Proc. Natl. Acad. Sci. USA. 2011, 108, 12675–12680. [Google Scholar] [CrossRef] [Green Version]
- Nagiec, M.J.; McCarter, P.C.; Kelley, J.B.; Dixit, G.; Elston, T.C.; Dohlman, H.G. Signal inhibition by a dynamically regulated pool of monophosphorylated MAPK. Mol. Biol. Cell. 2015, 26, 3359–3371. [Google Scholar] [CrossRef]
- English, J.G.; Shellhammer, J.P.; Malahe, M.; McCarter, P.C.; Elston, T.C.; Dohlman, H.G. MAPK feedback encodes a switch and timer for tunable stress adaptation in yeast. Sci. Signal. 2015, 8, ra5. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, B.; Soto, T.; del Dedo, J.E.; Franco, A.; Vicente, J.; Hidalgo, E.; Gacto, M.; Cansado, J.; Madrid, M. Distinct biological activity of threonine monophosphorylated MAPK isoforms during the stress response in fission yeast. Cell. Signal. 2015, 27, 2534–2542. [Google Scholar] [CrossRef]
- Martín, H.; Flández, M.; Nombela, C.; Molina, M. Protein phosphatases in MAPK signalling: We keep learning from yeast. Mol. Microbiol. 2005, 58, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rubio, G.; Fernandez-Acero, T.; Martin, H.; Molina, M. Mitogen-Activated Protein Kinase Phosphatases (MKPs) in Fungal Signaling: Conservation, Function, and Regulation. Int. J. Mol. Sci. 2019, 20, 1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugden, P.H.; Markou, T.; Fuller, S.J.; Tham el, L.; Molkentin, J.D.; Paterson, H.F.; Clerk, A. Monophosphothreonyl extracellular signal-regulated kinases 1 and 2 (ERK1/2) are formed endogenously in intact cardiac myocytes and are enzymically active. Cell. Signal. 2011, 23, 468–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askari, N.; Beenstock, J.; Livnah, O.; Engelberg, D. p38alpha is active in vitro and in vivo when monophosphorylated at threonine 180. Biochemistry 2009, 48, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Mei, Z.Q.; Wu, J.W.; Wang, Z.X. Enzymatic activity and substrate specificity of mitogen-activated protein kinase p38alpha in different phosphorylation states. J. Biol. Chem. 2008, 283, 26591–26601. [Google Scholar] [CrossRef] [Green Version]
- Bell, M.; Engelberg, D. Phosphorylation of Tyr-176 of the yeast MAPK Hog1/p38 is not vital for Hog1 biological activity. J. Biol. Chem. 2003, 278, 14603–14606. [Google Scholar] [CrossRef] [Green Version]
- Bardwell, L.; Cook, J.G.; Voora, D.; Baggott, D.M.; Martinez, A.R.; Thorner, J. Repression of yeast Ste12 transcription factor by direct binding of unphosphorylated Kss1 MAPK and its regulation by the Ste7 MEK. Genes Dev. 1998, 12, 2887–2898. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, R.P.; Reményi, A.; Good, M.C.; Bashor, C.J.; Falick, A.M.; Lim, W.A. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science 2006, 311, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Oppermann, F.S.; Gnad, F.; Olsen, J.V.; Hornberger, R.; Greff, Z.; Kéri, G.; Mann, M.; Daub, H. Large-scale proteomics analysis of the human kinome. Mol. Cell. Proteom. MCP 2009, 8, 1751–1764. [Google Scholar] [CrossRef] [Green Version]
- Daub, H.; Olsen, J.V.; Bairlein, M.; Gnad, F.; Oppermann, F.S.; Körner, R.; Greff, Z.; Kéri, G.; Stemmann, O.; Mann, M. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell 2008, 31, 438–448. [Google Scholar] [CrossRef]
- Dephoure, N.; Zhou, C.; Villén, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.; Pelech, S. Regulatory roles of conserved phosphorylation sites in the activation T-loop of the MAP kinase ERK1. Mol. Biol. Cell 2016, 27, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: The cell wall integrity signaling pathway. Genetics 2011, 189, 1145–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Irie, K.; Gotoh, Y.; Watanabe, Y.; Araki, H.; Nishida, E.; Matsumoto, K.; Levin, D.E. A yeast mitogen-activated protein kinase homolog (Mpk1p) mediates signalling by protein kinase C. Mol. Cell Biol. 1993, 13, 3067–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, H.; Rodríguez-Pachón, J.M.; Ruiz, C.; Nombela, C.; Molina, M. Regulatory mechanisms for modulation of signaling through the cell integrity Slt2-mediated pathway in Saccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- De Nobel, H.; Ruiz, C.; Martin, H.; Morris, W.; Brul, S.; Molina, M.; Klis, F.M. Cell wall perturbation in yeast results in dual phosphorylation of the Slt2/Mpk1 MAP kinase and in an Slt2-mediated increase in FKS2-lacZ expression, glucanase resistance and thermotolerance. Microbiology 2000, 146(pt 9), 2121–2132. [Google Scholar] [CrossRef] [Green Version]
- Tatjer, L.; Sacristán-Reviriego, A.; Casado, C.; González, A.; Rodríguez-Porrata, B.; Palacios, L.; Canadell, D.; Serra-Cardona, A.; Martín, H.; Molina, M.; et al. Wide-Ranging Effects of the Yeast Ptc1 Protein Phosphatase Acting Through the MAPK Kinase Mkk1. Genetics 2016, 202, 141–156. [Google Scholar] [CrossRef]
- Flández, M.; Cosano, I.C.; Nombela, C.; Martín, H.; Molina, M. Reciprocal regulation between Slt2 MAPK and isoforms of Msg5 dual-specificity protein phosphatase modulates the yeast cell integrity pathway. J. Biol. Chem. 2004, 279, 11027–11034. [Google Scholar] [CrossRef] [Green Version]
- Sacristán-Reviriego, A.; Madrid, M.; Cansado, J.; Martín, H.; Molina, M. A conserved non-canonical docking mechanism regulates the binding of dual specificity phosphatases to cell integrity mitogen-activated protein kinases (MAPKs) in budding and fission yeasts. PLoS ONE 2014, 9, e85390. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Gutierrez, E.; Alegria-Carrasco, E.; Sellers-Moya, A.; Molina, M.; Martin, H. Not just the wall: The other ways to turn the yeast CWI pathway on. Int. Microbiol. 2020, 23, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Martín, H.; Arroyo, J.; Sánchez, M.; Molina, M.; Nombela, C. Activity of the yeast MAP kinase homologue Slt2 is critically required for cell integrity at 37 degrees C. Mol. Gen. Genet. 1993, 241, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Marín, M.J.; Flández, M.; Bermejo, C.; Arroyo, J.; Martín, H.; Molina, M. Different modulation of the outputs of yeast MAPK-mediated pathways by distinct stimuli and isoforms of the dual-specificity phosphatase Msg5. Mol. Genet. Genom. 2009, 281, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, M.; Cid, V.J.; Molina, M. Retrophosphorylation of Mkk1 and Mkk2 MAPKKs by the Slt2 MAPK in the yeast cell integrity pathway. J. Biol. Chem. 2007, 282, 31174–31185. [Google Scholar] [CrossRef] [Green Version]
- García, R.; Sanz, A.B.; Rodríguez-Peña, J.M.; Nombela, C.; Arroyo, J. Rlm1 mediates positive autoregulatory transcriptional feedback that is essential for Slt2-dependent gene expression. J. Cell Sci. 2016, 129, 1649–1660. [Google Scholar] [CrossRef] [Green Version]
- García, R.; Rodríguez-Peña, J.M.; Bermejo, C.; Nombela, C.; Arroyo, J. The high osmotic response and cell wall integrity pathways cooperate to regulate transcriptional responses to zymolyase-induced cell wall stress in Saccharomyces cerevisiae. J. Biol. Chem. 2009, 284, 10901–10911. [Google Scholar] [CrossRef] [Green Version]
- Palacios, L.; Dickinson, R.J.; Sacristan-Reviriego, A.; Didmon, M.P.; Marin, M.J.; Martin, H.; Keyse, S.M.; Molina, M. Distinct docking mechanisms mediate interactions between the Msg5 phosphatase and mating or cell integrity mitogen-activated protein kinases (MAPKs) in Saccharomyces cerevisiae. J. Biol. Chem. 2011, 286, 42037–42050. [Google Scholar] [CrossRef] [Green Version]
- Levin-Salomon, V.; Maayan, I.; Avrahami-Moyal, L.; Marbach, I.; Livnah, O.; Engelberg, D. When expressed in yeast, mammalian mitogen-activated protein kinases lose proper regulation and become spontaneously phosphorylated. Biochem. J. 2009, 417, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, K.; Schmitt, J.P.; Schmitteckert, E.M.; Lohse, M.J. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat. Med. 2009, 15, 75–83. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, X.; Wang, J.; Wang, X.; Liu, X.; Chen, Y.; Xu, W.; Wang, Y. Nitration-induced ubiquitination and degradation control quality of ERK1. Biochem. J. 2019, 476, 1911–1926. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Takiyama, K.; Koike, T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteom. MCP 2006, 5, 749–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardwell, L.; Shah, K. Analysis of mitogen-activated protein kinase activation and interactions with regulators and substrates. Methods 2006, 40, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Zhang, Z.Y. The activity of the extracellular signal-regulated kinase 2 is regulated by differential phosphorylation in the activation loop. J. Biol. Chem. 2002, 277, 13889–13899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittelstadt, P.R.; Yamaguchi, H.; Appella, E.; Ashwell, J.D. T cell receptor-mediated activation of p38{alpha} by mono-phosphorylation of the activation loop results in altered substrate specificity. J. Biol. Chem. 2009, 284, 15469–15474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winters, M.J.; Pryciak, P.M. Analysis of the thresholds for transcriptional activation by the yeast MAP kinases Fus3 and Kss1. Mol. Biol. Cell 2018, 29, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Zhou, G.; Wu, L.; Zhao, R.; Yuvaniyama, J.; Saper, M.A.; Dixon, J.E. The purification and characterization of a human dual-specific protein tyrosine phosphatase. J. Biol. Chem. 1995, 270, 3796–3803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, Z.Y. The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. J. Biol. Chem. 2001, 276, 32382–32391. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Lee, J.W.; Kang, G.Y.; Park, S.H.; Kim, K.P. Quantification of the Dynamic Phosphorylation Process of ERK Using Stable Isotope Dilution Selective Reaction Monitoring Mass Spectrometry. Proteomics 2019, 19, e1900086. [Google Scholar] [CrossRef]
- Patwardhan, P.; Miller, W.T. Processive phosphorylation: Mechanism and biological importance. Cell. Signal. 2007, 19, 2218–2226. [Google Scholar] [CrossRef] [Green Version]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef] [Green Version]
- Tárrega, C.; Ríos, P.; Cejudo-Marín, R.; Blanco-Aparicio, C.; van den Berk, L.; Schepens, J.; Hendriks, W.; Tabernero, L.; Pulido, R. ERK2 shows a restrictive and locally selective mechanism of recognition by its tyrosine phosphatase inactivators not shared by its activator MEK1. J. Biol. Chem. 2005, 280, 37885–37894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caunt, C.J.; McArdle, C.A. Stimulus-induced uncoupling of extracellular signal-regulated kinase phosphorylation from nuclear localization is dependent on docking domain interactions. J. Cell Sci. 2010, 123, 4310–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, U.S.; Sobering, A.K.; Romeo, M.J.; Levin, D.E. Regulation of the yeast Rlm1 transcription factor by the Mpk1 cell wall integrity MAP kinase. Mol. Microbiol. 2002, 46, 781–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikorski, R.S.; Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Rodriguez, E.; Fernandez-Pinar, P.; Sacristan-Reviriego, A.; Molina, M.; Martin, H. An Analog-sensitive Version of the Protein Kinase Slt2 Allows Identification of Novel Targets of the Yeast Cell Wall Integrity Pathway. J. Biol. Chem. 2016, 291, 5461–5472. [Google Scholar] [CrossRef] [Green Version]
- Cid, V.J.; Adamíková, L.; Cenamor, R.; Molina, M.; Sánchez, M.; Nombela, C. Cell integrity and morphogenesis in a budding yeast septin mutant. Microbiology 1998, 144 Pt 12, 3463–3474. [Google Scholar] [CrossRef] [Green Version]
- Martin, H.; Shales, M.; Fernandez-Piñar, P.; Wei, P.; Molina, M.; Fiedler, D.; Shokat, K.M.; Beltrao, P.; Lim, W.; Krogan, N.J. Differential genetic interactions of yeast stress response MAPK pathways. Mol. Syst. Biol. 2015, 11, 800. [Google Scholar] [CrossRef]
- Leskoske, K.L.; Roelants, F.M.; Emmerstorfer-Augustin, A.; Augustin, C.M.; Si, E.P.; Hill, J.M.; Thorner, J. Phosphorylation by the stress-activated MAPK Slt2 down-regulates the yeast TOR complex 2. Genes Dev. 2018, 32, 1576–1590. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, E.; Kinoshita-Kikuta, E. Improved Phos-tag SDS-PAGE under neutral pH conditions for advanced protein phosphorylation profiling. Proteomics 2011, 11, 319–323. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Source |
|---|---|
| pRS316 | [54] |
| pRS316-SLT2 | [55] |
| YCp50[mpk1Y192F] | [25] |
| YCp50[mpk1T190A] | [25] |
| pRS315 | [54] |
| pRS315-SLT2 | This study |
| pRS315-slt2T190A | This study |
| pRS315-slt2Y192F | This study |
| pRS316-slt2T190A | This study |
| pRS316-slt2Y192F | This study |
| pRS316-slt2T190A Y192A | This study |
| pRS316-slt2ΔC374 | This study |
| pRS316-slt2CD3 | This study |
| pHR3 | [32] |
| pRS316-slt2KD | This study |
| pRS316-slt2T195V | This study |
| pRS316-slt2Y198F | This study |
| pRS316-slt2T195V Y198F | This study |
| YEp352GST | [30] |
| YEp352MSG5GST | [30] |
| Yp352MSG5C319AGST | [30] |
| pMLP1-LacZ | Dr. Javier Arroyo |
| pLA10 | [56] |
| pRS316-6×His-Slt2 | This study |
| pRS316-GFP-SLT2 | This study |
| pRS316-GFP-slt2T195V | This study |
| pRS316-GFP-slt2T195V Y198F | This study |
| pRS305-MKK1-6MYC | [34] |
| pRS305-MKK2-6MYC | [34] |
| Strain | Genotype | Reference |
|---|---|---|
| Y00993 | BY4741; slt2Δ::KanMx4 | Euroscarf |
| Y02487 | BY4741; mkk1Δ:: KanMx4 | Euroscarf |
| Y02112 | BY4741; mkk2Δ:: KanMx4 | Euroscarf |
| Y07373 | BY4741; msg5Δ:: KanMx4 | Euroscarf |
| BY4741 slt2Δ-RLM1Myc | BY4741; slt2Δ:: KanMx4; RLM1- 6MYC:: HIS3 | [35] |
| YMJ30 | BY4741, slt2Δ:: KanMx4, MKK1::6MYC::LEU2 | Dr. Jiménez-Sánchez |
| YMJ31 | BY4741, slt2Δ:: KanMx4, MKK2::6MYC::LEU2 | Dr. Jiménez-Sánchez |
| YSTH4 | Y3656; slt2:: NatMx4 | [57] |
| YGGR32 | BY4741, mkk1Δ:: KanMx4, slt2:: NatMx4 | This study |
| YGGR33 | BY4741, mkk2Δ:: KanMx4, slt2:: NatMx4 | This study |
| YGGR34 | BY4741, msg5Δ:: KanMx4, slt2:: NatMx4 | This study |
| Name | Catalog Number | Species | Dilution Factor a | Manufacturer |
|---|---|---|---|---|
| Mpk1 (E9) | sc-133189 | Mouse mC | WB: 1:1000, IP: 1:20 | Santa Cruz, Inc |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | 4370 | Rabbit mC | WB: 1:1000 | Cell signaling |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | 4377 | Rabbit mC | WB: 1:250 | Cell signaling |
| Diphosphorylated ERK-1&2 | M8159 | Mouse mC | WB: 1:1000 | SIGMA |
| GST | sc459 | Rabbit pC | WB: 1:1000 | Santa Cruz, Inc. |
| Myc 4A6 | 05-724 | Mouse mC | WB: 1:1000 | Merck Millipore |
| C-Myc | PLA0001 | Rabbit pC | WB: 1:1000 | Sigma-Aldrich |
| Actin C4 | 69100 | Mouse mC | WB: 1:1000 | MP Biomedicals |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Rubio, G.; Sellers-Moya, Á.; Martín, H.; Molina, M. Differential Role of Threonine and Tyrosine Phosphorylation in the Activation and Activity of the Yeast MAPK Slt2. Int. J. Mol. Sci. 2021, 22, 1110. https://doi.org/10.3390/ijms22031110
González-Rubio G, Sellers-Moya Á, Martín H, Molina M. Differential Role of Threonine and Tyrosine Phosphorylation in the Activation and Activity of the Yeast MAPK Slt2. International Journal of Molecular Sciences. 2021; 22(3):1110. https://doi.org/10.3390/ijms22031110
Chicago/Turabian StyleGonzález-Rubio, Gema, Ángela Sellers-Moya, Humberto Martín, and María Molina. 2021. "Differential Role of Threonine and Tyrosine Phosphorylation in the Activation and Activity of the Yeast MAPK Slt2" International Journal of Molecular Sciences 22, no. 3: 1110. https://doi.org/10.3390/ijms22031110
APA StyleGonzález-Rubio, G., Sellers-Moya, Á., Martín, H., & Molina, M. (2021). Differential Role of Threonine and Tyrosine Phosphorylation in the Activation and Activity of the Yeast MAPK Slt2. International Journal of Molecular Sciences, 22(3), 1110. https://doi.org/10.3390/ijms22031110