
Escape from X Chromosome Inactivation and the Female Predominance in Autoimmune Diseases


Abstract
:1. Introduction
2. The Role of X Chromosome in Sex Bias in Autoimmune Diseases
3. Candidate X-Linked Genes Escaping from XCI with Possible Contribution in Autoimmune Diseases
3.1. The Intracellular ssRNA Sensors: TLR7 and TLR8
3.2. The IRF-5 Adaptor Molecule CXorf21/TASL.
3.3. Immune Cell Homing and Third Signal Delivery: CXCR3 and CD40L
3.4. The Histone Demethylase KDM6a (Lysine Demethylase 6A, also Known as Utx)
4. Role of XIST RNA Localization on the Xi in AID?
5. Interactions between Sex and Genetic Polymorphism: the Case of the rs179008 Polymorphism of TLR7
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| XCI | X chromosome inactivation |
| Xa | active X chromosome |
| Xi | inactive X chromosome |
| SLE | systemic lupus erythematosus |
| RA | rheumatoid arthritis |
| MS | multiple sclerosis |
| IFN-I | Interferon type I |
| pDC | plasmacytoid dendritic cell |
| TLR | Toll-like receptor |
| IRF | interferon regulatory factor |
| EAE | experimental autoimmune encephalomyelitis |
| HIV | human immunodeficiency virus |
References
- Laffont, S.; Guery, J.C. Deconstructing the sex bias in allergy and autoimmunity: From sex hormones and beyond. Adv. Immunol. 2019, 142, 35–64. [Google Scholar]
- NIH Autoimmune Diseases Coordinating Committee (ADCC). Progress in Autoimmune Diseases Research: Report to Congress. NIH publication number N° 05-5140 2005. Available online: https://www.niaid.nih.gov/sites/default/files/adccfinal.pdf (accessed on 1 March 2005).
- Billi, A.C.; Kahlenberg, J.M.; Gudjonsson, J.E. Sex bias in autoimmunity. Curr. Opin. Rheumatol. 2019, 31, 53–61. [Google Scholar] [CrossRef]
- Fink, A.L.; Klein, S.L. The evolution of greater humoral immunity in females than males: Implications for vaccine efficacy. Curr. Opin. Physiol. 2018, 6, 16–20. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Zuk, M. The sicker sex. PLoS Pathog. 2009, 5, e1000267. [Google Scholar] [CrossRef] [Green Version]
- Bach, J.F. The hygiene hypothesis in autoimmunity: The role of pathogens and commensals. Nat. Rev. Immunol. 2018, 18, 105–120. [Google Scholar] [CrossRef]
- Libert, C.; Dejager, L.; Pinheiro, I. The X chromosome in immune functions: When a chromosome makes the difference. Nat. Rev. Immunol. 2010, 10, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Smith-Bouvier, D.L.; Divekar, A.A.; Sasidhar, M.; Du, S.; Tiwari-Woodruff, S.K.; King, J.K.; Arnold, A.P.; Singh, R.R.; Voskuhl, R.R. A role for sex chromosome complement in the female bias in autoimmune disease. J. Exp. Med. 2008, 205, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasidhar, M.V.; Itoh, N.; Gold, S.M.; Lawson, G.W.; Voskuhl, R.R. The XX sex chromosome complement in mice is associated with increased spontaneous lupus compared with XY. Ann. Rheum. Dis. 2012, 71, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Scofield, R.H.; Bruner, G.R.; Namjou, B.; Kimberly, R.P.; Ramsey-Goldman, R.; Petri, M.; Reveille, J.D.; Alarcon, G.S.; Vila, L.M.; Reid, J.; et al. Klinefelter’s syndrome (47,XXY) in male systemic lupus erythematosus patients: Support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheumatol. 2008, 58, 2511–2517. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.M.; Sharma, R.; Cavett, J.; Kurien, B.T.; Liu, K.; Koelsch, K.A.; Rasmussen, A.; Radfar, L.; Lewis, D.; Stone, D.U.; et al. Klinefelter’s syndrome (47,XXY) is in excess among men with Sjögren’s syndrome. Clin. Immunol. 2016, 168, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Disteche, C.M.; Berletch, J.B. X Inactivation and Escape: Epigenetic and Structural Features. Front. Cell Dev. Biol. 2019, 7, 219. [Google Scholar] [CrossRef]
- Graves, J.A.; Wakefield, M.J.; Toder, R. The origin and evolution of the pseudoautosomal regions of human sex chromosomes. Hum. Mol. Genet. 1998, 7, 1991–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, E.N. The X-files in immunity: Sex-based differences predispose immune responses. Nat. Rev. Immunol. 2008, 8, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, I.; Dejager, L.; Libert, C. X-chromosome-located microRNAs in immunity: Might they explain male/female differences? The X chromosome-genomic context may affect X-located miRNAs and downstream signaling, thereby contributing to the enhanced immune response of females. Bioessays 2011, 33, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, L.B.; Ben-Ali, M.; Quach, H.; Laval, G.; Patin, E.; Pickrell, J.K.; Bouchier, C.; Tichit, M.; Neyrolles, O.; Gicquel, B.; et al. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 2009, 5, e1000562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants among Young Men With Severe COVID-19. JAMA 2020, 324, 663–673. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Tukiainen, T.; Villani, A.C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Syrett, C.M.; Kramer, M.C.; Basu, A.; Atchison, M.L.; Anguera, M.C. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc. Natl. Acad. Sci. USA 2016, 113, E2029–E2038. [Google Scholar] [CrossRef] [Green Version]
- Hagen, S.H.; Henseling, F.; Hennesen, J.; Savel, H.; Delahaye, S.; Richert, L.; Ziegler, S.M.; Altfeld, M. Heterogeneous Escape from X Chromosome Inactivation Results in Sex Differences in Type I IFN Responses at the Single Human pDC Level. Cell Rep. 2020, 33, 108485. [Google Scholar] [CrossRef] [PubMed]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejia, J.E.; Guery, J.C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018, 3, eaap8855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, A.; Carrel, L.; Pennisi, D.; Philippe, C.; Quaderi, N.; Siggers, P.; Steiner, K.; Tam, P.P.; Monaco, A.P.; Willard, H.F.; et al. The UTX gene escapes X inactivation in mice and humans. Hum. Mol. Genet. 1998, 7, 737–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, L.X.; Lee, J.; Kapoor, U.; Kartnig, F.; Sedlyarov, V.; Papakostas, K.; Cesar-Razquin, A.; Essletzbichler, P.; Goldmann, U.; Stefanovic, A.; et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7-9. Nature 2020, 581, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Souyris, M.; Mejia, J.E.; Chaumeil, J.; Guery, J.C. Female predisposition to TLR7-driven autoimmunity: Gene dosage and the escape from X chromosome inactivation. Semin. Immunopathol. 2019, 41, 153–164. [Google Scholar] [CrossRef]
- de Marcken, M.; Dhaliwal, K.; Danielsen, A.C.; Gautron, A.S.; Dominguez-Villar, M. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci. Signal. 2019, 12, eaaw1347. [Google Scholar] [CrossRef]
- Ah Kioon, M.D.; Tripodo, C.; Fernandez, D.; Kirou, K.A.; Spiera, R.F.; Crow, M.K.; Gordon, J.K.; Barrat, F.J. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci. Transl. Med. 2018, 10, eaam8458. [Google Scholar] [CrossRef] [Green Version]
- Pisitkun, P.; Deane, J.A.; Difilippantonio, M.J.; Tarasenko, T.; Satterthwaite, A.B.; Bolland, S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 2006, 312, 1669–1672. [Google Scholar] [CrossRef]
- Deane, J.A.; Pisitkun, P.; Barrett, R.S.; Feigenbaum, L.; Town, T.; Ward, J.M.; Flavell, R.A.; Bolland, S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 2007, 27, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Guiducci, C.; Gong, M.; Cepika, A.M.; Xu, Z.; Tripodo, C.; Bennett, L.; Crain, C.; Quartier, P.; Cush, J.J.; Pascual, V.; et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med. 2013, 210, 2903–2919. [Google Scholar] [CrossRef]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theofilopoulos, A.N.; Kono, D.H.; Baccala, R. The multiple pathways to autoimmunity. Nat. Immunol 2017, 18, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Azar, P.; Mejia, J.E.; Cenac, C.; Shaiykova, A.; Youness, A.; Laffont, S.; Essat, A.; Izopet, J.; Passaes, C.; Muller-Trutwin, M.; et al. TLR7 dosage polymorphism shapes interferogenesis and HIV-1 acute viremia in women. JCI Insight 2020, 5, e136047. [Google Scholar] [CrossRef] [PubMed]
- Cotton, A.M.; Lam, L.; Affleck, J.G.; Wilson, I.M.; Peñaherrera, M.S.; McFadden, D.E.; Kobor, M.S.; Lam, W.L.; Robinson, W.P.; Brown, C.J. Chromosome-wide DNA methylation analysis predicts human tissue-specific X inactivation. Hum. Genet. 2011, 130, 187–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odhams, C.A.; Roberts, A.L.; Vester, S.K.; Duarte, C.S.T.; Beales, C.T.; Clarke, A.J.; Lindinger, S.; Daffern, S.J.; Zito, A.; Chen, L.; et al. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in Systemic Lupus Erythematosus. Nat. Commun. 2019, 10, 2164. [Google Scholar] [CrossRef]
- Mackay, M.; Oswald, M.; Sanchez-Guerrero, J.; Lichauco, J.; Aranow, C.; Kotkin, S.; Korsunsky, I.; Gregersen, P.K.; Diamond, B. Molecular signatures in systemic lupus erythematosus: Distinction between disease flare and infection. Lupus Sci. Med. 2016, 3, e000159. [Google Scholar] [CrossRef] [Green Version]
- Oghumu, S.; Varikuti, S.; Stock, J.C.; Volpedo, G.; Saljoughian, N.; Terrazas, C.A.; Satoskar, A.R. Cutting Edge: CXCR3 Escapes X Chromosome Inactivation in T Cells during Infection: Potential Implications for Sex Differences in Immune Responses. J. Immunol. 2019, 203, 789–794. [Google Scholar] [CrossRef]
- Le Coz, C.; Trofa, M.; Syrett, C.M.; Martin, A.; Jyonouchi, H.; Jyonouchi, S.; Anguera, M.C.; Romberg, N. CD40LG duplication-associated autoimmune disease is silenced by nonrandom X-chromosome inactivation. J. Allergy Clin. Immunol. 2018, 141, 2308–2311.e7. [Google Scholar] [CrossRef]
- Berletch, J.B.; Ma, W.; Yang, F.; Shendure, J.; Noble, W.S.; Disteche, C.M.; Deng, X. Escape from X inactivation varies in mouse tissues. PLoS Genet. 2015, 11, e1005079. [Google Scholar] [CrossRef]
- Itoh, Y.; Golden, L.C.; Itoh, N.; Matsukawa, M.A.; Ren, E.; Tse, V.; Arnold, A.P.; Voskuhl, R.R. The X-linked histone demethylase Kdm6a in CD4+ T lymphocytes modulates autoimmunity. J. Clin. Investig. 2019, 130, 3852–3863. [Google Scholar] [CrossRef] [Green Version]
- Dunford, A.; Weinstock, D.M.; Savova, V.; Schumacher, S.E.; Cleary, J.P.; Yoda, A.; Sullivan, T.J.; Hess, J.M.; Gimelbrant, A.A.; Beroukhim, R.; et al. Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat. Genet. 2017, 49, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.J.; Willard, H.F. The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature 1994, 368, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Csankovszki, G.; Nagy, A.; Jaenisch, R. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J. Cell Biol. 2001, 153, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Savarese, F.; Flahndorfer, K.; Jaenisch, R.; Busslinger, M.; Wutz, A. Hematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Mol. Cell Biol. 2006, 26, 7167–7177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syrett, C.M.; Sindhava, V.; Hodawadekar, S.; Myles, A.; Liang, G.; Zhang, Y.; Nandi, S.; Cancro, M.; Atchison, M.; Anguera, M.C. Loss of Xist RNA from the inactive X during B cell development is restored in a dynamic YY1-dependent two-step process in activated B cells. PLoS Genet. 2017, 13, e1007050. [Google Scholar] [CrossRef] [Green Version]
- Syrett, C.M.; Paneru, B.; Sandoval-Heglund, D.; Wang, J.; Banerjee, S.; Sindhava, V.; Behrens, E.M.; Atchison, M.; Anguera, M.C. Altered X-chromosome inactivation in T cells may promote sex-biased autoimmune diseases. JCI Insight 2019, 4, e126751. [Google Scholar] [CrossRef] [Green Version]
- Syrett, C.M.; Sierra, I.; Beethem, Z.T.; Dubin, A.H.; Anguera, M.C. Loss of epigenetic modifications on the inactive X chromosome and sex-biased gene expression profiles in B cells from NZB/W F1 mice with lupus-like disease. J. Autoimmun. 2019, 107, 102357. [Google Scholar] [CrossRef]
- Adrianse, R.L.; Smith, K.; Gatbonton-Schwager, T.; Sripathy, S.P.; Lao, U.; Foss, E.J.; Boers, R.G.; Boers, J.B.; Gribnau, J.; Bedalov, A. Perturbed maintenance of transcriptional repression on the inactive X-chromosome in the mouse brain after Xist deletion. Epigenetics Chromatin 2018, 11, 50. [Google Scholar] [CrossRef] [Green Version]
- Ober, C.; Loisel, D.A.; Gilad, Y. Sex-specific genetic architecture of human disease. Nat. Rev. Genet. 2008, 9, 911–922. [Google Scholar] [CrossRef] [Green Version]
- Laffont, S.; Rouquie, N.; Azar, P.; Seillet, C.; Plumas, J.; Aspord, C.; Guery, J.C. X-Chromosome complement and estrogen receptor signaling independently contribute to the enhanced TLR7-mediated IFN-alpha production of plasmacytoid dendritic cells from women. J. Immunol. 2014, 193, 5444–5452. [Google Scholar] [CrossRef] [Green Version]
- dos Santos, B.P.; Valverde, J.V.; Rohr, P.; Monticielo, O.A.; Brenol, J.C.; Xavier, R.M.; Chies, J.A. TLR7/8/9 polymorphisms and their associations in systemic lupus erythematosus patients from southern Brazil. Lupus 2012, 21, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, M.; Kats, A.; Cavallo, M.; Shoenfeld, Y. Clinical and molecular evidence for association of SLE with parvovirus B19. Lupus 2010, 19, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Quintial, R.; Nguyen, A.; Kono, D.H.; Oldstone, M.B.A.; Theofilopoulos, A.N.; Baccala, R. Lupus acceleration by a MAVS-activating RNA virus requires endosomal TLR signaling and host genetic predisposition. PLoS ONE 2018, 13, e0203118. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, M.; Ziegler, S.; Laffont, S.; Smith, N.; Chauveau, L.; Tomezsko, P.; Sharei, A.; Kourjian, G.; Porichis, F.; Hart, M.; et al. Sex Differences in Plasmacytoid Dendritic Cell Levels of IRF5 Drive Higher IFN-alpha Production in Women. J. Immunol. 2015, 195, 5327–5336. [Google Scholar] [CrossRef] [Green Version]



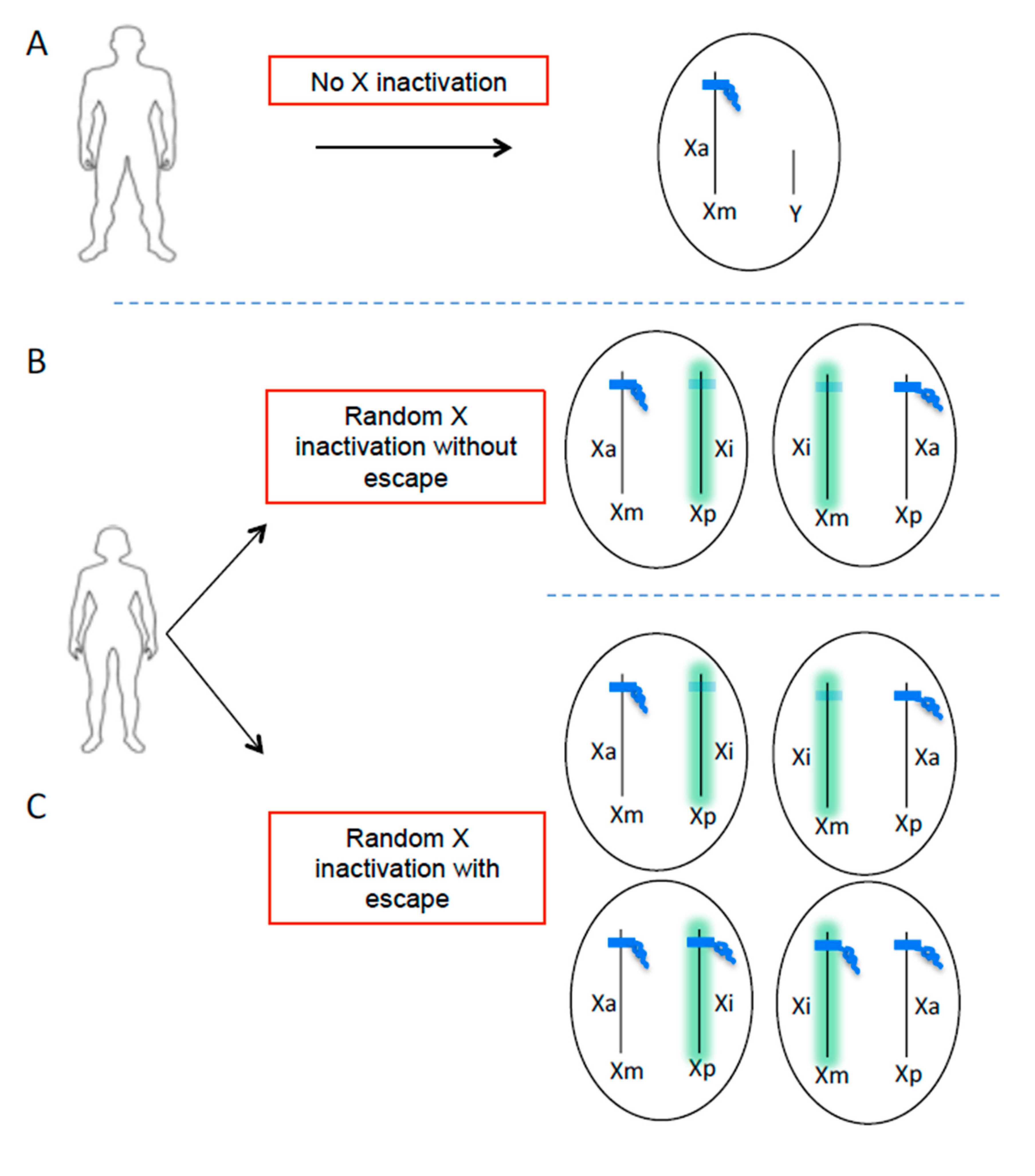{kind=link}
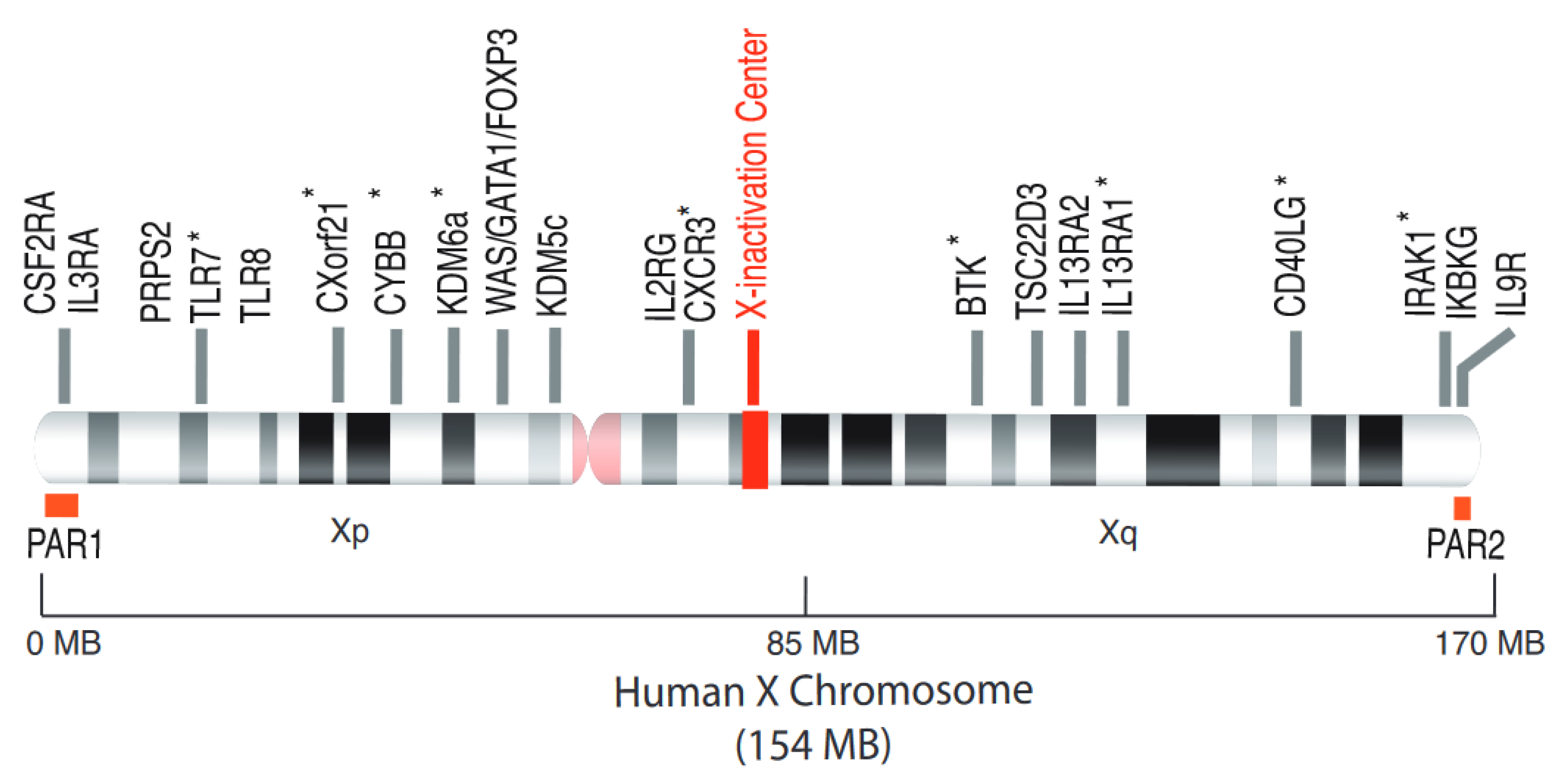{kind=link}
| Gene Symbol | Gene Nomenclature | Cell Type | Reference |
|---|---|---|---|
| IRAK1 | interleukin 1 receptor associated kinase 1 | variable escapes in primary fibroblast cell lines | [19] |
| CD40LG | CD40 ligand | escape in activated T cells and immortalized B-cell lines generated from pediatric SLE patients or healthy females | [21] |
| CXCR3 | C-X-C motif chemokine receptor 3 | ||
| IL13RA1 | interleukin 13 receptor subunit alpha 1 | escape in pDC from healthy women | [22] |
| CYBB | cytochrome b-245 beta chain | ||
| TLR7 | toll like receptor 7 | escapes in monocyte, lymphocyte B and pDC from healthy women and Klinefelter syndrome males (XXY) | [23] |
| escapes in pDC from healthy women | [22] | ||
| KDM6a | lysine demethylase 6A | escapes in mouse-human somatic cell hybrids | [24] |
| BTK | Burton tyrosine kinase | escapes in pDC from healthy women | [22] |
| CXorf21/TASL | chromosome X open reading frame 21; also known as TASL (TLR adaptor interacting with SLC15A4 on the Lysosome) [25] | variable escapes in primary fibroblast cell lines | [19] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youness, A.; Miquel, C.-H.; Guéry, J.-C. Escape from X Chromosome Inactivation and the Female Predominance in Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 1114. https://doi.org/10.3390/ijms22031114
Youness A, Miquel C-H, Guéry J-C. Escape from X Chromosome Inactivation and the Female Predominance in Autoimmune Diseases. International Journal of Molecular Sciences. 2021; 22(3):1114. https://doi.org/10.3390/ijms22031114
Chicago/Turabian StyleYouness, Ali, Charles-Henry Miquel, and Jean-Charles Guéry. 2021. "Escape from X Chromosome Inactivation and the Female Predominance in Autoimmune Diseases" International Journal of Molecular Sciences 22, no. 3: 1114. https://doi.org/10.3390/ijms22031114
APA StyleYouness, A., Miquel, C. -H., & Guéry, J. -C. (2021). Escape from X Chromosome Inactivation and the Female Predominance in Autoimmune Diseases. International Journal of Molecular Sciences, 22(3), 1114. https://doi.org/10.3390/ijms22031114