
The Central Role of Fibrinolytic Response in COVID-19—A Hematologist’s Perspective


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
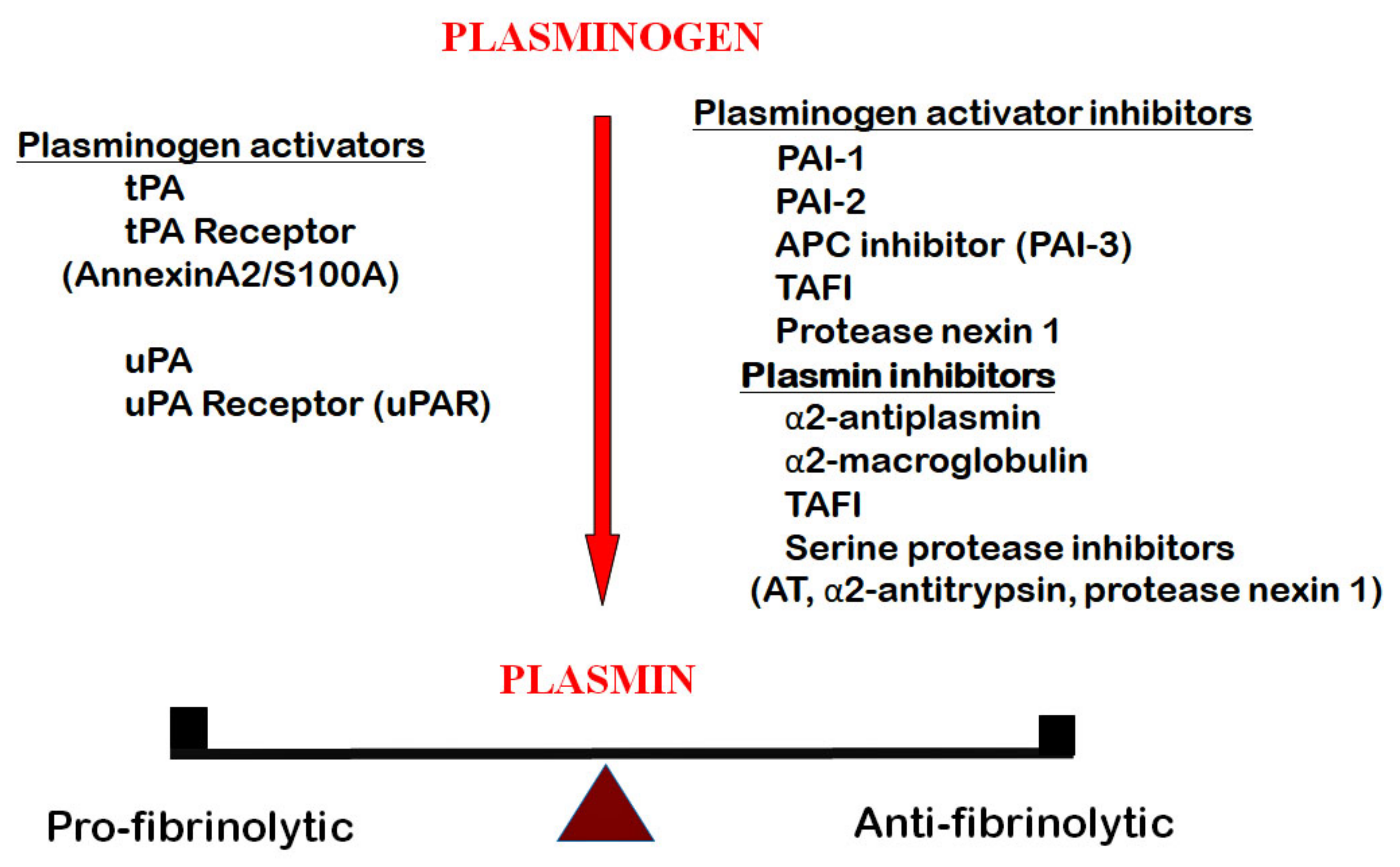
2. The Fibrinolytic System (Aka Plasminogen-Plasmin System)
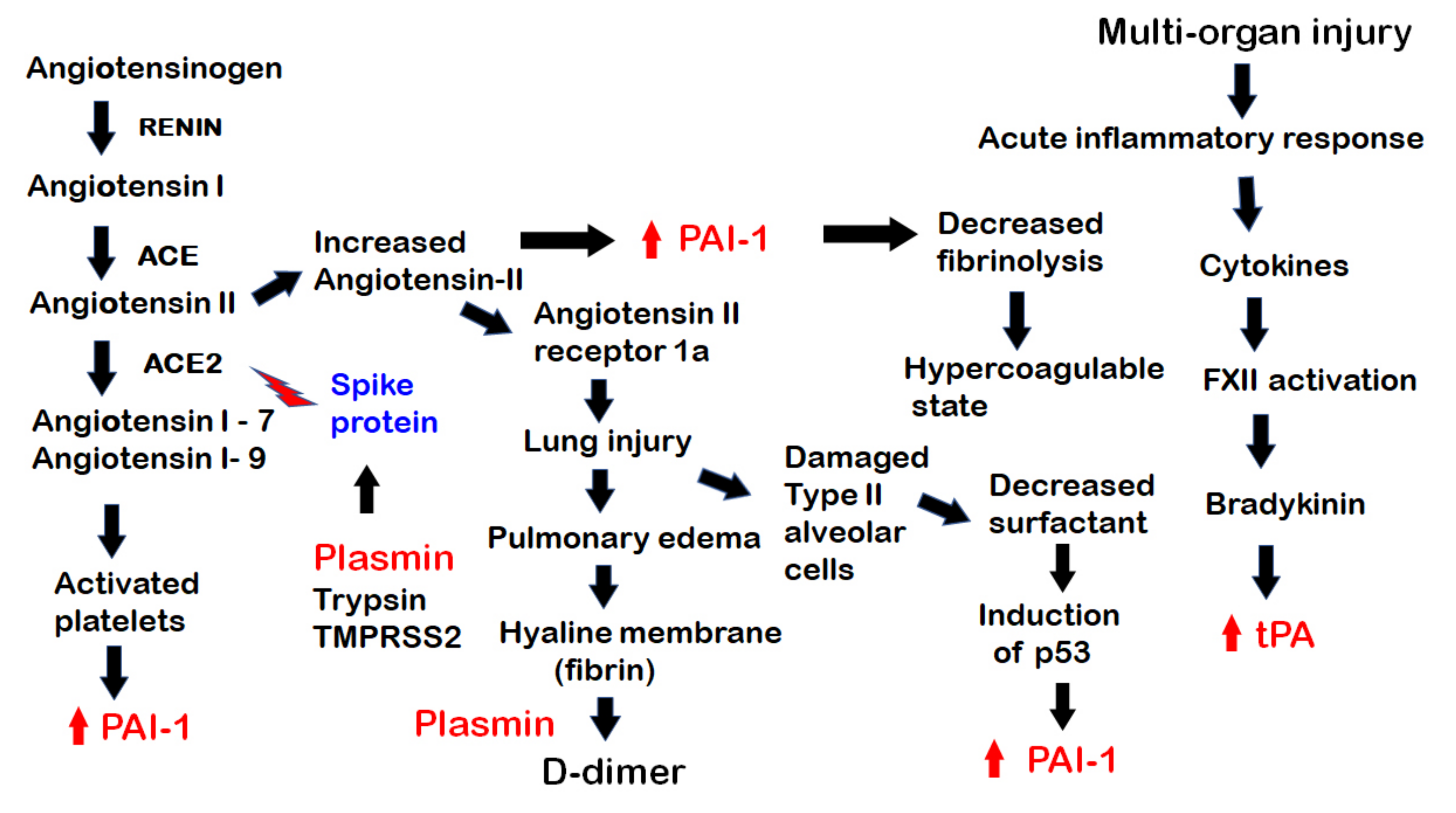
3. Invasion of SARS-CoV-2 into Host Cells and Subsequent Events
4. Hypercoagulability
5. Role of Platelets
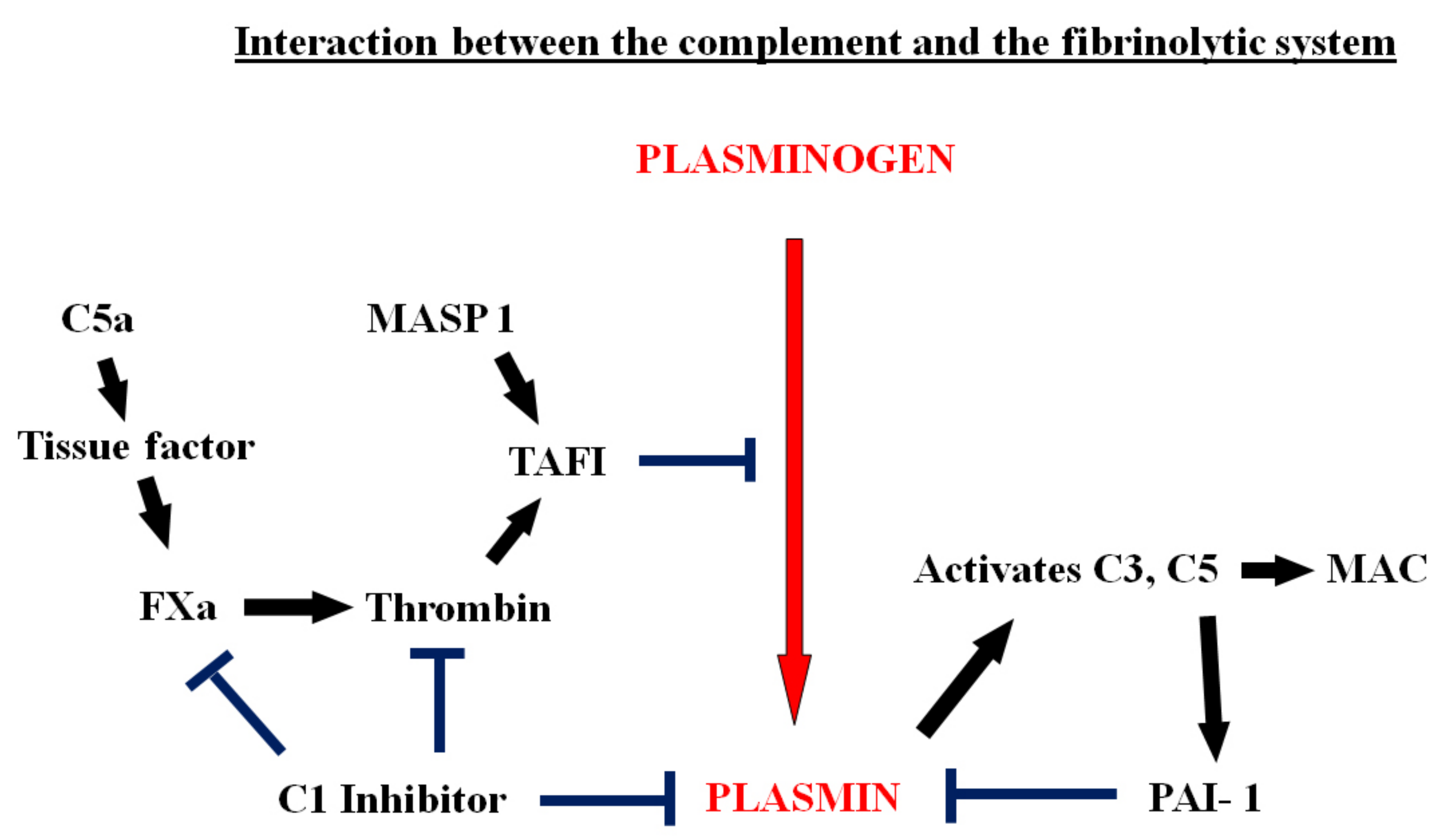
6. Activation of the Complement System
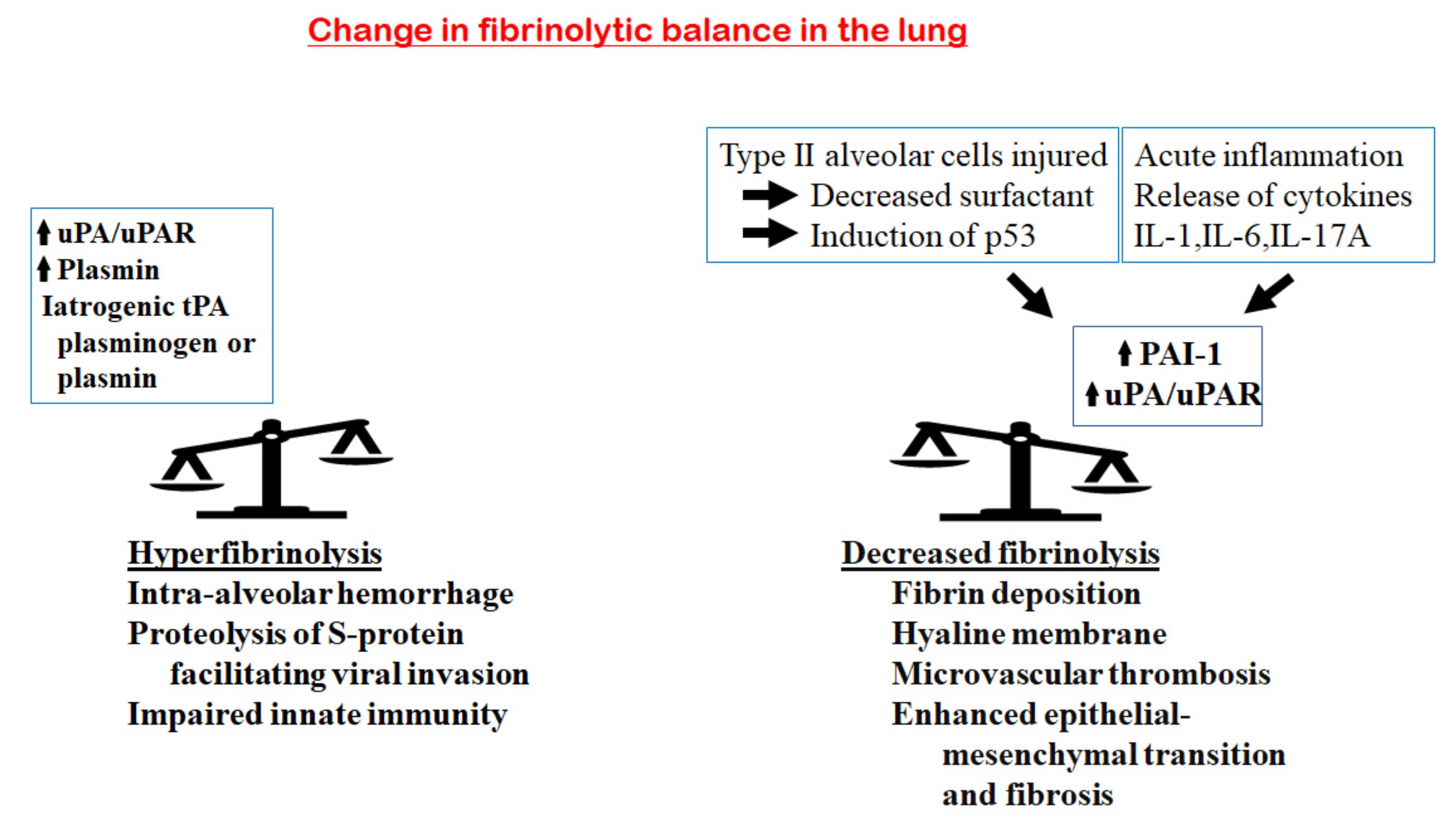
7. Fibrinolytic Balance
8. Bleeding Complications
9. Therapeutic Targets
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Kiu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, Y.L.; Peiris, J.S. Pathogenesis of severe acute respiratory syndrome. Curr. Opin. Immunol. 2005, 17, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wunderink, R.G. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology 2018, 23, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Zhang, X.; Qu, J. Coronavirus disease 2019 (COVID-19): A clinical update. Front. Med. 2020, 14, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, L.M.; Duval, E.J.; Stroberg, E.; Ghosh, S.; Mukhopadhyay, S. COVID-19 Autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020, 153, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Tian, S.; Xiong, Y.; Liu, H.; Niu, L.; Guo, J.; Liao, M.; Xiao, S.-Y. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod. Pathol. 2020, 33, 1007–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauter, J.L.; Baine, M.K.; Butnor, K.J.; Buonocore, D.J.; Chang, J.C.; Jungbluth, A.A.; Szabolcs, M.J.; Morjaria, S.; Mount, S.L.; Rekhtman, N.; et al. Insights into pathogenesis of fatal COVID-19 pneumonia from histopathology with immunohistochemical and viral RNA studies. Histopathology 2020, 77, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, J.M.; Poon, L.L.; Lee, K.C.; Ng, W.F.; Lai, S.T.; Leung, C.Y.; Chu, C.M.; Hui, P.K.; Mak, K.L.; Lim, W.; et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet 2003, 361, 1773–1778. [Google Scholar] [CrossRef] [Green Version]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Edler, C.; Schroder, A.S.; Aepfelbacher, M.; Fitzek, A.; Heinemann, A.; Heinrich, F.; Klein, A.; Langenwalder, F.; Lutgehetmann, M.; Meisner, K.; et al. Dying with SARS-CoV-2 infection-an autopsy study of the first consecutive 80 cases in Hamburg, Germany. Int. J. Leg. Med. 2020, 134, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Kwaan, H.C. From fibrinolysis to the plasminogen-plasmin system and beyond: A remarkable growth of knowledge, with personal observations on the history of fibrinolysis. Semin. Thromb. Hemost. 2014, 40, 585–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pechet, L. Fibrinolysis. N. Engl. J. Med. 1965, 273, 1024–1034. [Google Scholar] [CrossRef]
- Hajjar, K.A.; Menell, J.S. Annexin II: A novel mediator of cell surface plasmin generation. Ann. N. Y. Acad. Sci. 1997, 811, 337–349. [Google Scholar] [CrossRef]
- Surette, A.P.; Madureira, P.A.; Phipps, K.D.; Miller, V.A.; Svenningsson, P.; Waisman, D.M. Regulation of fibrinolysis by S100A10 in vivo. Blood 2011, 118, 3172–3181. [Google Scholar] [CrossRef] [Green Version]
- Ploug, M.; Ronne, E.; Behrendt, N.; Jensen, A.L.; Blasi, F.; Dano, K. Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J. Biol. Chem. 1991, 266, 1926–1933. [Google Scholar] [CrossRef]
- Abdul, S.; Leebeek, F.W.; Rijken, D.C.; Uitte de Willige, S. Natural heterogeneity of alpha2-antiplasmin: Functional and clinical consequences. Blood 2016, 127, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Van De Craen, B.; Declerck, P.J.; Gils, A. The Biochemistry, Physiology and Pathological roles of PAI-1 and the requirements for PAI-1 inhibition in vivo. Thromb. Res. 2012, 130, 576–585. [Google Scholar] [CrossRef]
- Hattori, N.; Sisson, T.H.; Xu, Y.; Desai, T.J.; Simon, R.H. Participation of urokinase-type plasminogen activator receptor in the clearance of fibrin from the lung. Am. J. Physiol. 1999, 277, L573–L579. [Google Scholar] [CrossRef] [PubMed]
- Idell, S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit. Care Med. 2003, 31, S213–S220. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M. The renin-angiotensin system: Importance in physiology and pathology. J. Cardiovasc. Pharmcol. 1990, 15 (Suppl. 3), S1–S5. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Patel, A.B.; Verma, A. COVID-19 and Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers: What Is the Evidence? JAMA 2020, 323, 1769–1770. [Google Scholar] [CrossRef]
- Turner, A.J.; Hiscox, J.A.; Hooper, N.M. ACE2: From vasopeptidase to SARS virus receptor. Trends Pharmcol. Sci. 2004, 25, 291–294. [Google Scholar] [CrossRef]
- Zhang, H.; Rostami, M.R.; Leopold, P.L.; Mezey, J.G.; O’Beirne, S.L.; Strulovici-Barel, Y.; Crystal, R.G. Expression of the SARS-CoV-2 ACE2 Receptor in the Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 2020, 202, 219–229. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.B.; Lyu, J.R.; Lei, X.M.; Li, W.; Wu, G.; Lyu, J.; Dai, Z.M. Specific ACE2 expression in small intestinal enterocytes may cause gastrointestinal symptoms and injury after 2019-nCoV infection. Int. J. Infect. Dis. 2020, 96, 19–24. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kam, Y.W.; Okumura, Y.; Kido, H.; Ng, L.F.; Bruzzone, R.; Altmeyer, R. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS ONE 2009, 4, e7870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Pohlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Hamming, I.; Cooper, M.E.; Haagmans, B.L.; Hooper, N.M.; Korstanje, R.; Osterhaus, A.D.; Timens, W.; Turner, A.J.; Navis, G.; van Goor, H. The emerging role of ACE2 in physiology and disease. J. Pathol. 2007, 212, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dijkman, R.; Jebbink, M.F.; Deijs, M.; Milewska, A.; Pyrc, K.; Buelow, E.; van der Bijl, A.; van der Hoek, L. Replication-dependent downregulation of cellular angiotensin-converting enzyme 2 protein expression by human coronavirus NL63. J. Gen. Virol. 2012, 93, 1924–1929. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.A.; Skorecki, K.; Heyman, S.N.; Kinaneh, S.; Armaly, Z. Covid-19 infection and mortality: A physiologist’s perspective enlightening clinical features and plausible interventional strategies. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L1020–L1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franks, T.J.; Chong, P.Y.; Chui, P.; Galvin, J.R.; Lourens, R.M.; Reid, A.H.; Selbs, E.; McEvoy, C.P.; Hayden, C.D.; Fukuoka, J.; et al. Lung pathology of severe acute respiratory syndrome (SARS): A study of 8 autopsy cases from Singapore. Hum. Pathol. 2003, 34, 743–748. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Bankhead, A., 3rd; Jeng, S.; Menachery, V.D.; Proll, S.; Belisle, S.E.; Matzke, M.; Webb-Robertson, B.J.; Luna, M.L.; Shukla, A.K.; et al. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, J.J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Sturzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef] [PubMed]
- Hayiroglu, M.I.; Cinar, T.; Tekkesin, A.I. Fibrinogen and D-dimer variances and anticoagulation recommendations in Covid-19: Current literature review. Rev. Assoc. Med. Bras. 2020, 66, 842–848. [Google Scholar] [CrossRef]
- Weitz, J.I.; Fredenburgh, J.C.; Eikelboom, J.W. A Test in Context: D-Dimer. J. Am. Coll Cardiol. 2017, 70, 2411–2420. [Google Scholar] [CrossRef]
- Vaughan, D.E.; Lazos, S.A.; Tong, K. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J. Clin. Investig. 1995, 95, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, D.E. Endothelial function, fibrinolysis, and angiotensin-converting enzyme inhibition. Clin. Cardiol. 1997, 20, II-34–II-37. [Google Scholar] [CrossRef]
- Nakamura, S.; Nakamura, I.; Ma, L.; Vaughan, D.E.; Fogo, A.B. Plasminogen activator inhibitor-1 expression is regulated by the angiotensin type 1 receptor in vivo. Kidney Int. 2000, 58, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Bhandary, Y.P.; Shetty, S.K.; Marudamuthu, A.S.; Ji, H.L.; Neuenschwander, P.F.; Boggaram, V.; Morris, G.F.; Fu, J.; Idell, S.; Shetty, S. Regulation of lung injury and fibrosis by p53-mediated changes in urokinase and plasminogen activator inhibitor-1. Am. J. Pathol. 2013, 183, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Puthusseri, B.; Marudamuthu, A.; Tiwari, N.; Fu, J.; Idell, S.; Shetty, S. Regulation of p53-mediated changes in the uPA-fibrinolytic system and in lung injury by loss of surfactant protein C expression in alveolar epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L783–L796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, M.; Naito, Y.; Urano, T.; Takada, A.; Takada, Y. L-158,809 and (D-Ala(7))-angiotensin I/II (1-7) decrease PAI-1 release from human umbilical vein endothelial cells. Thromb. Res. 2002, 105, 531–536. [Google Scholar] [CrossRef]
- Mogielnicki, A.; Kramkowski, K.; Hermanowicz, J.M.; Leszczynska, A.; Przyborowski, K.; Buczko, W. Angiotensin-(1-9) enhances stasis-induced venous thrombosis in the rat because of the impairment of fibrinolysis. J. Renin Angiotensin Aldosterone Syst. 2014, 15, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flevaris, P.; Vaughan, D. The Role of Plasminogen Activator Inhibitor Type-1 in Fibrosis. Semin. Thromb. Hemost. 2017, 43, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Marudamuthu, A.S.; Bhandary, Y.P.; Shetty, S.K.; Fu, J.; Sathish, V.; Prakash, Y.; Shetty, S. Role of the urokinase-fibrinolytic system in epithelial-mesenchymal transition during lung injury. Am. J. Pathol. 2015, 185, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagunduz, P.; Tabak, F.; Atagunduz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef]
- Rega, G.; Kaun, C.; Weiss, T.W.; Demyanets, S.; Zorn, G.; Kastl, S.P.; Steiner, S.; Seidinger, D.; Kopp, C.W.; Frey, M.; et al. Inflammatory cytokines interleukin-6 and oncostatin m induce plasminogen activator inhibitor-1 in human adipose tissue. Circulation 2005, 111, 1938–1945. [Google Scholar] [CrossRef] [Green Version]
- Declerck, P.J.; Gils, A. Three decades of research on plasminogen activator inhibitor-1: A multifaceted serpin. Semin. Thromb. Hemost. 2013, 39, 356–364. [Google Scholar]
- Vaughan, D.E. PAI-1 and atherothrombosis. J. Thromb. Haemost. 2005, 3, 1879–1883. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Ghosh, A.K.; Eren, M.; Miyata, T.; Vaughan, D.E. PAI-1 contributes to homocysteine-induced cellular senescence. Cell Signal. 2019, 64, 109394. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Kojima, T.; Adachi, T.; Hayashi, M.; Matsushita, T.; Takamatsu, J.; Loskutoff, D.J.; Saito, H. Obesity enhances the induction of plasminogen activator inhibitor-1 by restraint stress: A possible mechanism of stress-induced renal fibrin deposition in obese mice. J. Thromb. Haemost. 2005, 3, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Saito, H. A pathological role of increased expression of plasminogen activator inhibitor-1 in human or animal disorders. Int. J. Hematol. 1998, 68, 371–385. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takeshita, K.; Kojima, T.; Takamatsu, J.; Saito, H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: Implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc. Res. 2005, 66, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Liang, W.H.; Zhao, Y.; Liang, H.R.; Chen, Z.S.; Li, Y.M.; Liu, X.Q.; Chen, R.C.; Tang, C.L.; Wang, T.; et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: A nationwide analysis. Eur. Respir. J. 2020, 55, 2000547. [Google Scholar] [CrossRef] [Green Version]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef]
- Paramo, J.A. Pulmonary Embolism, Pulmonary Microvascular Thrombosis, or Both in COVID-19? Clin. Appl. Thromb. Hemost. 2020, 26. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef]
- Middeldorp, S.; Coppens, M.; van Haaps, T.F.; Foppen, M.; Vlaar, A.P.; Muller, M.C.A.; Bouman, C.C.S.; Beenen, L.F.M.; Kootte, R.S.; Heijmans, J.; et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1995–2002. [Google Scholar] [CrossRef]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S.; et al. Pulmonary Embolism in COVID-19 Patients: Awareness of an Increased Prevalence. Circulation 2020, 142, 184–186. [Google Scholar] [CrossRef]
- Guagliumi, G.; Sonzogni, A.; Pescetelli, I.; Pellegrini, D.; Finn, A.V. Microthrombi and ST-Segment Elevation Myocardial Infarction in COVID-19. Circulation 2020, 142, 804–809. [Google Scholar] [CrossRef]
- Pun, M.; Haggerty-Skeans, J.; Pratt, D.; Fudym, Y.; Al-Holou, W.N.; Camelo-Piragua, S.; Venneti, S. H3K27M-mutant diffuse midline glioma with extensive intratumoral microthrombi in a young adult with COVID-19-associated coagulopathy. Acta Neuropathol. 2020, 140, 227–229. [Google Scholar] [CrossRef]
- Tee, A.; Wong, A.; Yusuf, G.T.; Rao, D.; Sidhu, P.S. Contrast-enhanced ultrasound (CEUS) of the lung reveals multiple areas of microthrombi in a COVID-19 patient. Intensive Care Med. 2020, 46, 1660–1662. [Google Scholar] [CrossRef]
- Tutiya, C.T.; Siaulys, M.M.; Kondo, M.M.; Miglioli-Galvao, L.C.A.; Galvao, E.; Pinheiro, C.C.; Torloni, M.R.; de Mello, F.B. Possible formation of pulmonary microthrombi in the early puerperium of pregnant women critically ill with COVID-19: Two case reports. Case Rep. Womens Health 2020, 27, e00237. [Google Scholar] [CrossRef]
- Batlle, D.; Soler, M.J.; Sparks, M.A.; Hiremath, S.; South, A.M.; Welling, P.A.; Swaminathan, S. COVID-19 and ACE2 in Cardiovascular, Lung, and Kidney Working Group. Acute Kidney Injury in COVID-19: Emerging Evidence of a Distinct Pathophysiology. J. Am. Soc. Nephrol. 2020, 31, 1380–1383. [Google Scholar] [CrossRef]
- Su, H.; Yang, M.; Wan, C.; Yi, L.X.; Tang, F.; Zhu, H.Y.; Yi, F.; Yang, H.C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef]
- Hanley, B.; Lucas, S.B.; Youd, E.; Swift, B.; Osborn, M. Autopsy in suspected COVID-19 cases. J. Clin. Pathol. 2020, 73, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Gencer, S.; Lacy, M.; Atzler, D.; van der Vorst, E.P.C.; Doring, Y.; Weber, C. Immunoinflammatory, Thrombohaemostatic, and Cardiovascular Mechanisms in COVID-19. Thromb. Haemost. 2020, 120, 1629–1641. [Google Scholar] [CrossRef]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.M.; Charytan, D.M.; Gasmi, B.; et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine 2020, 24, 100434. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Blasi, A.; von Meijenfeldt, F.A.; Adelmeijer, J.; Calvo, A.; Ibanez, C.; Perdomo, J.; Reverter, J.C.; Lisman, T. In vitro hypercoagulability and ongoing in vivo activation of coagulation and fibrinolysis in COVID-19 patients on anticoagulation. J. Thromb. Haemost. 2020, 18, 2646–2653. [Google Scholar] [CrossRef]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Dowton, S.B.; Colten, H.R. Acute phase reactants in inflammation and infection. Semin. Hematol. 1988, 25, 84–90. [Google Scholar]
- Bi, X.; Su, Z.; Yan, H.; Du, J.; Wang, J.; Chen, L.; Peng, M.; Chen, S.; Shen, B.; Li, J. Prediction of severe illness due to COVID-19 based on an analysis of initial Fibrinogen to Albumin Ratio and Platelet count. Platelets 2020, 31, 674–679. [Google Scholar] [CrossRef]
- Wright, F.L.; Vogler, T.O.; Moore, E.E.; Moore, H.B.; Wohlauer, M.V.; Urban, S.; Nydam, T.L.; Moore, P.K.; McIntyre, R.C., Jr. Fibrinolysis Shutdown Correlation with Thromboembolic Events in Severe COVID-19 Infection. J. Am. Coll. Surg. 2020, 231, 193–203. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in Intensive Care Unit. A Report of Thromboelastography Findings and other Parameters of Hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef]
- Ibanez, C.; Perdomo, J.; Calvo, A.; Ferrando, C.; Reverter, J.C.; Tassies, D.; Blasi, A. High D dimers and low global fibrinolysis coexist in COVID19 patients: What is going on in there? J. Thromb. Thrombolysis 2020. [Google Scholar] [CrossRef]
- Medcalf, R.L.; Keragala, C.B.; Myles, P.S. Fibrinolysis and COVID-19: A plasmin paradox. J. Thromb. Haemost. 2020, 18, 2118–2122. [Google Scholar] [CrossRef]
- Prabhakaran, P.; Ware, L.B.; White, K.E.; Cross, M.T.; Matthay, M.A.; Olman, M.A. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L20–L28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs-Buder, T.; de Moerloose, P.; Ricou, B.; Reber, G.; Vifian, C.; Nicod, L.; Romand, J.A.; Suter, P.M. Time course of procoagulant activity and D dimer in bronchoalveolar fluid of patients at risk for or with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1996, 153, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, H.; Gabazza, E.C.; Hataji, O.; Yuda, H.; D’Alessandro-Gabazza, C.N.; Nakano, M.; Franco, O.E.; Hayashi, T.; Suzuki, K.; Adachi, Y.; et al. Thrombin-activatable fibrinolysis inhibitor and protein C inhibitor in interstitial lung disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Bertozzi, P.; Astedt, B.; Zenzius, L.; Lynch, K.; LeMaire, F.; Zapol, W.; Chapman, H.A., Jr. Depressed bronchoalveolar urokinase activity in patients with adult respiratory distress syndrome. N. Engl. J. Med. 1990, 322, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, W.; Jiang, W.; Xiao, M.; Li, Y.; Tang, N.; Liu, Z.; Yan, X.; Zhao, Y.; Li, T.; et al. Profile of natural anticoagulant, coagulant factor and anti-phospholipid antibody in critically ill COVID-19 patients. J. Thromb. Thromb. 2020, 50, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Bowles, L.; Platton, S.; Yartey, N.; Dave, M.; Lee, K.; Hart, D.P.; MacDonald, V.; Green, L.; Sivapalaratnam, S.; Pasi, K.J.; et al. Lupus Anticoagulant and Abnormal Coagulation Tests in Patients with Covid-19. N. Engl. J. Med. 2020, 383, 288–290. [Google Scholar] [CrossRef]
- Idell, S.; Kueppers, F.; Lippmann, M.; Rosen, H.; Niederman, M.; Fein, A. Angiotensin converting enzyme in bronchoalveolar lavage in ARDS. Chest 1987, 91, 52–56. [Google Scholar] [CrossRef]
- Konings, J.; Hoving, L.R.; Ariens, R.S.; Hethershaw, E.L.; Ninivaggi, M.; Hardy, L.J.; de Laat, B.; Ten Cate, H.; Philippou, H.; Govers-Riemslag, J.W. The role of activated coagulation factor XII in overall clot stability and fibrinolysis. Thromb. Res. 2015, 136, 474–480. [Google Scholar] [CrossRef]
- Kruse, J.M.; Magomedov, A.; Kurreck, A.; Munch, F.H.; Koerner, R.; Kamhieh-Milz, J.; Kahl, A.; Gotthardt, I.; Piper, S.K.; Eckardt, K.U.; et al. Thromboembolic complications in critically ill COVID-19 patients are associated with impaired fibrinolysis. Crit. Care 2020, 24, 676. [Google Scholar] [CrossRef]
- Pavoni, V.; Gianesello, L.; Pazzi, M.; Stera, C.; Meconi, T.; Frigieri, F.C. Evaluation of coagulation function by rotation thromboelastometry in critically ill patients with severe COVID-19 pneumonia. J. Thromb. Thrombolysis 2020, 50, 281–286. [Google Scholar] [CrossRef]
- van Veenendaal, N.; Scheeren, T.W.L.; Meijer, K.; van der Voort, P.H.J. Rotational thromboelastometry to assess hypercoagulability in COVID-19 patients. Thromb. Res. 2020, 196, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Spiezia, L.; Boscolo, A.; Poletto, F.; Cerruti, L.; Tiberio, I.; Campello, E.; Navalesi, P.; Simioni, P. COVID-19-Related Severe Hypercoagulability in Patients Admitted to Intensive Care Unit for Acute Respiratory Failure. Thromb. Haemost. 2020, 120, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Slomka, A.; Kowalewski, M.; Zekanowska, E. Haemostasis in coronavirus disease 2019—Lesson from viscoelastic methods—A systemic review. Thromb. Haemost. 2021. [Google Scholar] [CrossRef]
- Moore, H.B.; Moore, E.E.; Neal, M.D.; Sheppard, F.R.; Kornblith, L.Z.; Draxler, D.F.; Walsh, M.; Medcalf, R.L.; Cohen, M.J.; Cotton, B.A.; et al. Fibrinolysis Shutdown in Trauma: Historical Review and Clinical Implications. Anesth. Analg. 2019, 129, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregates formation trigger tissue factor expression in severe COVID-19 patients. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet Gene Expression and Function in COVID-19 Patients. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Huebner, B.R.; Moore, E.E.; Moore, H.B.; Stettler, G.R.; Nunns, G.R.; Lawson, P.; Sauaia, A.; Kelher, M.; Banerjee, A.; Silliman, C.C. Thrombin Provokes Degranulation of Platelet alpha-Granules Leading to the Release of Active Plasminogen Activator Inhibitor-1 (PAI-1). Shock 2018, 50, 671–676. [Google Scholar] [CrossRef]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Fletcher-Sandersjoo, A.; Bellander, B.M. Is COVID-19 associated thrombosis caused by overactivation of the complement cascade? A literature review. Thromb. Res. 2020, 194, 36–41. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, G.; Song, N.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; Du, L.; Jiang, S.; Guo, R.; et al. Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg. Microbes Infect. 2018, 7, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosnier, L.O.; Bouma, B.N. Regulation of fibrinolysis by thrombin activatable fibrinolysis inhibitor, an unstable carboxypeptidase B that unites the pathways of coagulation and fibrinolysis. Arter. Thromb. Vasc. Biol. 2006, 26, 2445–2453. [Google Scholar] [CrossRef] [PubMed]
- Kozarcanin, H.; Lood, C.; Munthe-Fog, L.; Sandholm, K.; Hamad, O.A.; Bengtsson, A.A.; Skjoedt, M.O.; Huber-Lang, M.; Garred, P.; Ekdahl, K.N.; et al. The lectin complement pathway serine proteases (MASPs) represent a possible crossroad between the coagulation and complement systems in thromboinflammation. J. Thromb. Haemost. 2016, 14, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.; Ajjan, R.; Phoenix, F.; Dobo, J.; Gal, P.; Schroeder, V. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS ONE 2012, 7, e35690. [Google Scholar] [CrossRef]
- Davis, A.E., 3rd; Lu, F.; Mejia, P. C1 inhibitor, a multi-functional serine protease inhibitor. Thromb. Haemost. 2010, 104, 886–893. [Google Scholar]
- Brown, E.W.; Ravindran, S.; Patston, P.A. The reaction between plasmin and C1-inhibitor results in plasmin inhibition by the serpin mechanism. Blood Coagul. Fibrinolysis 2002, 13, 711–714. [Google Scholar] [CrossRef]
- Foley, J.H.; Walton, B.L.; Aleman, M.M.; O’Byrne, A.M.; Lei, V.; Harrasser, M.; Foley, K.A.; Wolberg, A.S.; Conway, E.M. Complement Activation in Arterial and Venous Thrombosis is Mediated by Plasmin. EBioMedicine 2016, 5, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Wojta, J.; Huber, K.; Valent, P. New aspects in thrombotic research: Complement induced switch in mast cells from a profibrinolytic to a prothrombotic phenotype. Pathophysiol. Haemost. Thromb. 2003, 33, 438–441. [Google Scholar] [CrossRef]
- Fletcher-Sandersjoo, A.; Maegele, M.; Bellander, B.M. Does Complement-Mediated Hemostatic Disturbance Occur in Traumatic Brain Injury? A Literature Review and Observational Study Protocol. Int. J. Mol. Sci. 2020, 21, 1596. [Google Scholar] [CrossRef] [Green Version]
- Sauter, R.J.; Sauter, M.; Obrich, M.; Emschermann, F.N.; Nording, H.; Patzelt, J.; Wendel, H.P.; Reil, J.C.; Edlich, F.; Langer, H.F. Anaphylatoxin Receptor C3aR Contributes to Platelet Function, Thrombus Formation and In Vivo Haemostasis. Thromb. Haemost. 2019, 119, 179–182. [Google Scholar] [CrossRef]
- Sauter, R.J.; Sauter, M.; Reis, E.S.; Emschermann, F.N.; Nording, H.; Ebenhoch, S.; Kraft, P.; Munzer, P.; Mauler, M.; Rheinlaender, J.; et al. Functional Relevance of the Anaphylatoxin Receptor C3aR for Platelet Function and Arterial Thrombus Formation Marks an Intersection Point Between Innate Immunity and Thrombosis. Circulation 2018, 138, 1720–1735. [Google Scholar] [CrossRef]
- Trimarchi, H.; Gianserra, R.; Lampo, M.; Monkowski, M.; Lodolo, J. Eculizumab, SARS-CoV-2 and atypical hemolytic uremic syndrome. Clin. Kidney J. 2020, 13, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, K.; Pfefferle, S.; Bertram, S.; Glowacka, I.; Drosten, C.; Pohlmann, S.; Simmons, G. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J. Virol. 2010, 84, 8753–8764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisson, T.H.; Simon, R.H. The plasminogen activation system in lung disease. Curr. Drug Targets 2007, 8, 1016–1029. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Padijnayayveetil, J.; Tucker, T.; Stankowska, D.; Idell, S. The fibrinolytic system and the regulation of lung epithelial cell proteolysis, signaling, and cellular viability. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L967–L975. [Google Scholar] [CrossRef] [PubMed]
- Raghavendran, K.; Willson, D.; Notter, R.H. Surfactant therapy for acute lung injury and acute respiratory distress syndrome. Crit. Care Clin. 2011, 27, 525–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouda, M.M.; Shaikh, S.B.; Bhandary, Y.P. Inflammatory and Fibrinolytic System in Acute Respiratory Distress Syndrome. Lung 2018, 196, 609–616. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef]
- Desborough, M.J.R.; Doyle, A.J.; Griffiths, A.; Retter, A.; Breen, K.A.; Hunt, B.J. Image-proven thromboembolism in patients with severe COVID-19 in a tertiary critical care unit in the United Kingdom. Thromb. Res. 2020, 193, 1–4. [Google Scholar] [CrossRef]
- Fraisse, M.; Logre, E.; Pajot, O.; Mentec, H.; Plantefeve, G.; Contou, D. Thrombotic and hemorrhagic events in critically ill COVID-19 patients: A French monocenter retrospective study. Crit. Care 2020, 24, 275. [Google Scholar] [CrossRef] [PubMed]
- Musoke, N.; Lo, K.B.; Albano, J.; Peterson, E.; Bhargav, R.; Gul, F.; DeJoy, R., 3rd; Salacup, G.; Pelayo, J.; Tipparaju, P.; et al. Anticoagulation and bleeding risk in patients with COVID-19. Thromb. Res. 2020, 196, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Stillson, J.E.; Bunch, C.M.; Gillespie, L.; Khan, R.; Wierman, J.; Pulvirenti, J.; Phyu, H.; Anderson, S.; Al-Fadhl, M.; Thomas, A.V.; et al. Thromboelastography guided management of the anticoagulated COVID-19 patient. Semin. Thromb. Hematol. 2021, 47. in press. [Google Scholar]
- Liu, C.; Ma, Y.; Su, Z.; Zhao, R.; Zhao, X.; Nie, H.-G.; Ping, X.; Zhu, L.; Zhang, M.; Li, X.; et al. Meta-Analysis of Preclinical Studies of Fibrinolytic Therapy for Acute Lung Injury. Front. Immunol. 2018, 9, 1898. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, T.; Guo, C.; Zhang, D.; Ge, X.; Huang, Z.; Zhou, X.; Li, Y.; Peng, Q.; Li, J. Plasminogen improves lung lesions and hypoxemia in patients with COVID-19. QJM 2020, 113, 539–545. [Google Scholar] [CrossRef]
- Choudhury, R.; Barrett, C.D.; Moore, H.B.; Moore, E.E.; McIntyre, R.C.; Moore, P.K.; Talmor, D.S.; Nydam, T.L.; Yaffe, M.B. Salvage use of tissue plasminogen activator (tPA) in the setting of acute respiratory distress syndrome (ARDS) due to COVID-19 in the USA: A Markov decision analysis. World J. Emerg. Surg. 2020, 15, 29. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hajizadeh, N.; Moore, E.E.; McIntyre, R.C.; Moore, P.K.; Veress, L.A.; Yaffe, M.B.; Moore, H.B.; Barrett, C.D. Tissue plasminogen activator (tPA) treatment for COVID-19 associated acute respiratory distress syndrome (ARDS): A case series. J. Thromb. Haemost. 2020, 18, 1752–1755. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Rai, R.; Park, K.E.; Eren, M.; Miyata, T.; Wilsbacher, L.D.; Vaughan, D.E. A small molecule inhibitor of PAI-1 protects against doxorubicin-induced cellular senescence. Oncotarget 2016, 7, 72443–72457. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwaan, H.C.; Lindholm, P.F. The Central Role of Fibrinolytic Response in COVID-19—A Hematologist’s Perspective. Int. J. Mol. Sci. 2021, 22, 1283. https://doi.org/10.3390/ijms22031283
Kwaan HC, Lindholm PF. The Central Role of Fibrinolytic Response in COVID-19—A Hematologist’s Perspective. International Journal of Molecular Sciences. 2021; 22(3):1283. https://doi.org/10.3390/ijms22031283
Chicago/Turabian StyleKwaan, Hau C., and Paul F. Lindholm. 2021. "The Central Role of Fibrinolytic Response in COVID-19—A Hematologist’s Perspective" International Journal of Molecular Sciences 22, no. 3: 1283. https://doi.org/10.3390/ijms22031283
APA StyleKwaan, H. C., & Lindholm, P. F. (2021). The Central Role of Fibrinolytic Response in COVID-19—A Hematologist’s Perspective. International Journal of Molecular Sciences, 22(3), 1283. https://doi.org/10.3390/ijms22031283