
Molecular Mechanism of Lipid Accumulation and Metabolism of Oleaginous Chlorococcum sphacosum GD from Soil under Salt Stress



Abstract
:1. Introduction
2. Results
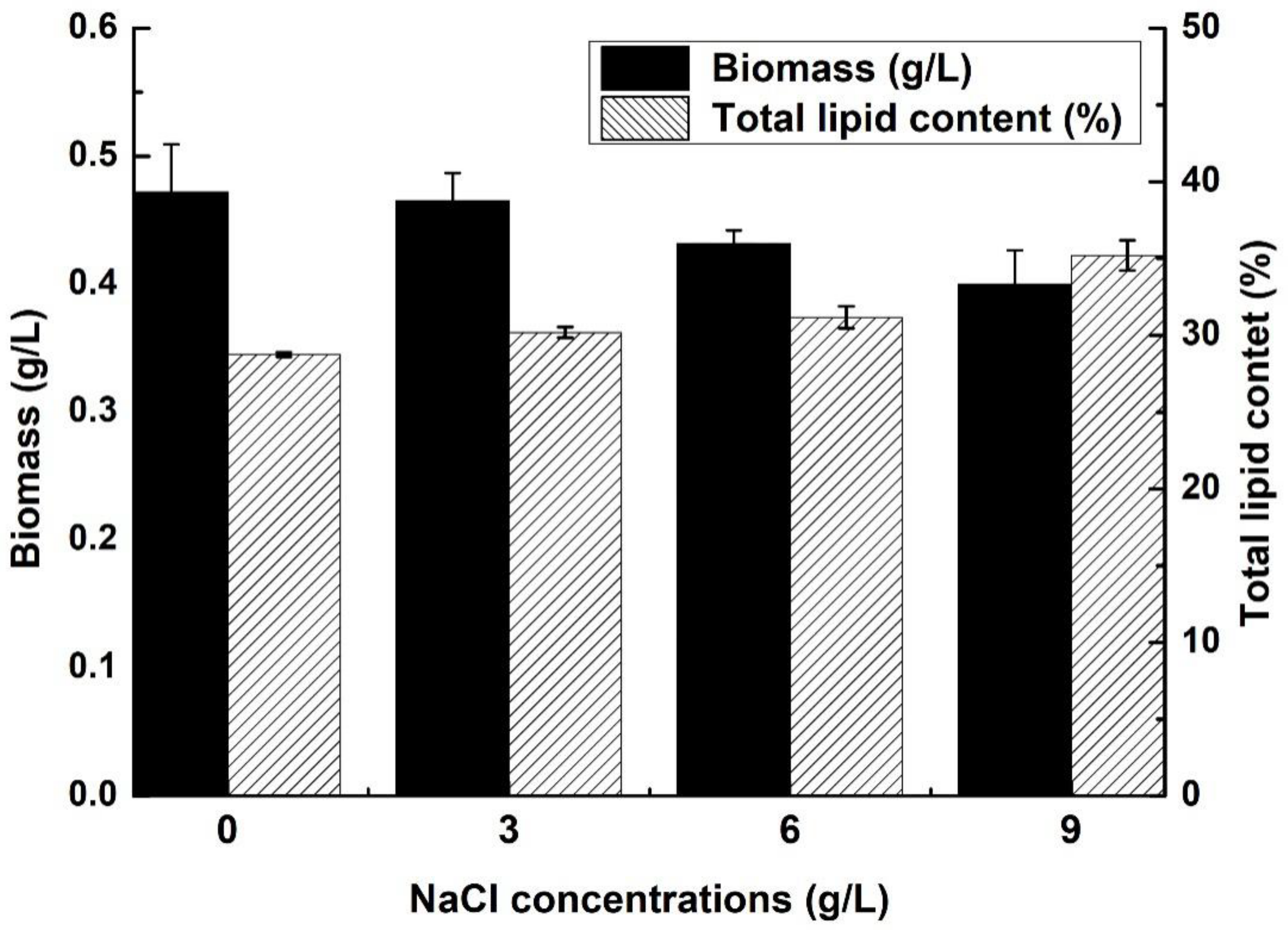
2.1. Biomass and Lipid Content of C. sphacosum GD with Different NaCl Concentrations
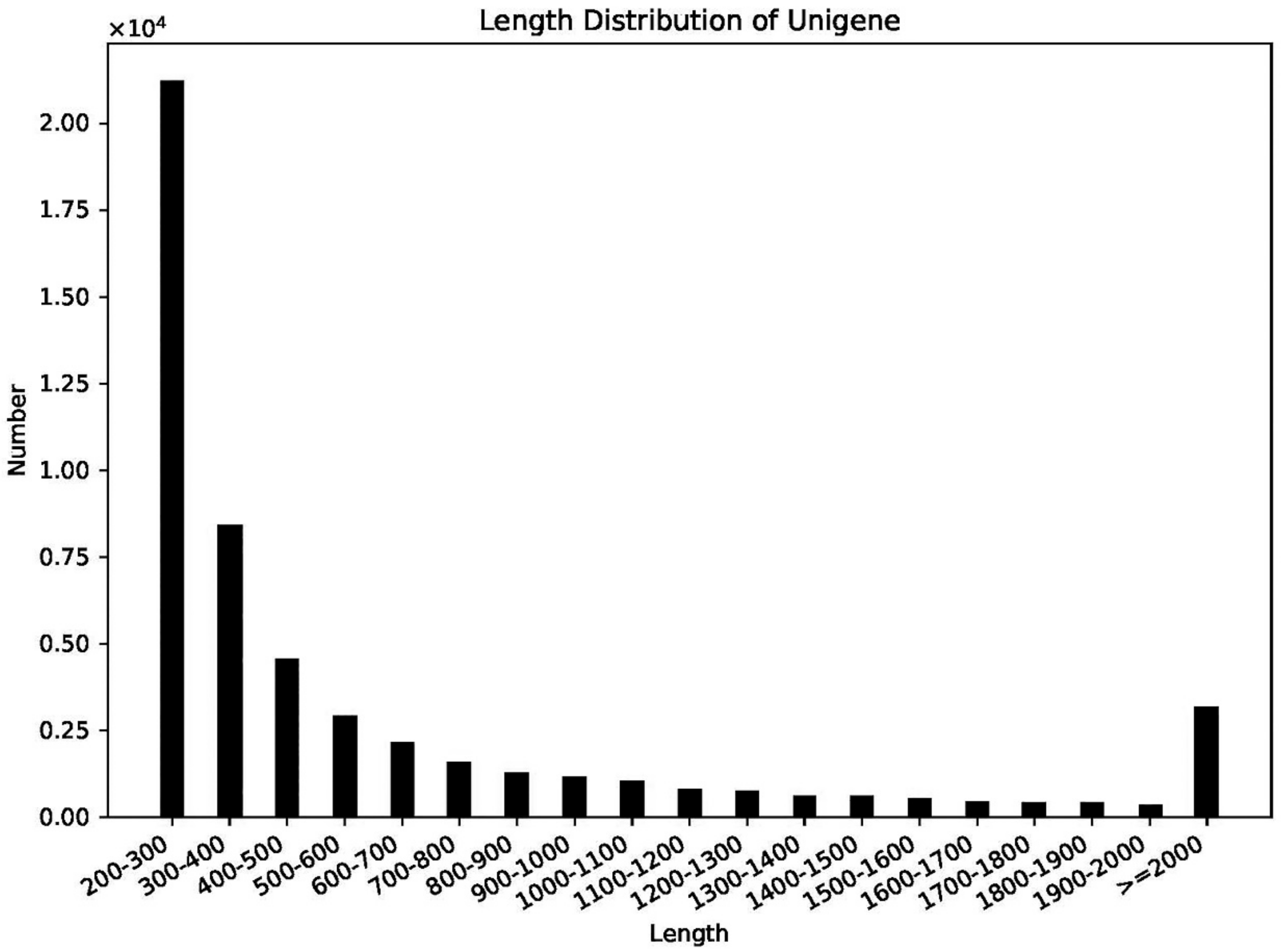
2.2. Transcriptome Assembly
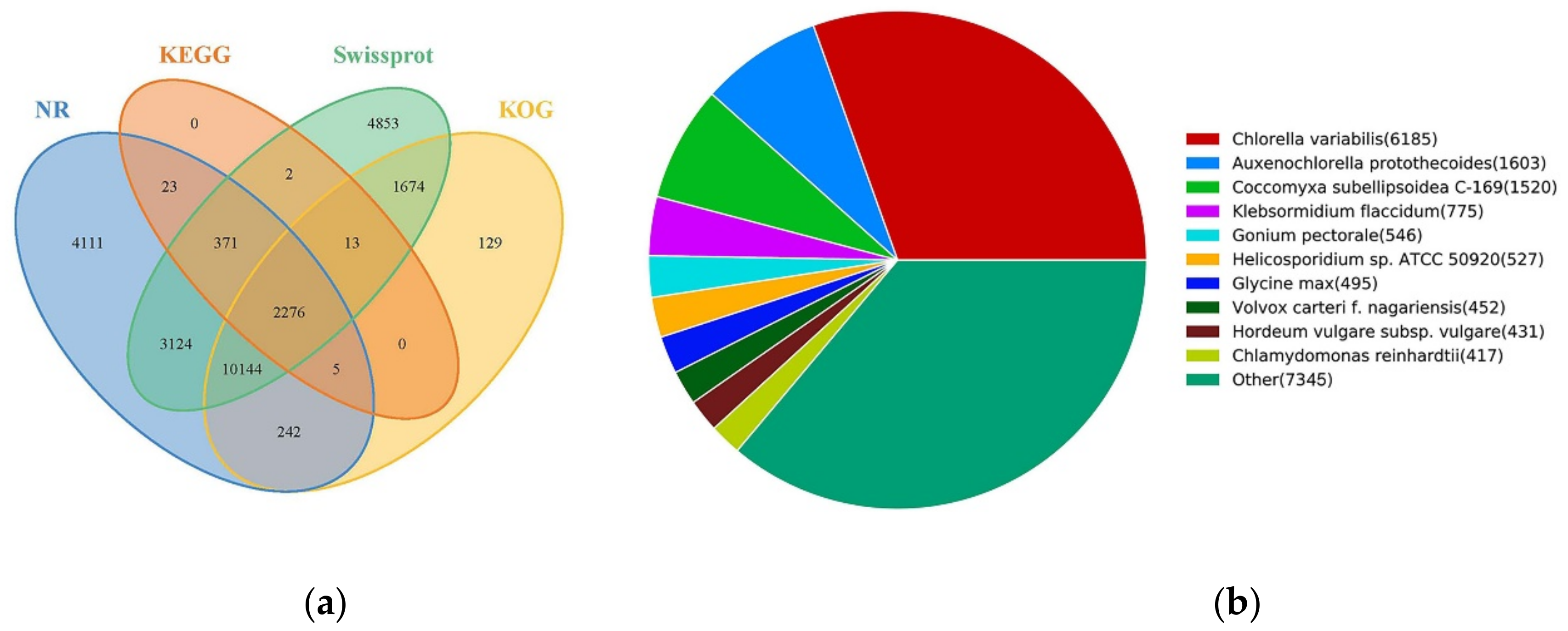
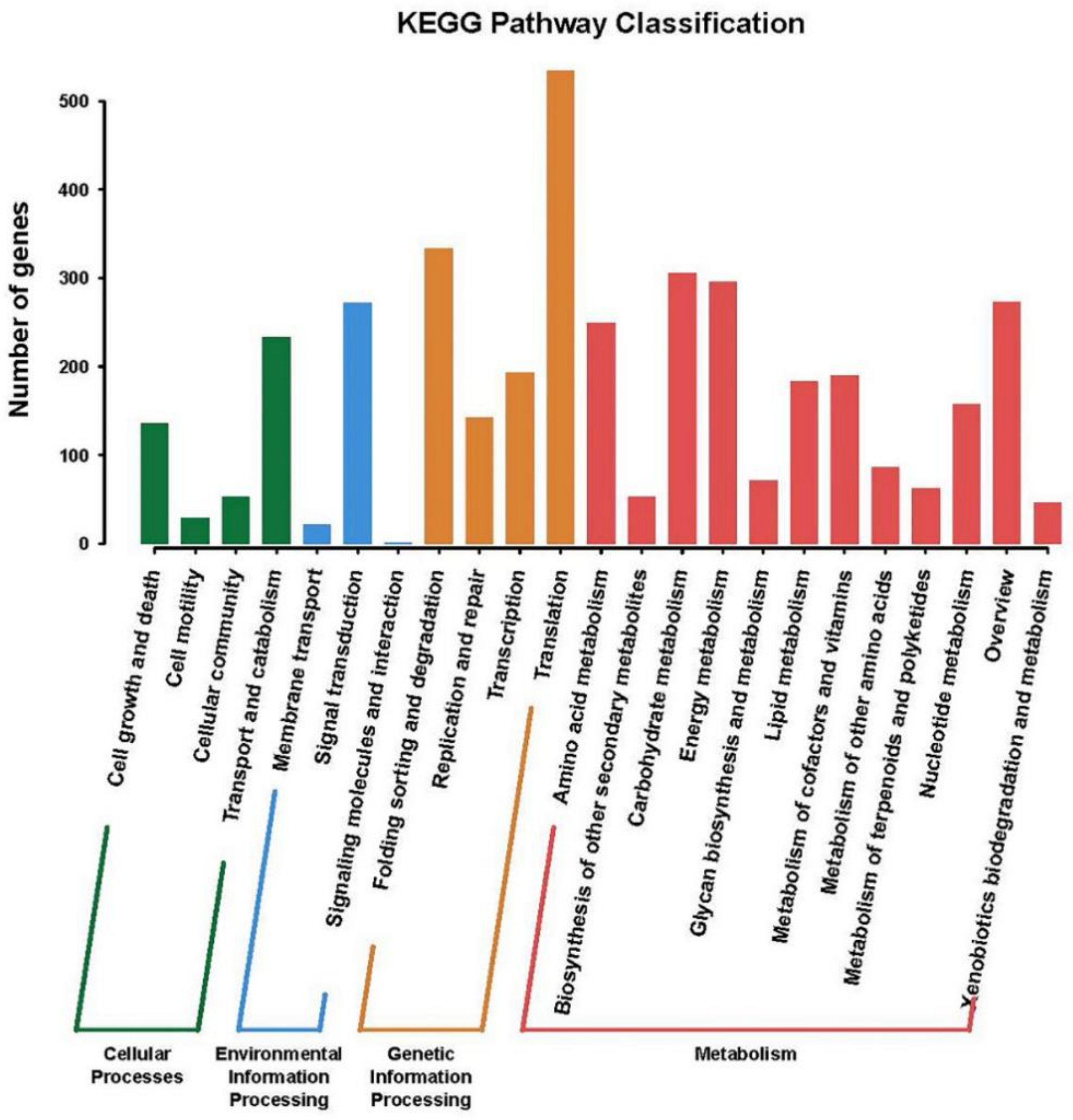
2.3. Gene Functional Annotation
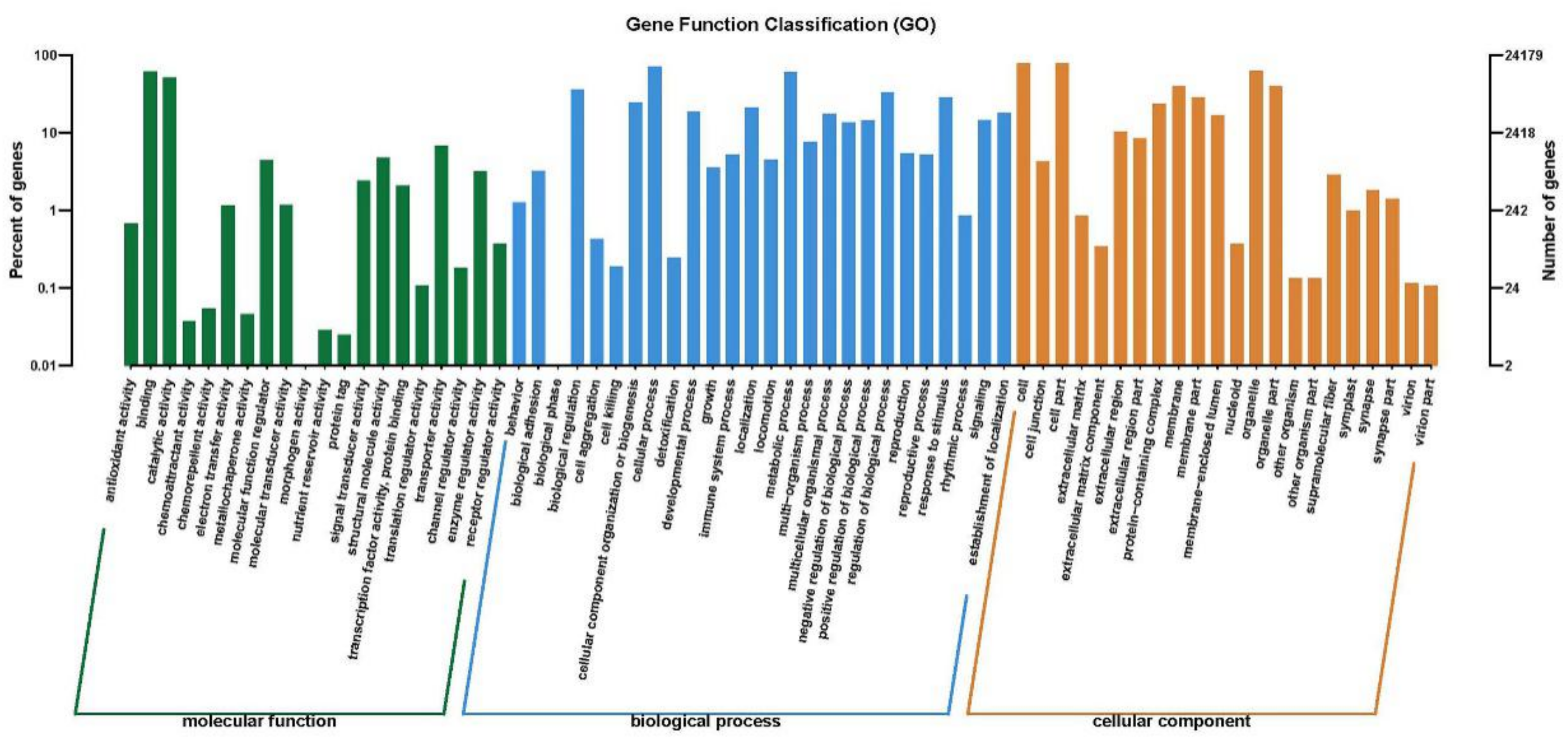
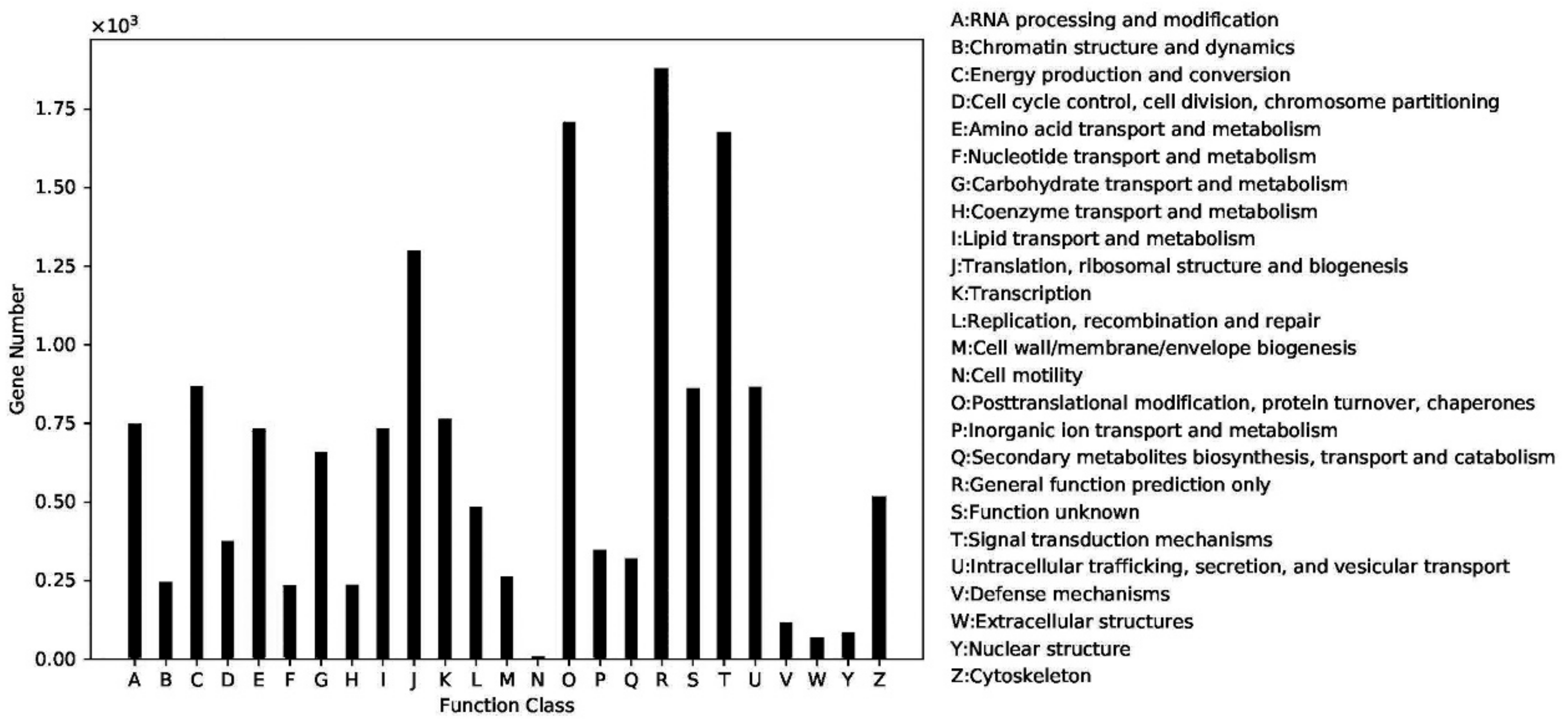
2.4. Gene Function Classification
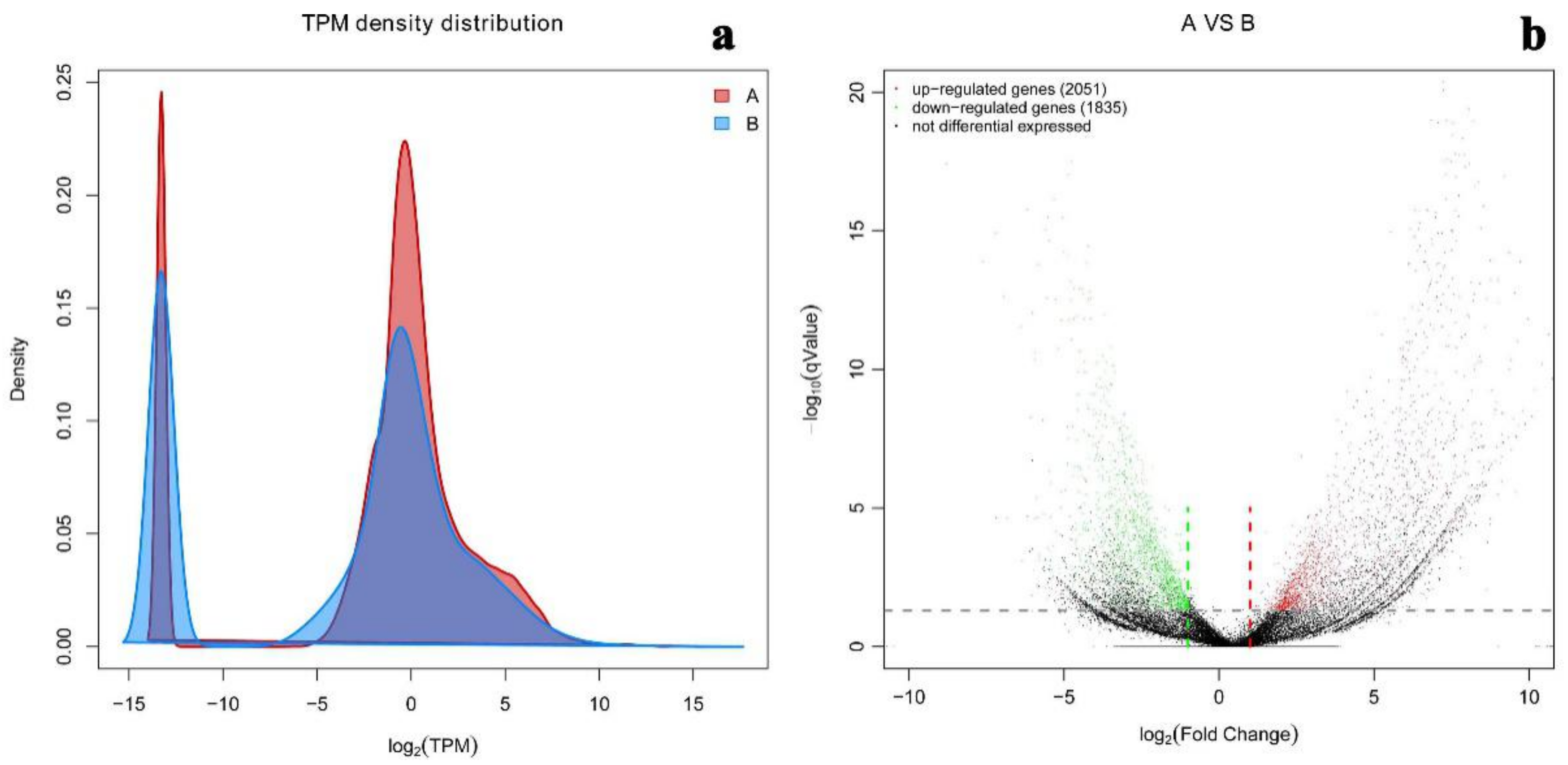
2.5. Differential Gene Expression Level
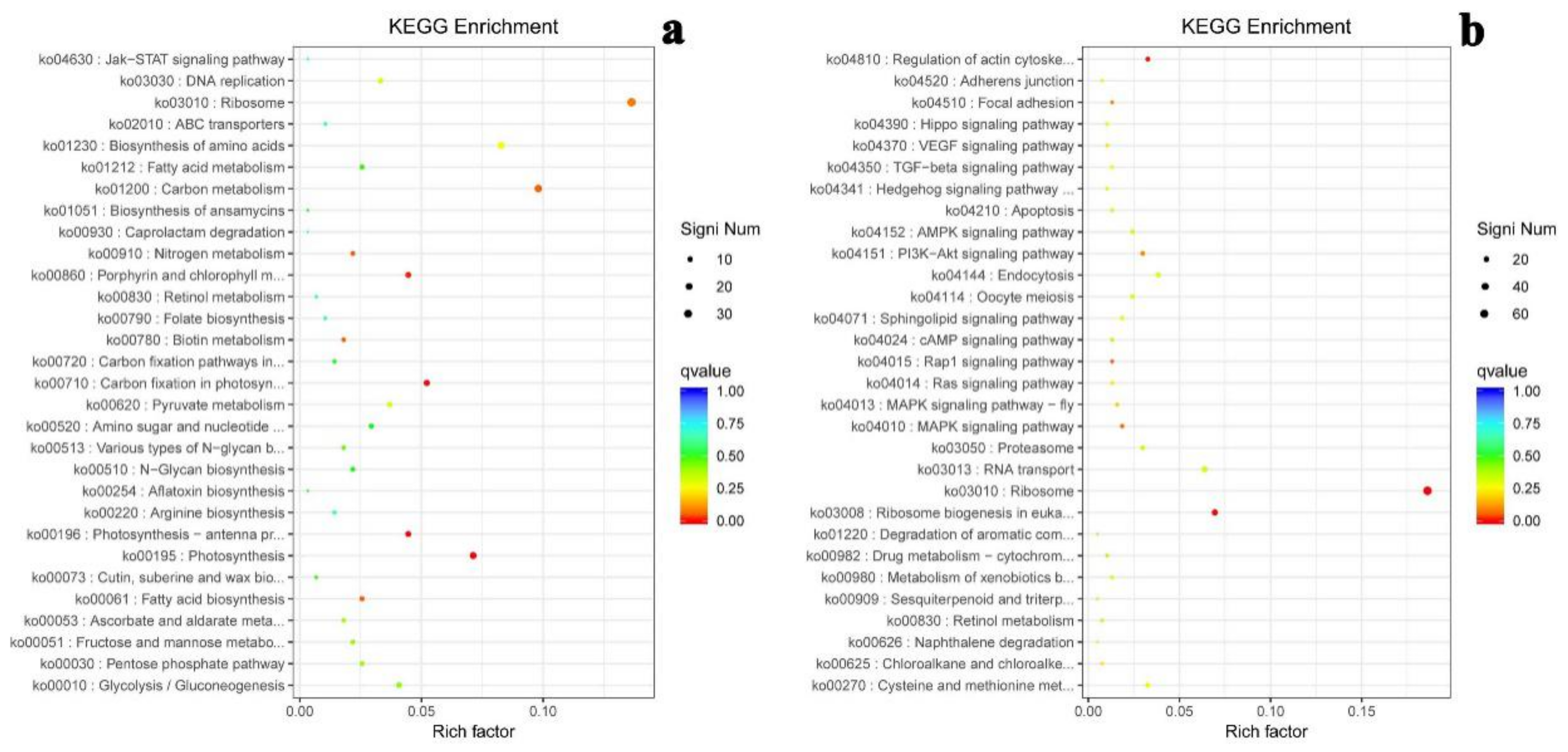
2.6. KEGG Enrichment of Differential Gene Expression
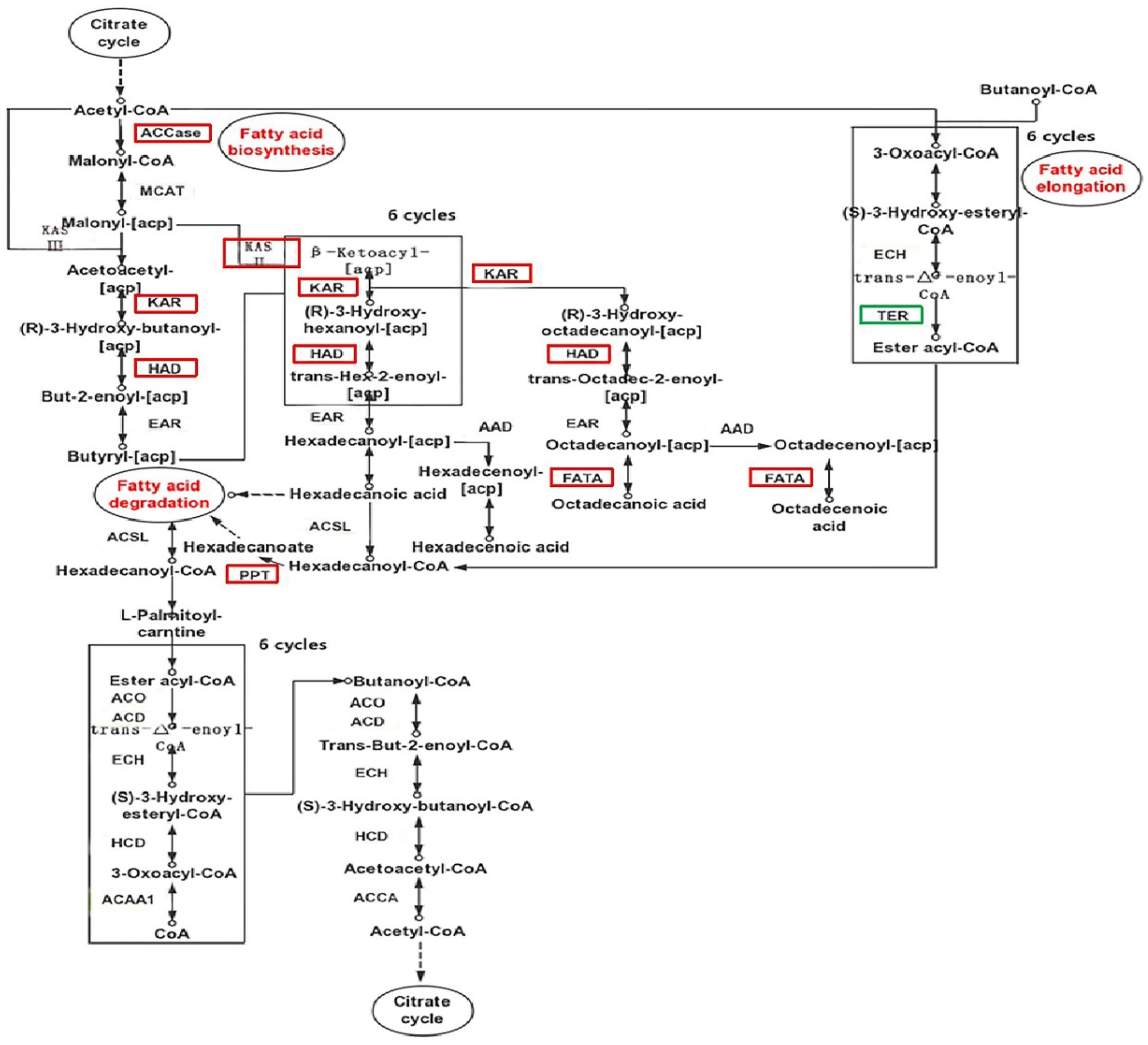
2.7. Lipid-Related Metabolic Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. Microalga and Culture Conditions
4.2. Physiological and Biochemical Analyses
4.3. RNA Extraction, cDNA Library Construction and Illumina Sequencing
4.4. RNA-Seq Data Filtering and Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koonin, S.E. Getting Serious About Biofuels. Science 2006, 311, 435. [Google Scholar] [CrossRef] [Green Version]
- Francisco, É.C.; Franco, T.T.; Maroneze, M.M.; Zepka, L.Q.; Jacob-Lopes, E. Third generation biodiesel production from microalgae. Cienc. Rural 2015, 45, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Wijffels, R.H.; Barbosa, M.J. An Outlook on Microalgal Biofuels. Science 2010, 329, 796–799. [Google Scholar] [CrossRef] [Green Version]
- Chisti, Y. Biodiesel from microalgae. Biotechnol. Adv. 2007, 25, 294–306. [Google Scholar] [CrossRef]
- Michels, M.H.A.; Van Der Goot, A.J.; Vermuë, M.H.; Wijffels, R.H. Cultivation of shear stress sensitive and tolerant microalgal species in a tubular photobioreactor equipped with a centrifugal pump. J. Appl. Phycol. 2016, 28, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doan, T.T.Y.; Sivaloganathan, B.; Obbard, J.P. Screening of marine microalgae for biodiesel feedstock. Biomass Bioenergy 2011, 35, 2534–2544. [Google Scholar] [CrossRef]
- Hu, Q.; Sommerfeld, M.; Jarvis, E.; Ghirardi, M.; Posewitz, M.; Seibert, M.; Darzins, A. Microalgal triacylglycerols as feed-stocks for biofuel production: Perspectives and advances. Plant J. 2008, 54, 621–639. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Yang, H.; Hu, C. Effects of temperature and its combination with high light intensity on lipid production of Monoraphidium dybowskii Y2 from semi-arid desert areas. Bioresour. Technol. 2018, 265, 407–414. [Google Scholar] [CrossRef]
- Ruangsomboon, S. Effect of light, nutrient, cultivation time and salinity on lipid production of newly isolated strain of the green microalga, Botryococcus braunii KMITL 2. Bioresour. Technol. 2012, 109, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; He, Q.; Hu, C. Lipid accumulation by NaCl induction at different growth stages and concentrations in pho-toautotrophic two-step cultivation of Monoraphidium dybowskii LB50. Bioresour. Technol. 2015, 187, 221–227. [Google Scholar] [CrossRef]
- Yang, H.; He, Q.; Rong, J.; Xia, L.; Hu, C. Rapid neutral lipid accumulation of the alkali-resistant oleaginous Monoraphidium dybowskii LB50 by NaCl induction. Bioresour. Technol. 2014, 172, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Kaewkannetra, P.; Enmak, P.; Chiu, T.Y. The effect of CO2 and salinity on the cultivation of Scenedesmus obliquus for bio-diesel production. Biotechnol. Bioprocess Eng. 2012, 17, 591–597. [Google Scholar] [CrossRef]
- Kaplan, E.; Sayar, N.A.; Kazan, D.; Sayar, A.A. Assessment of different carbon and salinity level on growth kinetics, lipid, and starch composition of Chlorella vulgaris SAG 211-12. Int. J. Green Energy 2020, 17, 290–300. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Nan, F.; Feng, J.; Lv, J.; Liu, Q.; Xie, S. Transcriptome analysis of the typical freshwater rhodophytes Sheathia arcuata grown under different light intensities. PLoS ONE 2018, 13, e0197729. [Google Scholar] [CrossRef]
- Lv, J.; Zhao, F.; Feng, J.; Liu, Q.; Nan., F.R.; Liu, Q.; Xie, S.L. Transcriptomic analysis reveals the mechanism on the response of Chlorococcum sp. GD to glucose concentration in mixotrophic cultivation. Bioresour. Technol. 2019, 288, 121568. [Google Scholar] [CrossRef]
- Shang, C.; Bi, G.; Qi, W.; Wang, Z.M.; Xie, J. Discovery of genes for production of biofuels through transcriptome sequencing of Dunaliella parva. Algal Res. 2016, 13, 318–326. [Google Scholar] [CrossRef]
- Gwak, Y.; Wang, Y.S.; Wang, B.B.; Kim, M.; Jeong, J.; Lee, C.G.; Hu, Q.; Han, D.X.; Jin, E.S. Comparative analyses of lipidomes and transcriptomes reveal a concerted action of multiple defensive systems against photooxidative stress in Haematococcus pluvialis. J. Exp. Bot. 2014, 65, 4317–4334. [Google Scholar] [CrossRef]
- Wang, H.; Gao, L.; Shao, H.; Zhou, W.; Liu, T. Lipid accumulation and metabolic analysis based on transcriptome se-quencing of filamentous oleaginous microalgae Tribonema minus at different growth phases. Bioprocess Biosyst. Eng. 2017, 40, 1327–1335. [Google Scholar] [CrossRef]
- Fang, L.; Sun, D.; Xu, Z.; He, J.; Qi, S.; Chen, X.; Chew, W.; Liu, J. Transcriptomic analysis of a moderately growing subisolate Botryococcus braunii 779 (Chlorophyta) in response to nitrogen deprivation. Biotechnol. Biofuels 2015, 8, 130. [Google Scholar] [CrossRef] [Green Version]
- Rismani-Yazdi, H.; Haznedaroglu, B.Z.; Bibby, K.; Peccia, J. Transcriptome sequencing and annotation of the microalgae Dunaliella tertiolecta: Pathway description and gene discovery for production of next-generation biofuels. BMC Genom. 2011, 12, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Rong, J.; Yang, H.; He, Q.; Zhang, D.; Hu, C. NaCl as an effective inducer for lipid accumulation in freshwater microalgae Desmodesmus abundans. Bioresour. Technol. 2014, 161, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.; Zhou, W.; Leksawasdi, N.; Liu, T. Mechanism and enhancement of lipid accumulation in filamentous oleaginous microalgae Tribonema minus under heterotrophic condition. Biotechnol. Biofuels 2018, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Singh, P. Effect of temperature and light on the growth of algae species: A review. Renew. Sustain. Energy Rev. 2015, 50, 431–444. [Google Scholar] [CrossRef]
- Pal, D.; Khozin-Goldberg, I.; Cohen, Z.; Boussiba, S. The effect of light, salinity, and nitrogen availability on lipid pro-duction by Nannochloropsis sp. Appl. Microbiol. Biotechnol. 2011, 90, 1429–1441. [Google Scholar] [CrossRef]
- Feng, J.; Guo, Y.; Zhang, X.; Wang, G.; Lv, J.; Liu, Q.; Xie, S. Identification and characterization of a symbiotic alga from soil bryophyte for lipid profiles. Biol. Open 2016, 5, 1317–1323. [Google Scholar] [CrossRef] [Green Version]
- Leyva, L.; Bashan, Y.; de-Bashan, L. Activity of acetyl-CoA carboxylase is not directly linkedto accumulation of lipids when Chlorella vulgaris is co-immobilised with Azospirillum brasilense in alginate under autotrophic and heterotrophic conditions. Ann. Microbiol. 2015, 65, 339–349. [Google Scholar] [CrossRef]
- Yoon, K.; Han, D.; Li, Y.; Sommerfeld, M.; Hu, Q. Phospholipid: Diacylglycerol Acyltransferase is a multifunctional en-zyme involved in membrane lipid turnover and degradation while synthesizing triacylglycerol in the unicellular green microalga Chlamydomonas reinhardtii. Plant Cell 2012, 24, 3708–3724. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.; Liang, Y.; Légeret, B.; Beyly-Adriano, A.; Blangy, S.; Haslam, R.P.; Napier, J.A.; Beisson, F.; Peltier, G.; Li-Beisson, Y. Chlamydomonas carries out fatty acid β-oxidation in ancestral peroxisomes using a bona fide acyl-CoA oxidase. Plant J. Cell Mol. Biol. 2017, 90, 358. [Google Scholar] [CrossRef] [Green Version]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410–424. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Number of Genes | Percentage (%) |
|---|---|---|
| Annotated in CDD | 16,377 | 31.07 |
| Annotated in KOG | 14,483 | 27.47 |
| Annotated in NR | 20,296 | 38.50 |
| Annotated in NT | 16,189 | 30.71 |
| Annotated in PFAM | 11,440 | 21.70 |
| Annotated in Swiss-Prot | 22,457 | 42.60 |
| Annotated in TrEMBL | 20,223 | 38.36 |
| Annotated in GO | 24,179 | 45.87 |
| Annotated in KEGG | 2690 | 5.10 |
| Annotated in at least one database | 30,964 | 58.74 |
| Annotated in all database | 1071 | 2.03 |
| Total genes | 52,715 | 100.00 |
| ID | Pathway Term | Gene Number | Rich Factor | Q-Value |
|---|---|---|---|---|
| ko03010 | Ribosome | 102 | 0.41 | 0.00000001 |
| ko00196 | Photosynthesis—antenna proteins | 12 | 0.52 | 0.138575956 |
| ko04010 | MAPK signaling pathway | 9 | 0.56 | 0.182751548 |
| ko00710 | Carbon fixation in photosynthetic organisms | 18 | 0.40 | 0.240032566 |
| ko04810 | Regulation of actin cytoskeleton | 12 | 0.41 | 0.279504446 |
| ko04510 | Focal adhesion | 6 | 0.60 | 0.279504446 |
| ko04015 | Rap1 signaling pathway | 5 | 0.62 | 0.279504446 |
| ko03008 | Ribosome biogenesis in eukaryotes | 26 | 0.33 | 0.279504446 |
| ko00830 | Retinol metabolism | 5 | 0.62 | 0.279504446 |
| ko00195 | Photosynthesis | 21 | 0.36 | 0.279504446 |
| ko04151 | PI3K-Akt signaling pathway | 14 | 0.38 | 0.358512098 |
| ko00980 | Metabolism of xenobiotics by cytochrome P450 | 7 | 0.47 | 0.375967376 |
| ko00270 | Cysteine and methionine metabolism | 18 | 0.35 | 0.375967376 |
| ko00010 | Glycolysis/Gluconeogenesis | 22 | 0.33 | 0.375967376 |
| ko01220 | Degradation of aromatic compounds | 3 | 0.75 | 0.398007486 |
| Enzyme | KEGG | Symbol | EC Number | Fold Change |
|---|---|---|---|---|
| Fatty acid biosynthesis (ko00061) | ||||
| acetyl-CoA carboxylase/biotin carboxylase 1 | K11262 | ACCase | 6.4.1.2 | −1.25 |
| 3-oxoacyl-[acyl-carrier-protein] synthase II | K09458 | KASII | 2.3.1.179 | −2.30 |
| 3-oxoacyl-[acyl-carrier protein] reductase | K00059 | KAR | 1.1.1.100 | −1.65 |
| 3-hydroxyacyl-dehydratase | K02372 | HAD | 4.2.1.59 | −2.19 |
| fatty acyl-ACP thioesterase A | K10782 | FATA | 3.1.2.14 | −2.01 |
| fatty acid elongation (ko00062) | ||||
| palmitoyl-protein thioesterase | K01074 | PPT | 3.1.2.22 | −1.10 |
| mitochondrial enoyl—[acyl-carrier protein] reductase/trans-2-enoyl-CoA | K07512 | TER | 1.3.1.38 | 6.74 |
| unsaturated fatty acid biosynthesis (ko01040) | ||||
| stearoyl-CoA desaturase (Delta-9 desaturase) | K00507 | Δ9-SCD | 1.14.19.1 | 5.30 |
| omega-6 fatty acid desaturase | K10256 | Δ12-FAD | 1.14.19.6 | −1.70 |
| fatty acid degradation (ko00071) | ||||
| S-(hydroxymethyl)glutathione dehydrogenase/alcohol dehydrogenase | K00121 | ADH5 | 1.1.1.284 | 9.01 |
| aldehyde dehydrogenase (NAD+) | K00128 | ALDH | 1.2.1.3 | 3.76 |
| aldehyde dehydrogenase family 7 member A1 | K14085 | ALDH7a1 | 1.2.1.31 | 5.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, H.; Feng, J.; Lv, J.; Liu, Q.; Nan, F.; Liu, X.; Xie, S. Molecular Mechanism of Lipid Accumulation and Metabolism of Oleaginous Chlorococcum sphacosum GD from Soil under Salt Stress. Int. J. Mol. Sci. 2021, 22, 1304. https://doi.org/10.3390/ijms22031304
Su H, Feng J, Lv J, Liu Q, Nan F, Liu X, Xie S. Molecular Mechanism of Lipid Accumulation and Metabolism of Oleaginous Chlorococcum sphacosum GD from Soil under Salt Stress. International Journal of Molecular Sciences. 2021; 22(3):1304. https://doi.org/10.3390/ijms22031304
Chicago/Turabian StyleSu, Hang, Jia Feng, Junping Lv, Qi Liu, Fangru Nan, Xudong Liu, and Shulian Xie. 2021. "Molecular Mechanism of Lipid Accumulation and Metabolism of Oleaginous Chlorococcum sphacosum GD from Soil under Salt Stress" International Journal of Molecular Sciences 22, no. 3: 1304. https://doi.org/10.3390/ijms22031304
APA StyleSu, H., Feng, J., Lv, J., Liu, Q., Nan, F., Liu, X., & Xie, S. (2021). Molecular Mechanism of Lipid Accumulation and Metabolism of Oleaginous Chlorococcum sphacosum GD from Soil under Salt Stress. International Journal of Molecular Sciences, 22(3), 1304. https://doi.org/10.3390/ijms22031304