
The Cardiovascular Phenotype in Fabry Disease: New Findings in the Research Field



Abstract
:1. Introduction
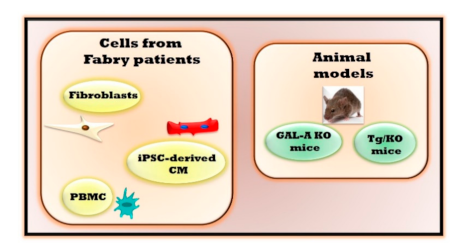
2. The Experimental Models of Fabry Disease
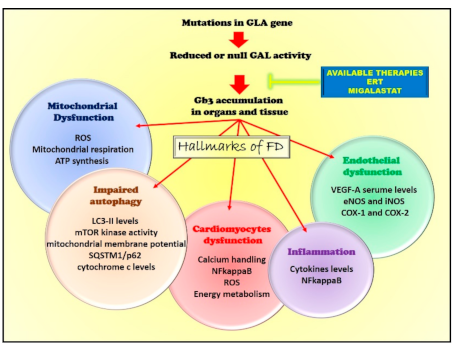
3. Pathogenetic Mechanisms
3.1. Endothelial Dysfunction
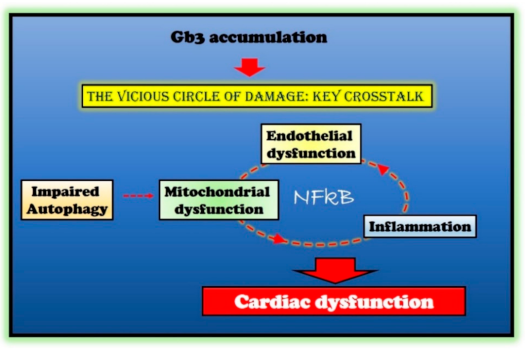
3.2. Impairment of Mitochondrial Quality Control
Alterations of Autophagy
3.3. Mitochondrial Dysfunction
3.4. The Inflammatory Phenotype
4. The Cardiac Phenotype
4.1. The Cardiac Variant
4.2. Molecular Mechanisms of Cardiac Dysfunction
5. Available and Potential Therapeutic Interventions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Germain, D. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Boutin, M.; Menkovic, I.; Martineau, T.; Vaillancourt-Lavigueur, V.; Toupin, A.; Auray-Blais, C. Separation and Analysis of Lactosylceramide, Galabiosylceramide, and Globotriaosylceramide by LC-MS/MS in Urine of Fabry Disease Patients. Anal. Chem. 2017, 89, 13382–13390. [Google Scholar] [CrossRef]
- Young, E.; Mills, K.; Morris, P.; Vellodi, A.; Lee, P.; Waldek, S.; Winchester, B. Is globotriaosylceramide a useful biomarker in Fabry disease? Acta Paediatr. Suppl. 2005, 94, 51–54. [Google Scholar]
- Young-Gqamana, B.; Brignol, N.; Chang, H.-H.; Khanna, R.; Soska, R.; Fuller, M.; Sitaraman, S.A.; Germain, D.P.; Giugliani, R.; Hughes, D.A.; et al. Migalastat HCl Reduces Globotriaosylsphingosine (Lyso-Gb3) in Fabry Transgenic Mice and in the Plasma of Fabry Patients. PLoS ONE 2013, 8, e57631. [Google Scholar] [CrossRef] [Green Version]
- Deegan, P.B.; Baehner, A.F.; Romero, M.-Á.B.; Hughes, D.A.; Kampmann, C.; Beck, M. Natural history of Fabry disease in females in the Fabry Outcome Survey. J. Med. Genet. 2006, 43, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Chevrier, M.; Brakch, N.; Celine, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Annie Laquerriere, A.L.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar]
- Das, A.M.; Naim, H. Chapter 3 Biochemical Basis of Fabry Disease with Emphasis on Mitochondrial Function and Protein Trafficking. Adv. Clin. Chem. 2009, 49, 57–71. [Google Scholar] [CrossRef]
- Lucke, T.; Hoppner, W.; Schmidt, E.; Illsinger, S.; Das, A.M. Fabry disease: Reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar]
- De Francesco, P.N.; Mucci, J.M.; Ceci, R.; Fossati, C.A.; Rozenfeld, P.A. Fabry disease peripheral blood immune cells release inflammatory cytokines: Role of globotriaosylceramide. Mol. Genet. Metab. 2013, 109, 93–99. [Google Scholar] [CrossRef]
- Ivanova, M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE 2019, 14, e0210617. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.J.; Ribeiro, D.; Santos, R.; Moreira, L.; Bragança, J.; Amaral, O. Induced pluripotent stem cell line (INSAi002-A) from a Fabry Disease patient hemizygote for the rare p.W287X mutation. Stem Cell Res. 2020, 45, 101794. [Google Scholar] [CrossRef]
- Itier, J.M.; Ret, G.; Viale, S.; Sweet, L.; Bangari, D.; Caron, A.; Le-Gall, F.; Benichou, B.; Leonard, J.; Deleuze, J.F.; et al. Effective clearance of GL-3 in a human iPSC-derived cardiomyocyte model of Fabry disease. J. Inherit. Metab. Dis. 2014, 37, 1013–1022. [Google Scholar]
- Ohshima, T.; Murray, G.J.; Swaim, W.D.; Longenecker, G.; Quirk, J.M.; Cardarelli, C.O.; Sugimoto, Y.; Pastan, I.; Gottesman, M.M.; Brady, R.O.; et al. alpha-Galactosidase A deficient mice: A model of Fabry disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2540–2544. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, Y.A.; Zeidner, K.M.; Gordon, R.E.; Desnick, R.J. Fabry disease: Preclinical studies demonstrate the effectiveness of alpha-galactosidase A replacement in enzyme-deficient mice. Am. J. Hum. Genet. 2001, 68, 14–25. [Google Scholar]
- Yoshimitsu, M.; Higuchi, K.; Ramsubir, S.; Nonaka, T.; Rasaiah, V.I.; Siatskas, C.; Liang, S.B.; Murray, G.J.; Brady, R.O.; Medin, J.A. Efficient correction of Fabry mice and patient cells mediated by lentiviral transduction of hematopoietic stem/progenitor cells. Gene Ther. 2007, 14, 256–265. [Google Scholar]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar]
- Germain, D.; Poenaru, L. Fabry Disease: Identification of Novel Alpha-Galactosidase A Mutations and Molecular Carrier Detection by Use of Fluorescent Chemical Cleavage of Mismatches. Biochem. Biophys. Res. Commun. 1999, 257, 708–713. [Google Scholar] [CrossRef]
- Khanna, R.; Soska, R.; Lun, Y.; Feng, J.; Frascella, M.; Young, B.; Brignol, N.; Pellegrino, L.; Sitaraman, S.A.; Desnick, R.J.; et al. The Pharmacological Chaperone 1-Deoxygalactonojirimycin Reduces Tissue Globotriaosylceramide Levels in a Mouse Model of Fabry Disease. Mol. Ther. 2010, 18, 23–33. [Google Scholar] [CrossRef]
- Gal, A.; Schafer, E.; Rohard, I. The genetic basis of Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford Press: Oxford, UK, 2006. [Google Scholar]
- Mehta, A.; Clarke, J.T.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G.; Investigators, F.O.S. Natural course of Fabry disease: Changing pattern of causes of death in FOS—Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552. [Google Scholar] [CrossRef] [Green Version]
- Lenders, M.; Weidemann, F.; Kurschat, C.; Canaan-Kühl, S.; Duning, T.; Stypmann, J.; Schmitz, B.; Reiermann, S.; Krämer, J.; Blaschke, D.; et al. Alpha-Galactosidase A p.A143T, a non-Fabry disease-causing variant. Orphanet J. Rare Dis. 2016, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- Cerkauskaite, A.; Cerkauskiene, R.; Miglinas, M.; Laurinavicius, A.; Ding, C.; Rolfs, A.; Venceviciene, L.; Barysiene, J.; Kazenaite, E.; Sadauskiene, E. Genotype(-)Phenotype Correlation in a New Fabry-Disease-Causing Mutation. Medicina 2019, 55, 122. [Google Scholar]
- Militaru, S.; Saftoiu, A.; Streubel, B.; Jurcut, R. New Fabry disease mutation confirms cardiomyopathy aetiology: A case report. Eur. Heart J. Case Rep. 2018, 2, yty133–yty135. [Google Scholar] [CrossRef]
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Robles, A.R.; Costa, M.A.; Pereira, O.; Vide, A.T.; Branco, G.C.; Simões, S.; Guimarães, M.J.; et al. Natural history of the late-onset phenotype of Fabry disease due to the p.F113L mutation. Mol. Genet. Metab. Rep. 2020, 22, 100565. [Google Scholar] [CrossRef]
- Germain, D.; Brand, E.; Burlina, A.; Cecchi, F.; Garman, S.C.; Kempf, J.; Laney, D.A.; Linhart, A.; Maródi, L.; Nicholls, K.; et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: A multicenter Fabry Registry study. Mol. Genet. Genom. Med. 2018, 6, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Ries, M.; Gal, A. Genotype-phenotype correlation in Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; NCBI: Bethesda, MD, USA, 2006. [Google Scholar]
- Hassan, S.; Sidransky, E.; Tayebi, N. The role of epigenetics in lysosomal storage disorders: Uncharted territory. Mol. Genet. Metab. 2017, 122, 10–18. [Google Scholar] [CrossRef]
- Altarescu, G.; Moore, D.F.; Schiffmann, R. Effect of genetic modifiers on cerebral lesions in Fabry disease. Neurology 2005, 64, 2148–2150. [Google Scholar]
- Rombach, S.M.; Bogaard, B.V.D.; De Groot, E.; Groener, J.E.; Poorthuis, B.J.; Linthorst, G.E.; Born, B.-J.H.V.D.; Hollak, C.E.; Aerts, J.M. Vascular Aspects of Fabry Disease in Relation to Clinical Manifestations and Elevations in Plasma Globotriaosylsphingosine. Hypertension 2012, 60, 998–1005. [Google Scholar] [CrossRef] [Green Version]
- Loso, J.; Lund, N.; Avanesov, M.; Muschol, N.; Lezius, S.; Cordts, K.; Schwedhelm, E.; Patten, M. Serum Biomarkers of Endothelial Dysfunction in Fabry Associated Cardiomyopathy. Front. Cardiovasc. Med. 2018, 5, 108. [Google Scholar] [CrossRef]
- Zampetti, A.; Gnarra, M.; Borsini, W.; Giurdanella, F.; Antuzzi, D.; Piras, A.; Smaldone, C.; Pieroni, M.; Cadeddu, C.; De Waure, C.; et al. Vascular Endothelial Growth Factor (VEGF-a) in Fabry disease: Association with cutaneous and systemic manifestations with vascular involvement. Cytokine 2013, 61, 933–939. [Google Scholar] [CrossRef]
- Shen, J.-S.; Meng, X.-L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Namdar, M.; Gebhard, C.; Studiger, R.; Shi, Y.; Mocharla, P.; Schmied, C.; Brugada, P.; Luscher, T.F.; Camici, G.G. Globotriaosylsphingosine accumulation and not alpha-galactosidase-A deficiency causes endothelial dysfunction in Fabry disease. PLoS ONE 2012, 7, e36373. [Google Scholar]
- Rombach, S.M.; Twickler, T.; Aerts, J.M.F.G.; Linthorst, G.E.; Wijburg, F.A.; Hollak, C.E.M. Vasculopathy in patients with Fabry disease: Current controversies and research directions. Mol. Genet. Metab. 2010, 99, 99–108. [Google Scholar] [CrossRef]
- Machann, W.; Breunig, F.; Weidemann, F.; Sandstede, J.; Hahn, D.; Köstler, H.; Neubauer, S.; Wanner, C.; Beer, M. Cardiac energy metabolism is disturbed in Fabry disease and improves with enzyme replacement therapy using recombinant human galactosidase A. Eur. J. Heart Fail. 2011, 13, 278–283. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, M. Altered Sphingolipids Metabolism Damaged Mitochondrial Functions: Lessons Learned From Gaucher and Fabry Diseases. J. Clin. Med. 2020, 9, 1116. [Google Scholar] [CrossRef] [Green Version]
- Liebau, M.C.; Braun, F.; Höpker, K.; Weitbrecht, C.; Bartels, V.; Müller, R.-U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated Autophagy Contributes to Podocyte Damage in Fabry’s Disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef] [Green Version]
- De La Mata, M.; Cotán, D.; Villanueva-Paz, M.; De Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Suárez-Rivero, J.M.; Tiscornia, G.; Oropesa-Ávila, M. Mitochondrial Dysfunction in Lysosomal Storage Disorders. Diseases 2016, 4, 31. [Google Scholar] [CrossRef] [Green Version]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef]
- Song, H.-Y.; Chien, C.-S.; Yarmishyn, A.A.; Chou, S.-J.; Yang, Y.-P.; Wang, M.-L.; Leu, H.-B.; Yu, W.-C.; Chang, Y.-L.; Chiou, S.-H. Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy. Cells 2019, 8, 327. [Google Scholar] [CrossRef] [Green Version]
- Strasberg, P.M.; Callahan, J.W. Lysosphingolipids and mitochondrial function. II. Deleterious effects of sphingosylphosphorylcholine. Biochem. Cell Biol. 1988, 66, 1322–1332. [Google Scholar]
- Hsing, L.C.; Rudensky, A.Y. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol. Rev. 2005, 207, 229–241. [Google Scholar] [CrossRef]
- Schmid, D.; Münz, C. Immune surveillance of intracellular pathogens via autophagy. Cell Death Differ. 2005, 12, 1519–1527. [Google Scholar] [CrossRef]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef]
- Park, E.S.; Choi, J.O.; Park, J.W.; Lee, M.H.; Park, H.Y.; Jung, S.C. Expression of genes and their responses to enzyme replacement therapy in a Fabry disease mouse model. Int. J. Mol. Med. 2009, 24, 401–407. [Google Scholar]
- Lee, M.H.; Choi, E.N.; Jeon, Y.J.; Jung, S.C. Possible role of transforming growth factor-beta1 and vascular endothelial growth factor in Fabry disease nephropathy. Int. J. Mol. Med. 2012, 30, 1275–1280. [Google Scholar]
- Linhart, A.; Paleček, T.; Bultas, J.; Ferguson, J.J.; Hrudová, J.; Karetová, D.; Zeman, J.; Ledvinová, J.; Poupětová, H.; Elleder, M.; et al. New insights in cardiac structural changes in patients with Fabry’s disease. Am. Heart J. 2000, 139, 1101–1108. [Google Scholar] [CrossRef]
- Linhart, A. The heart in Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; NCBI: Bethesda, MD, USA, 2006. [Google Scholar]
- Yenercag, M.; Arslan, U. Tp-e interval and Tp-e/QT ratio and their association with left ventricular diastolic dysfunction in Fabry disease without left ventricular hypertrophy. J. Electrocardiol. 2020, 59, 20–24. [Google Scholar]
- Kalliokoski, R.J.; Kalliokoski, K.; Sundell, J.; Engblom, E.; Penttinen, M.; Kantola, I.; Raitakari, O.; Knuuti, J.; Nuutila, P. Impaired myocardial perfusion reserve but preserved peripheral endothelial function in patients with Fabry disease. J. Inherit. Metab. Dis. 2005, 28, 563–573. [Google Scholar] [CrossRef]
- Pochis, W.T.; Litzow, J.T.; King, B.G.; Kenny, D. Electrophysiologic findings in Fabry’s disease with a short PR interval. Am. J. Cardiol. 1994, 74, 203–204. [Google Scholar]
- Shah, J.S.; Hughes, D.A.; Sachdev, B.; Tome, M.; Ward, D.; Lee, P.; Mehta, A.B.; Elliott, P.M. Prevalence and Clinical Significance of Cardiac Arrhythmia in Anderson-Fabry Disease. Am. J. Cardiol. 2005, 96, 842–846. [Google Scholar] [CrossRef]
- Shanks, M.; Thompson, R.B.; Paterson, I.D.; Putko, B.; Khan, A.; Chan, A.; Becher, H.; Oudit, G.Y. Systolic and Diastolic Function Assessment in Fabry Disease Patients Using Speckle-Tracking Imaging and Comparison with Conventional Echocardiographic Measurements. J. Am. Soc. Echocardiogr. 2013, 26, 1407–1414. [Google Scholar] [CrossRef]
- Linhart, A.; Lubanda, J.-C.; Palecek, T.; Bultas, J.; Karetová, D.; Ledvinová, J.; Elleder, M.; Aschermann, M. Cardiac manifestations in Fabry disease. J. Inherit. Metab. Dis. 2001, 24, 75–83. [Google Scholar] [CrossRef]
- Nordin, S.; Kozor, R.; Medina-Menacho, K.; Abdel-Gadir, A.; Baig, S.; Sado, D.M.; Lobascio, I.; Murphy, E.; Lachmann, R.H.; Mehta, A.; et al. Proposed Stages of Myocardial Phenotype Development in Fabry Disease. JACC Cardiovasc. Imaging 2019, 12, 1673–1683. [Google Scholar] [CrossRef]
- Csanyi, B.; Hategan, L.; Nagy, V.; Obal, I.; Varga, E.T.; Borbas, J.; Tringer, A.; Eichler, S.; Forster, T.; Rolfs, A.; et al. Identification of a Novel GLA Gene Mutation, p.Ile239Met, in Fabry Disease With a Predominant Cardiac Phenotype. Int. Heart J. 2017, 58, 454–458. [Google Scholar]
- Ogawa, K.; Sugamata, K.; Funamoto, N.; Abe, T.; Sato, T.; Nagashima, K.; Ohkawa, S. Restricted accumulation of globotriaosylceramide in the hearts of atypical cases of Fabry’s disease. Hum. Pathol. 1990, 21, 1067–1073. [Google Scholar]
- Ishii, S.; Nakao, S.; Minamikawa-Tachino, R.; Desnick, R.J.; Fan, J.-Q. Alternative Splicing in the α-Galactosidase A Gene: Increased Exon Inclusion Results in the Fabry Cardiac Phenotype. Am. J. Hum. Genet. 2002, 70, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Onishi, R.; Kanaoka, K.; Sugiura, J.; Tokunaga, M.; Takemoto, Y.; Onoue, K.; Yamamoto, Y.; Horii, M.; Saito, Y. A Cardiac Variant of Fabry Disease Diagnosed with Chance Urinary Mulberry Cells. Intern. Med. 2018, 57, 3385–3388. [Google Scholar] [CrossRef] [Green Version]
- Kuramoto, Y.; Naito, A.T.; Tojo, H.; Sakai, T.; Ito, M.; Shibamoto, M.; Nakagawa, A.; Higo, T.; Okada, K.; Yamaguchi, T.; et al. Generation of Fabry cardiomyopathy model for drug screening using induced pluripotent stem cell-derived cardiomyocytes from a female Fabry patient. J. Mol. Cell. Cardiol. 2018, 121, 256–265. [Google Scholar] [CrossRef]
- Birket, M.J.; Raibaud, S.; Lettieri, M.; Adamson, A.D.; Letang, V.; Cervello, P.; Redon, N.; Ret, G.; Viale, S.; Wang, B.; et al. A Human Stem Cell Model of Fabry Disease Implicates LIMP-2 Accumulation in Cardiomyocyte Pathology. Stem Cell Rep. 2019, 13, 380–393. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Shen, J.; Kong, M.; Brady, R.; Li, R.A.; Schiffmann, R. Abnormal intracellular calcium handling: A key pathogenic and therapeutic target of the cardiac manifestations in Fabry disease. Mol. Genet. Metab. 2014, 111, S77. [Google Scholar] [CrossRef]
- Ravarotto, V.; Simioni, F.; Carraro, G.; Bertoldi, G.; Pagnin, E.; Calò, L.A. Oxidative Stress and Cardiovascular-Renal Damage in Fabry Disease: Is There Room for a Pathophysiological Involvement? J. Clin. Med. 2018, 7, 409. [Google Scholar] [CrossRef] [Green Version]
- Ravarotto, V.; Carraro, G.; Pagnin, E.; Bertoldi, G.; Simioni, F.; Maiolino, G.; Martinato, M.; Landini, L.; Davis, P.A.; Calò, L.A. Oxidative stress and the altered reaction to it in Fabry disease: A possible target for cardiovascular-renal remodeling? PLoS ONE 2018, 13, e0204618. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar]
- Ciccarelli, M.; Sorriento, D.; Fiordelisi, A.; Gambardella, J.; Franco, A.; Del Giudice, C.; Sala, M.; Monti, M.G.; Bertamino, A.; Campiglia, P.; et al. Pharmacological inhibition of GRK2 improves cardiac metabolism and function in experimental heart failure. ESC Heart Fail. 2020, 7, 1571–1584. [Google Scholar] [CrossRef]
- Sorriento, D.; Ciccarelli, M.; Cipolletta, E.; Trimarco, B.; Iaccarino, G. “Freeze, Don’t Move”: How to Arrest a Suspect in Heart Failure—A Review on Available GRK2 Inhibitors. Front. Cardiovasc. Med. 2016, 3, 48. [Google Scholar]
- Sorriento, D.; Franco, A.; Rusciano, M.R.; Maione, A.S.; Soprano, M.; Illario, M.; Iaccarino, G.; Ciccarelli, M. Good at Heart: Preserving Cardiac Metabolism during aging. Curr. Diabetes Rev. 2015, 12, 90–99. [Google Scholar] [CrossRef]
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated Inflammatory Plasma Biomarkers in Patients With Fabry Disease: A Critical Link to Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e009098. [Google Scholar] [CrossRef] [Green Version]
- Frustaci, A.; Verardo, R.; Grande, C.; Galea, N.; Piselli, P.; Carbone, I.; Alfarano, M.; Russo, M.A.; Chimenti, C. Immune-Mediated Myocarditis in Fabry Disease Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e009052. [Google Scholar]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef] [Green Version]
- Sorriento, D.; Iaccarino, G. Inflammation and Cardiovascular Diseases: The Most Recent Findings. Int. J. Mol. Sci. 2019, 20, 3879. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-Garcia, C.; Valcarcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar]
- Sorriento, D.; Ciccarelli, M.; Santulli, G.; Campanile, A.; Altobelli, G.G.; Cimini, V.; Galasso, G.; Astone, D.; Piscione, F.; Pastore, L.; et al. The G-protein-coupled receptor kinase 5 inhibits NFkappaB transcriptional activity by inducing nuclear accumulation of IkappaB alpha. Proc. Natl. Acad. Sci. USA 2008, 105, 17818–17823. [Google Scholar]
- Sorriento, D.; Santulli, G.; Fusco, A.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertension 2010, 56, 696–704. [Google Scholar]
- Vedder, A.C.; Linthorst, G.E.; Houge, G.; Groener, J.E.; Ormel, E.E.; Bouma, B.J.; Aerts, J.M.; Hirth, A.; Hollak, C.E.M. Treatment of Fabry Disease: Outcome of a Comparative Trial with Agalsidase Alfa or Beta at a Dose of 0.2 mg/kg. PLoS ONE 2007, 2, e598. [Google Scholar] [CrossRef] [Green Version]
- Lenders, M.; Brand, E. Effects of Enzyme Replacement Therapy and Antidrug Antibodies in Patients with Fabry Disease. J. Am. Soc. Nephrol. 2018, 29, 2265–2278. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef]
- McCafferty, E.H.; Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 2019, 79, 543–554. [Google Scholar]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomized phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar]
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in Fabry disease: 30-month results from the open-label extension of the randomized, phase 3 ATTRACT study. Mol. Genet. Metab. 2020, 131, 219–228. [Google Scholar] [CrossRef]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Muntze, J.; Uceyler, N.; et al. Treatment of Fabry’s Disease With Migalastat: Outcome From a Prospective Observational Multicenter Study (FAMOUS). Clin. Pharmacol. Ther. 2020, 108, 326–337. [Google Scholar]
- Riccio, E.; AFFIINITY Group; Zanfardino, M.; Ferreri, L.; Santoro, C.; Cocozza, S.; Capuano, I.; Imbriaco, M.; Feriozzi, S.; Pisani, A. Switch from enzyme replacement therapy to oral chaperone migalastat for treating fabry disease: Real-life data. Eur. J. Hum. Genet. 2020, 28, 1662–1668. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| In Vivo Experimental Models | Main Features | Advantages | Limitations |
| Fibroblasts derived from Fabry patients | Alterations of autophagy [6], mitochondrial respiration and energy metabolism [7,8] | Optimal model to evaluate molecular mechanisms involved in cardiac fibrosis. | The in vitro model lacks a physiological relevance preventing the evaluations of different cells crosstalk that could interfere in the pathogenetic mechanism. |
| Induced pluripotent stem cells -derived cardiomyocytes (iPSC-derived CM) | Accumulation of Gb3 [12] Increased excitability with altered electrophysiology and calcium handling [12], accumulation of LIMP-2 cathepsin F and HSPA2/HSP70-2 [11] | Optimal model for evaluating cardiac alterations, cardiac safety and efficacy for evolving drugs. | |
| Peripheral blood mononuclear cells (PBMC) | Impaired mitochondrial function [10], inflammatory cytokines and regulators [9] | Alterations in PBMC can reflect the same ones in the heart | |
| In Vivo Experimental Models | Main Features | Advantages | Limitations |
| Mouse with deletion of endogenous galactosidase alpha gene (GAL-A KO) | Gb3 accumulation in kidneys, heart, and liver [13] | Optimal model to evaluate the effects of drugs on restoring GAL activity | No organ damage |
| Mouse lacking endogenous GLA but expressing a human R301Q GLA transgene transcriptionally regulated by the human GLA promoter (Tg/KO) | Age-dependent Gb3 accumulation in disease-relevant tissues [18] | No data are available yet to define the effectiveness and advantages of this experimental model | This model could reproduce only the clinical signs of Fabry patients which are associated with R301Q specific mutation. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorriento, D.; Iaccarino, G. The Cardiovascular Phenotype in Fabry Disease: New Findings in the Research Field. Int. J. Mol. Sci. 2021, 22, 1331. https://doi.org/10.3390/ijms22031331
Sorriento D, Iaccarino G. The Cardiovascular Phenotype in Fabry Disease: New Findings in the Research Field. International Journal of Molecular Sciences. 2021; 22(3):1331. https://doi.org/10.3390/ijms22031331
Chicago/Turabian StyleSorriento, Daniela, and Guido Iaccarino. 2021. "The Cardiovascular Phenotype in Fabry Disease: New Findings in the Research Field" International Journal of Molecular Sciences 22, no. 3: 1331. https://doi.org/10.3390/ijms22031331
APA StyleSorriento, D., & Iaccarino, G. (2021). The Cardiovascular Phenotype in Fabry Disease: New Findings in the Research Field. International Journal of Molecular Sciences, 22(3), 1331. https://doi.org/10.3390/ijms22031331