
KV1.5–KVβ1.3 Recycling Is PKC-Dependent

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
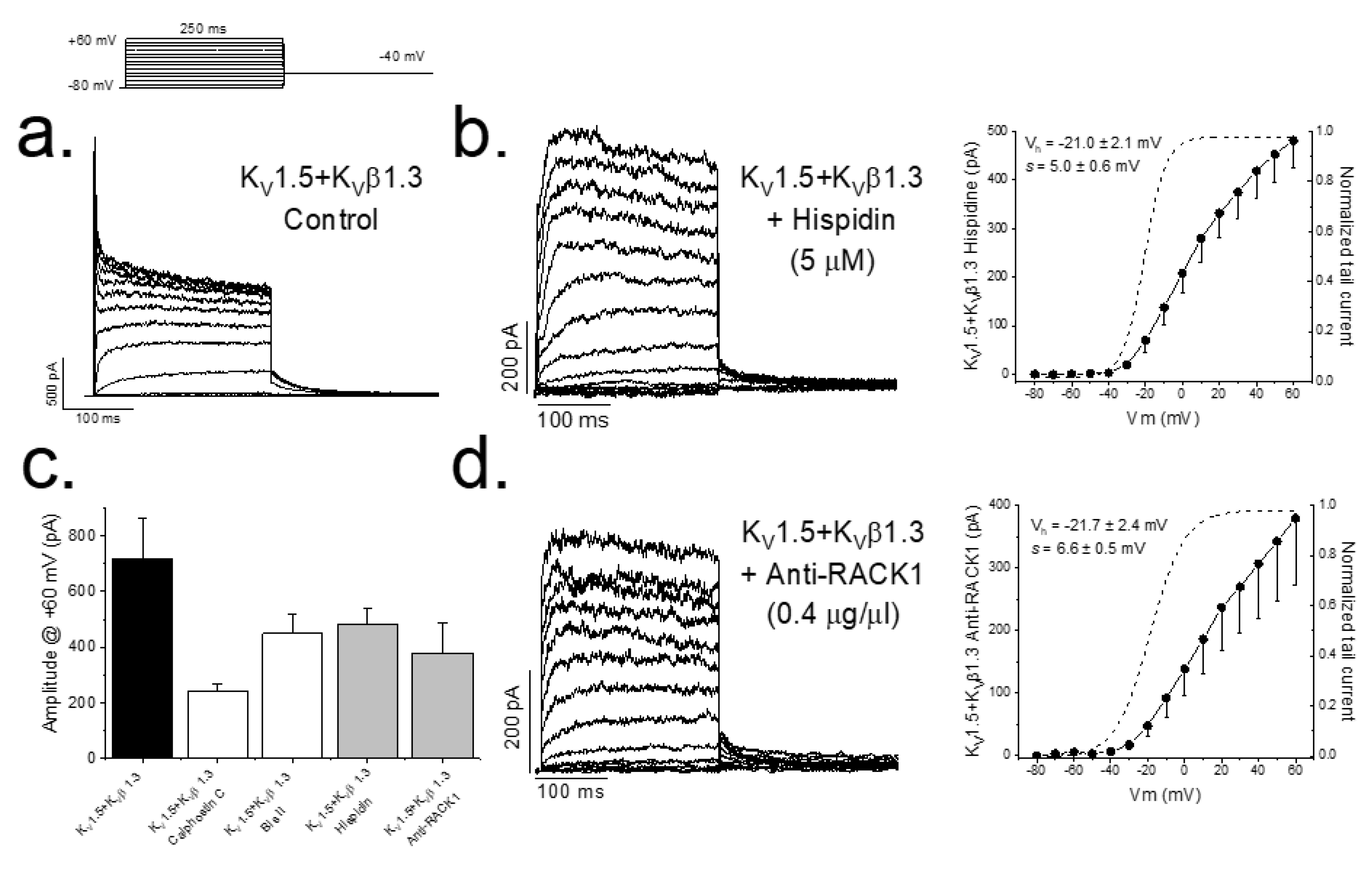
2. Results
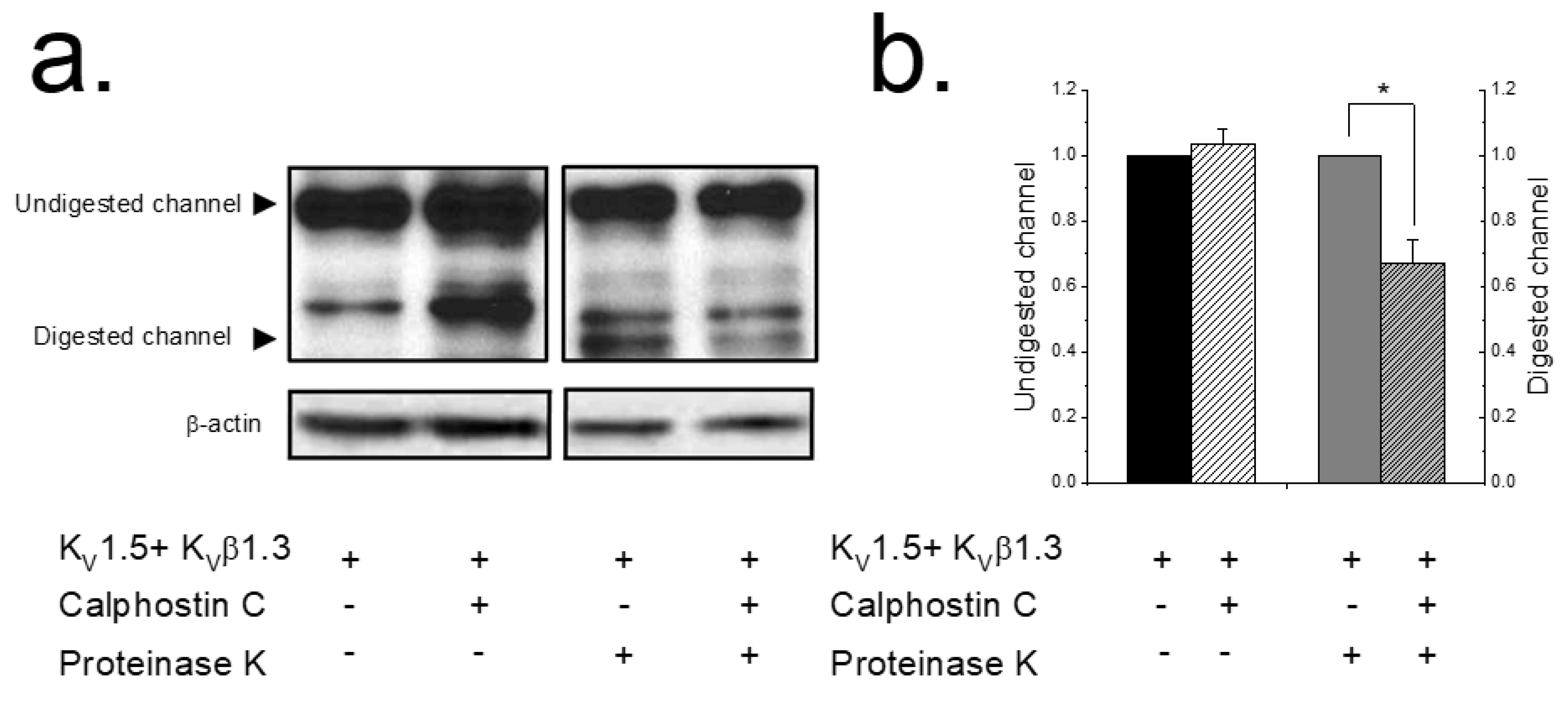
2.1. PKC Inhibition Reduces the Surface Amount of KV1.5 Channel
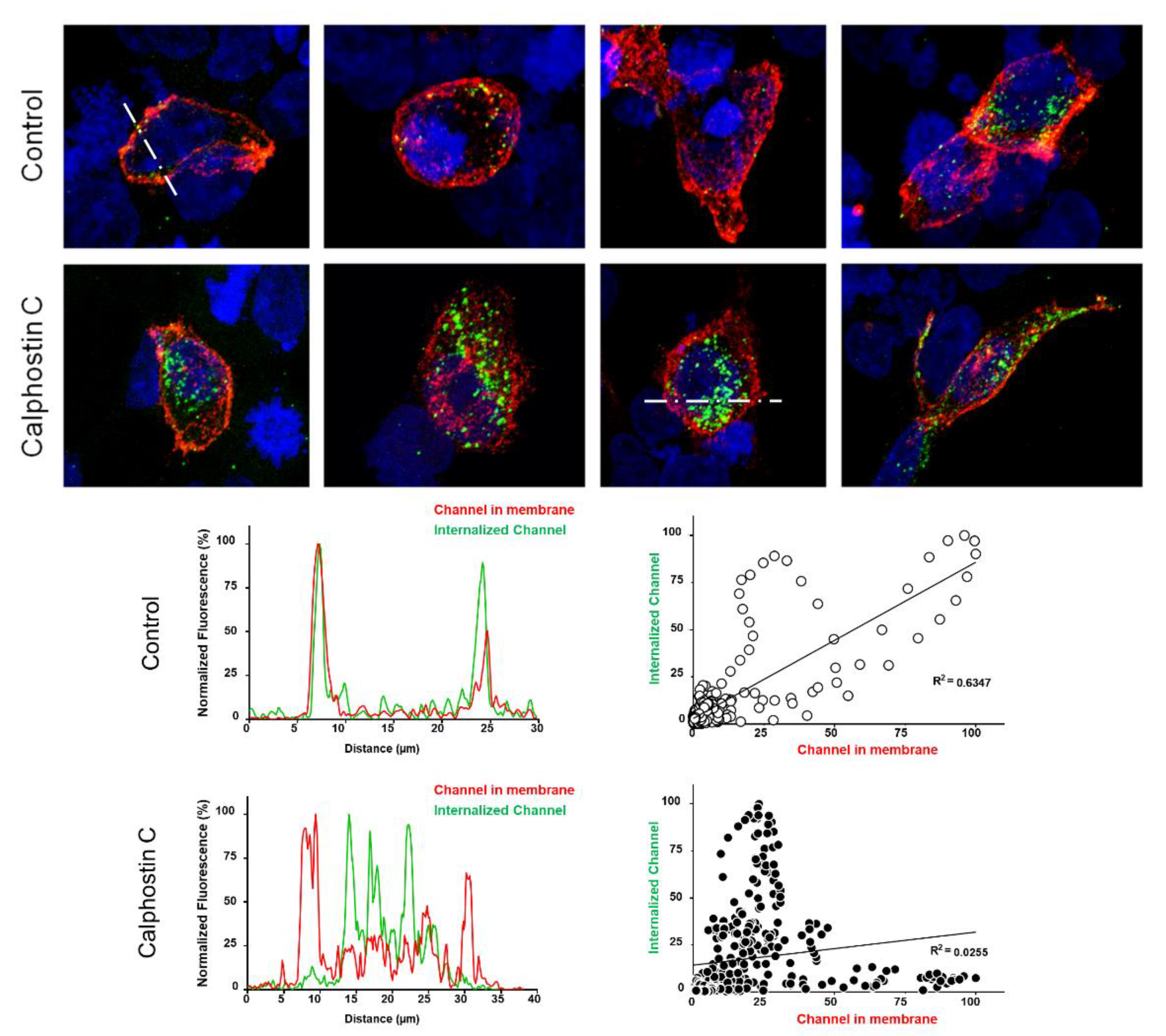
2.2. PKC Inhibition Increases the Internalized KV1.5–KVβ1.3 Channels
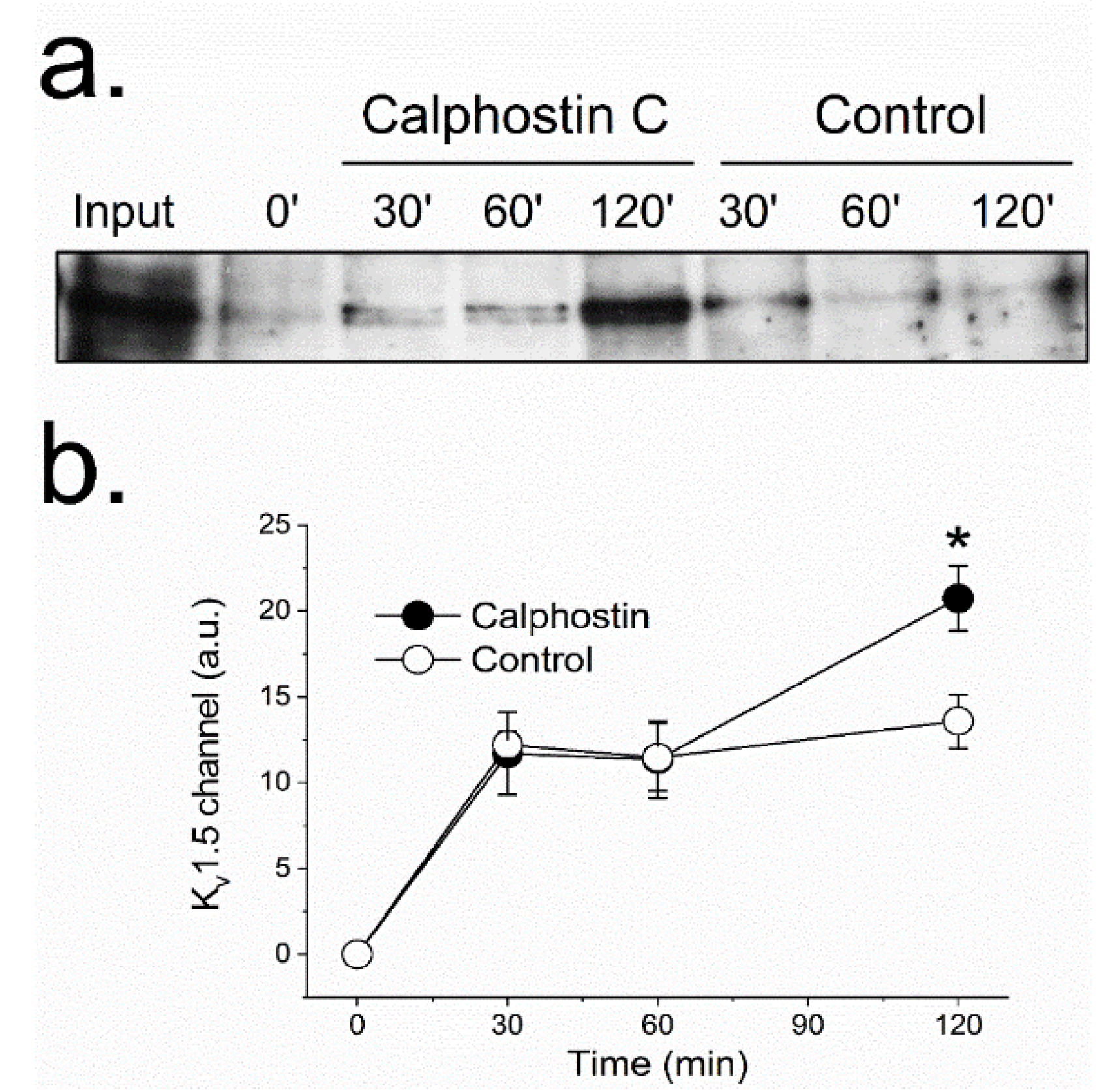
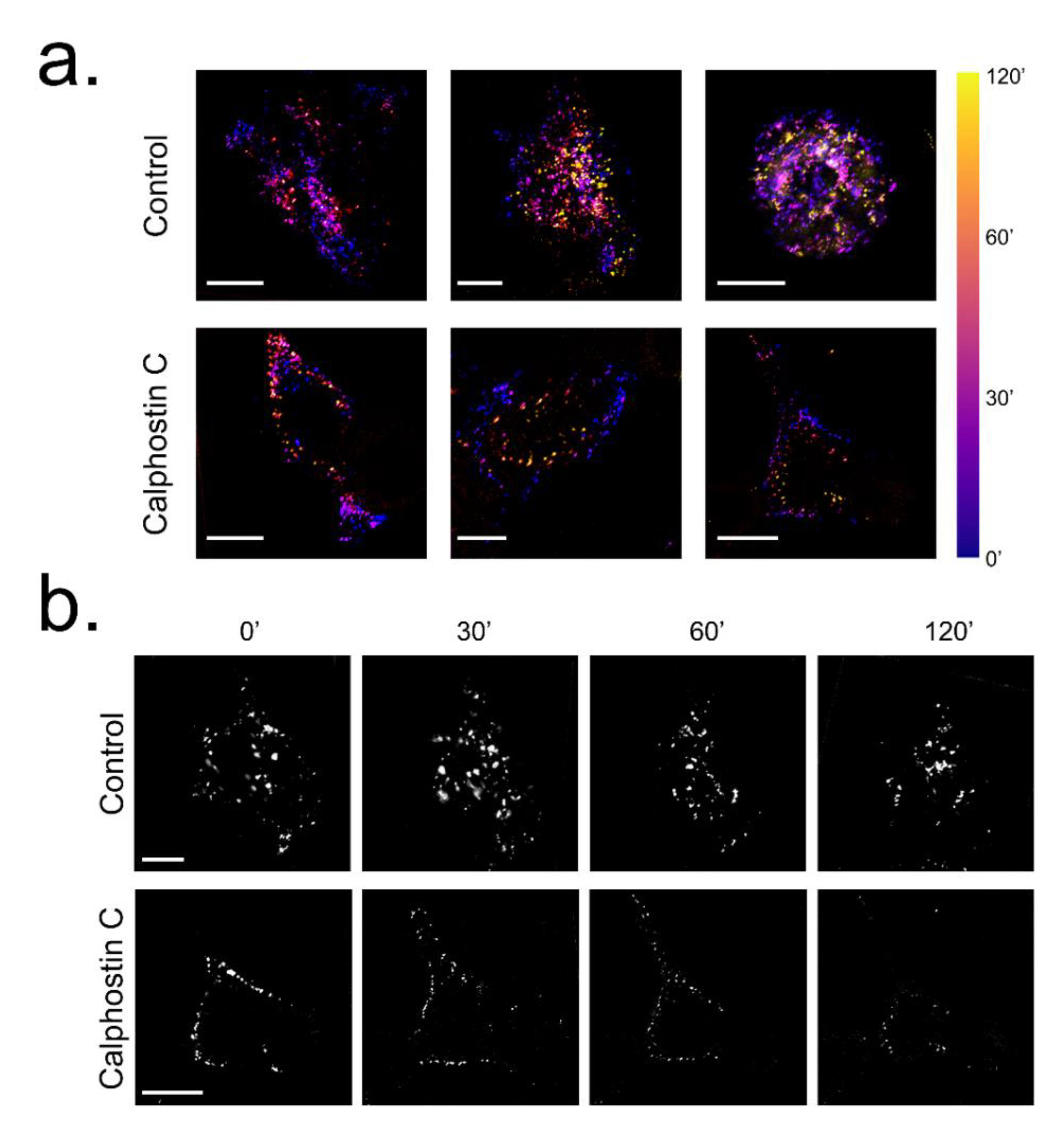
2.3. Inhibition of PKC Disrupts KV1.5–KVβ1.3 Channel Dynamics
3. Discussion
4. Materials and Methods
4.1. Plasmids, Cell Culture and Transfection
4.2. Internalization Assays by Biotinylation
4.3. Internalization Assays by Immunocytochemistry
4.4. Proteinase K Digestion Assay: Analysis of Cell Surface Protein
4.5. Electrophysiological Recordings and Data Acquisition
4.6. Live Cell Imaging
4.7. Drugs
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PKC | Protein kinase C |
| RACK1 | Receptor of activated protein C kinase 1 |
| HA | Hemagglutinin |
| PMA | Phorbol 12-myristate 13-acetate |
References
- Fedida, D.; Wible, B.; Wang, Z.; Fermini, B.; Faust, F.; Nattel, S.; Brown, A.M. Identity of a novel delayed rectifier current from human heart with a cloned K + channel current. Circ. Res. 1993, 73, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Snyders, D.J.; Tamkun, M.M.; Bennett, P.B. A rapidly activating and slowly inactivating potassium channel cloned from human heart: Functional analysis after stable mammalian cell culture expression. J. Gen. Physiol. 1993, 101, 513–543. [Google Scholar] [CrossRef]
- Uebele, V.N.; England, S.K.; Chaudhary, A.; Tamkun, M.M.; Snyders, D.J. Functional differences in Kv1.5 currents expressed in mammalian cell lines are due to the presence of endogenous Kvβ2.1 subunits. J. Biol. Chem. 1996, 271, 2406–2412. [Google Scholar] [CrossRef] [Green Version]
- Uebele, V.N.; England, S.K.; Galla gher, D.J.; Snyders, D.J.; Bennett, P.B.; Tamkun, M.M. Distinct domains of the voltage-gated K+ channel Kvβ1.3 β-subunit affect voltage-dependent gating. Am. J. Physiol. Cell Physiol. 1998, 274, 1485–1495. [Google Scholar] [CrossRef]
- Kwak, Y.G.; Navarro-Polanco, R.A.; Grobaski, T.; Gallagher, D.J.; Tamkun, M.M. Phosphorylation is required for alteration of Kv1.5 K+ channel function by the Kvβ1.3 subunit. J. Biol. Chem. 1999, 274, 25355–25361. [Google Scholar] [CrossRef] [Green Version]
- Arias, C.; Guizy, M.; David, M.; Marzian, S.; González, T.; Decher, N.; Valenzuela, C. Kvβ1.3 reduces the degree of stereoselective bupivacaine block of Kv1.5 channels. Anesthesiology 2007, 107, 641–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, M.; Macías, Á.; Moreno, C.; Prieto, Á.; Martínez-Mármol, R.; Vicente, R.; González, T.; Felipe, A.; Tamkun, M.M.; Valenzuela, C. Protein kinase C (PKC) activity regulates functional effects of K v β1.3 subunit on K V 1.5 channels. J. Biol. Chem. 2012, 287, 21416–21428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, T.; Navarro-polanco, R.; Arias, C.; Caballero, R.; Moreno, I.; Delpón, E.; Tamargo, J.; Tamkun, M.M.; Valenzuela, C. Assembly with the Kvβ1.3 subunit modulates drug block of hKv1.5 channels. Mol. Pharmacol. 2002, 62, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Decher, N.; Kumar, P.; Gonzalez, T.; Renigunta, V.; Sanguinetti, M.C. Structural basis for competition between drug binding and Kvβ1.3 accessory subunit-induced N-type inactivation of Kv1.5 channels. Mol. Pharmacol. 2005, 68, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Macías, A.; de la Cruz, A.; Prieto, A.; Peraza, D.A.; Tamkun, M.M.; González, T.; Valenzuela, C. PKC inhibition results in a K v 1.5 + K v β1.3 pharmacology closer to K v 1.5 channels. Br. J. Pharmacol. 2014, 171, 4914–4926. [Google Scholar] [CrossRef] [Green Version]
- Souroujon, M.C.; Mochly-Rosen, D. Peptide modulators of protein–Protein interactions in intracellular signaling. Nat. Biotechnol. 1998, 16, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Neer, E.J.; Schmidt, C.J.; Nambudripad, R.; Smith, T.F. The ancient regulatory-protein family of WD-repeat proteins. Nature 1994, 371, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Schechtman, D.; Mochly-Rosen, D. Adaptor proteins in protein kinase C-mediated signal transduction. Oncogene 2001, 20, 6339–6347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manganas, L.N.; Wang, Q.; Scannevin, R.H.; Antonucci, D.E.; Rhodes, K.J.; Trimmer, J.S. Identification of a trafficking determinant localized to the Kv1 potassium channel pore. Proc. Natl. Acad. Sci. USA 2001, 98, 14055–14059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.S.; Khurana, A.; Mathur, R.; Viswanathan, V.; Steele, D.F.; Fedida, D. Kv1.5 surface expression is modulated by retrograde trafficking of newly endocytosed channels by the dynein motor. Circ. Res. 2005, 97, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Macías, A. Characterization and Regulation of KV1.5-KVβ1.3 Complex. Ph.D. Thesis, Universidad Autónoma de Madrid, Madrid, Spain, March 2014. [Google Scholar]
- Andersen, M.N.; Skibsbye, L.; Tang, C.; Petersen, F.; MacAulay, N.; Rasmussen, H.B.; Jespersen, T. PKC and AMPK regulation of Kv1.5 potassium channels. Channels 2015, 9, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balse, E.; El-Haou, S.; Dillanian, G.; Dauphin, A.; Eldstrom, J.; Fedida, D.; Coulombe, A.; Hatem, S.N. Cholesterol modulates the recruitment of Kv1.5 channels from Rab11-associated recycling endosome in native atrial myocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 14681–14686. [Google Scholar] [CrossRef] [Green Version]
- Horgan, C.P.; McCaffrey, M.W. The dynamic Rab11-FIPs. Biochem. Soc. Trans. 2009, 37, 1032–1036. [Google Scholar] [CrossRef]
- Hales, C.M.; Vaerman, J.-P.; Goldenring, J.R. Rab11 family interacting protein 2 associates with myosin Vb and regulates plasma membrane recycling. J. Biol. Chem. 2002, 277, 50415–50421. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, F. PKC and calcium channel trafficking. Channels 2018, 12, 15–16. [Google Scholar] [CrossRef]
- Scheuer, T. Regulation of sodium channel activity by phosphorylation. Semin. Cell Dev. Biol. 2011, 22, 160–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aromolaran, A.S.; Chahine, M.; Boutjdir, M. Regulation of cardiac voltage-gated sodium channel by kinases: Roles of protein kinases A and C. In Handbook of Experimental Pharmacology; Springer: New York, NY, USA, 2017; pp. 161–184. [Google Scholar]
- Hallaq, H.; Wang, D.W.; Kunic, J.D.; George, A.L.; Wells, K.S.; Murray, K.T. Activation of protein kinase C alters the intracellular distribution and mobility of cardiac Na + channels. Am. J. Physiol. Circ. Physiol. 2012, 302, H782–H789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, X.X.; Ronzier, E.; Jin, O.; Lopes, C.M. Fluvastatin inhibits Rab5-mediated IKs internalization caused by chronic Ca2+-dependent PKC activation. J. Mol. Cell. Cardiol. 2019, 129, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.D.; Zhai, X.; Yin, W.; Jaggar, J.H. Impaired trafficking of β1 subunits inhibits BK channels in cerebral arteries of hypertensive rats. Hypertension 2018, 72, 765–775. [Google Scholar] [CrossRef]
- Zhai, X.; Leo, M.D.; Jaggar, J.H. Endothelin-1 stimulates vasoconstriction through Rab11A serine 177 phosphorylation. Circ. Res. 2017, 121, 650–661. [Google Scholar] [CrossRef]
- Idkowiak-Baldys, J.; Baldys, A.; Raymond, J.R.; Hannun, Y.A. Sustained receptor stimulation leads to sequestration of recycling endosomes in a classical protein kinase C- and phospholipase D-dependent manner. J. Biol. Chem. 2009, 284, 22322–22331. [Google Scholar] [CrossRef] [Green Version]
- Pavarotti, M.; Capmany, A.; Vitale, N.; Colombo, M.I.; Damiani, M.T. Rab11 is phosphorylated by classical and novel protein kinase C isoenzymes upon sustained phorbol ester activation. Biol. Cell 2012, 104, 102–115. [Google Scholar] [CrossRef]
- Dietrich, M.; Malik, M.S.; Nikolaysen, F.; Skeie, M.; Stang, E. Protein kinase C mediated internalization of ErbB2 is independent of clathrin, ubiquitination and Hsp90 dissociation. Exp. Cell Res. 2018, 371, 139–150. [Google Scholar] [CrossRef]
- Rahbek-Clemmensen, T.; Bay, T.; Eriksen, J.; Gether, U.; Jørgensen, T.N. The serotonin transporter undergoes constitutive internalization and is primarily sorted to late endosomes and lysosomal degradation. J. Biol. Chem. 2014, 289, 23004–23019. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Alroy, I.; Waterman, H.; Schejter, E.D.; Brodie, C.; Gruenberg, J.; Yarden, Y. Threonine phosphorylation diverts internalized epidermal growth factor receptors from a degradative pathway to the recycling endosome. J. Biol. Chem. 2000, 275, 26178–26186. [Google Scholar] [CrossRef] [Green Version]
- McEwen, D.P.; Schumacher, S.M.; Li, Q.; Benson, M.D.; Iñiguez-Lluhí, J.A.; Van Genderen, K.M.; Martens, J.R. Rab-GTPase-dependent endocytic recycling of KV1.5 in atrial myocytes. J. Biol. Chem. 2007, 282, 29612–29620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadeh, A.D.; Xu, H.; Loewen, M.E.; Noble, G.P.; Steele, D.F.; Fedida, D. Internalized Kv1.5 traffics via Rab-dependent pathways. J. Physiol. 2008, 586, 4793–4813. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, S.M.; Martens, J.R. Ion channel trafficking: A new therapeutic horizon for atrial fibrillation. Hear. Rhythm 2010, 7, 1309–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burg, E.D.; Platoshyn, O.; Tsigelny, I.F.; Lozano-Ruiz, B.; Rana, B.K.; Yuan, J.X.J. Tetramerization domain mutations in KCNA5 affect channel kinetics and cause abnormal trafficking patterns. Am. J. Physiol. Cell Physiol. 2010, 298, C496–C509. [Google Scholar] [CrossRef] [Green Version]
- Svoboda, L.K.; Reddie, K.G.; Zhang, L.; Vesely, E.D.; Williams, E.S.; Schumacher, S.M.; O’Connell, R.P.; Shaw, R.; Day, S.M.; Anumonwo, J.M.; et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ. Res. 2012, 111, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Mia, S.; Munoz, C.; Pakladok, T.; Siraskar, G.; Voelkl, J.; Alesutan, I.; Lang, F. Downregulation of Kv1.5 K + channels by the AMP-activated protein kinase. Cell. Physiol. Biochem. 2012, 30, 1039–1050. [Google Scholar] [CrossRef]
- Boycott, H.E.; Barbier, C.S.M.; Eichel, C.A.; Costa, K.D.; Martins, R.P.; Louault, F.; Dilanian, G.; Coulombe, A.; Hatem, S.N.; Balse, E. Shear stress triggers insertion of voltage-gated potassium channels from intracellular compartments in atrial myocytes. Proc. Natl. Acad. Sci. USA 2013, 110, E3955–E3964. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, S.M.; McEwen, D.P.; Zhang, L.; Arendt, K.L.; Van Genderen, K.M.; Martens, J.R. Antiarrhythmic drug–Induced internalization of the atrial-specific K + channel Kv1.5. Circ. Res. 2009, 104, 1390–1398. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, T.; David, M.; Moreno, C.; Macias, A.; Valenzuela, C. Kv1.5-Kvß; Interactions: Molecular determinants and pharmacological consequences. Mini Rev. Med. Chem. 2010, 10, 635–642. [Google Scholar] [CrossRef]
- Zhu, Y.; Jiang, X.; Zheng, Y.; Xiong, J.; Wei, D.; Zhang, D. Cardiac function modulation depends on the A-kinase anchoring protein complex. J. Cell. Mol. Med. 2019, 23, 7170–7179. [Google Scholar] [CrossRef]
- Higuchi, R.; Krummel, B.; Saiki, R. A general method of in vitro preparation and specific mutagenesis of DNA fragments: Study of protein and DNA interactions. Nucleic Acids Res. 1988, 16, 7351–7367. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N-term | |
| Forward | 5′cggctgggctcagcgatgggcccaaggagccggc 3′ |
| Reverse | 5′ccccttgtcatcgtcgtccttgtagtccccgccagaggcgggggccatgaccccgctgccg 3′ |
| C-term | |
| Forward | 5′ggggactacaaggacgacgatgacaaggggcctacggtggcaccgctcctgcccCGTACG 3′ |
| Reverse | 5′ gctcttccttaaggactgccggctcctcgtgatcc 3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macias, A.; de la Cruz, A.; Peraza, D.A.; Benito-Bueno, A.d.; Gonzalez, T.; Valenzuela, C. KV1.5–KVβ1.3 Recycling Is PKC-Dependent. Int. J. Mol. Sci. 2021, 22, 1336. https://doi.org/10.3390/ijms22031336
Macias A, de la Cruz A, Peraza DA, Benito-Bueno Ad, Gonzalez T, Valenzuela C. KV1.5–KVβ1.3 Recycling Is PKC-Dependent. International Journal of Molecular Sciences. 2021; 22(3):1336. https://doi.org/10.3390/ijms22031336
Chicago/Turabian StyleMacias, Alvaro, Alicia de la Cruz, Diego A. Peraza, Angela de Benito-Bueno, Teresa Gonzalez, and Carmen Valenzuela. 2021. "KV1.5–KVβ1.3 Recycling Is PKC-Dependent" International Journal of Molecular Sciences 22, no. 3: 1336. https://doi.org/10.3390/ijms22031336
APA StyleMacias, A., de la Cruz, A., Peraza, D. A., Benito-Bueno, A. d., Gonzalez, T., & Valenzuela, C. (2021). KV1.5–KVβ1.3 Recycling Is PKC-Dependent. International Journal of Molecular Sciences, 22(3), 1336. https://doi.org/10.3390/ijms22031336