
Artemisia annua L. Polyphenol-Induced Cell Death Is ROS-Independently Enhanced by Inhibition of JNK in HCT116 Colorectal Cancer Cells


 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
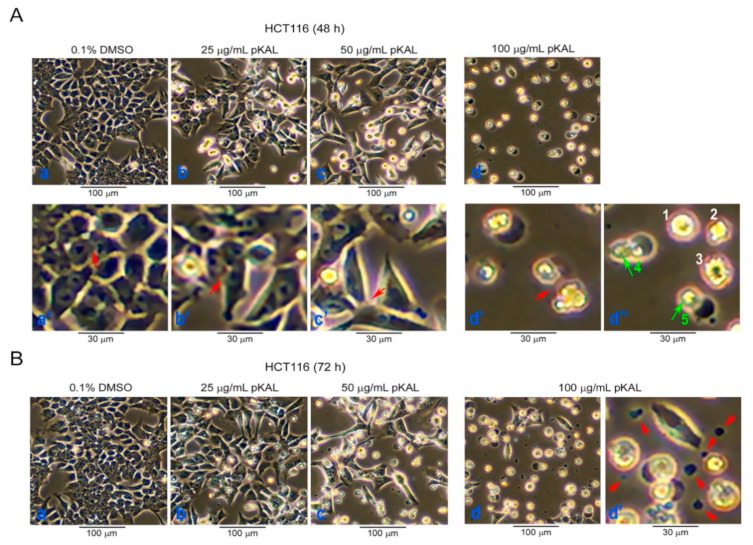
2.1. Effect of pKAL on the Regulation of Cell Morphology in p53 Wild-Type HCT116 Colorectal Cancer Cells
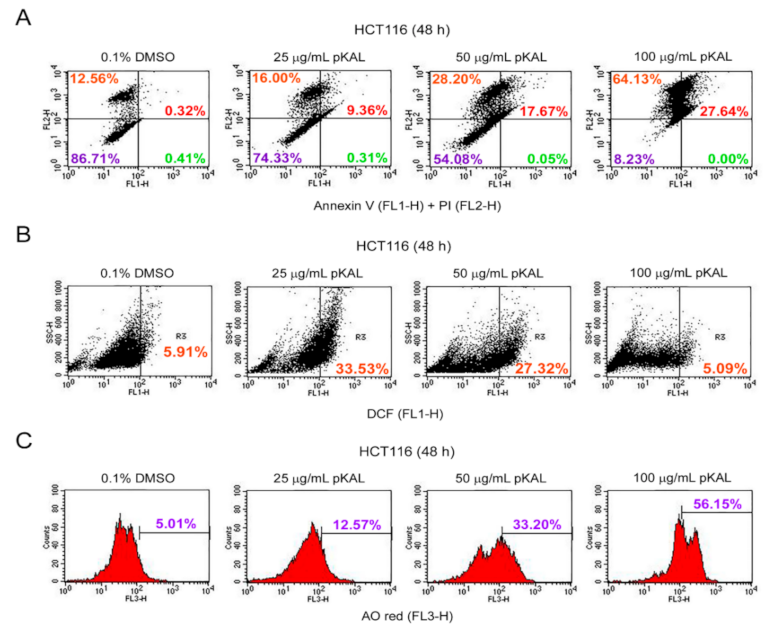
2.2. Effect of pKAL on the Regulation of PI Uptake, Apoptosis, ROS, and Acidic Vesicles
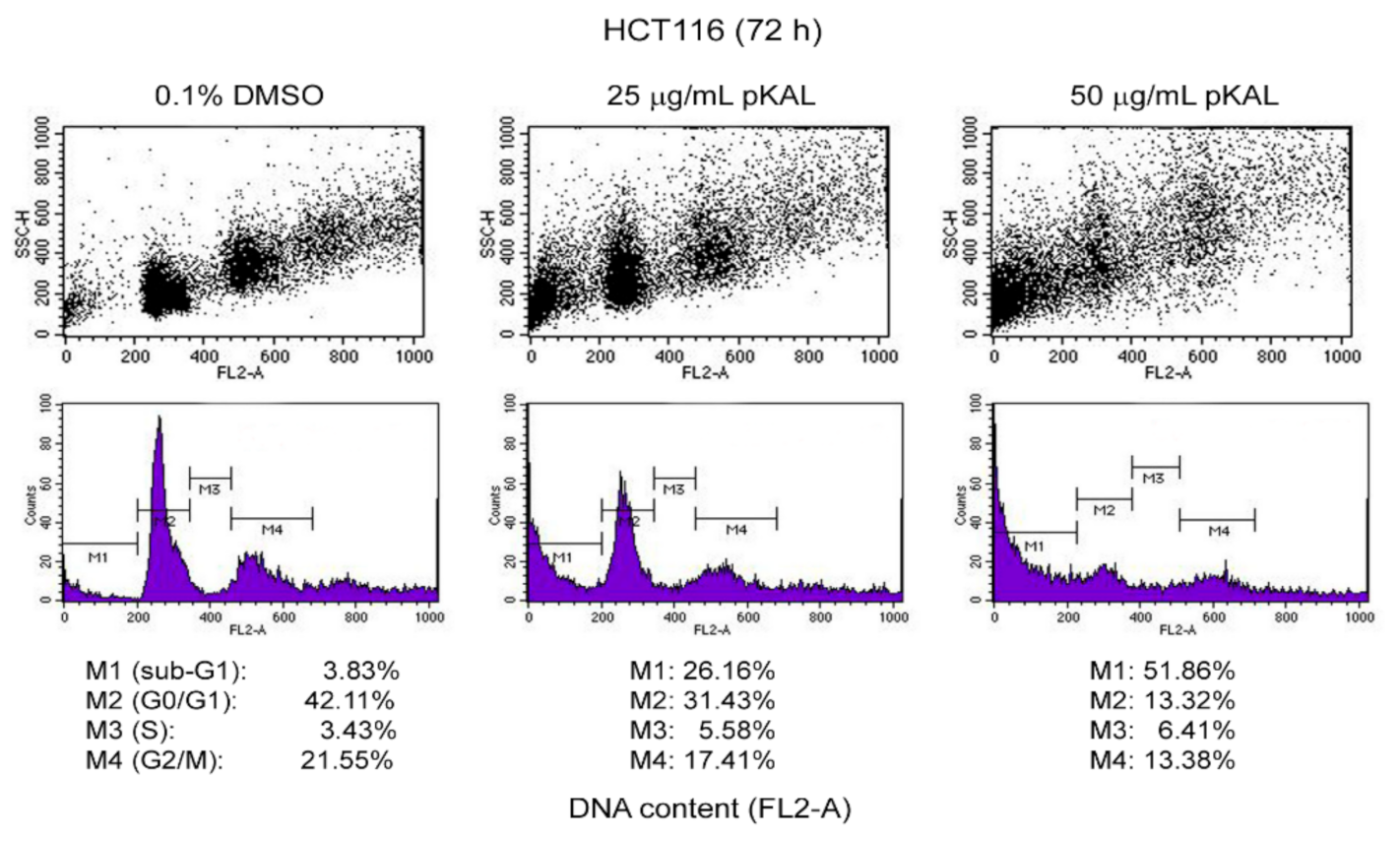
2.3. Effect of pKAL on the Regulation of DNA Content, Cell Cycle, and Intracellular Granularity
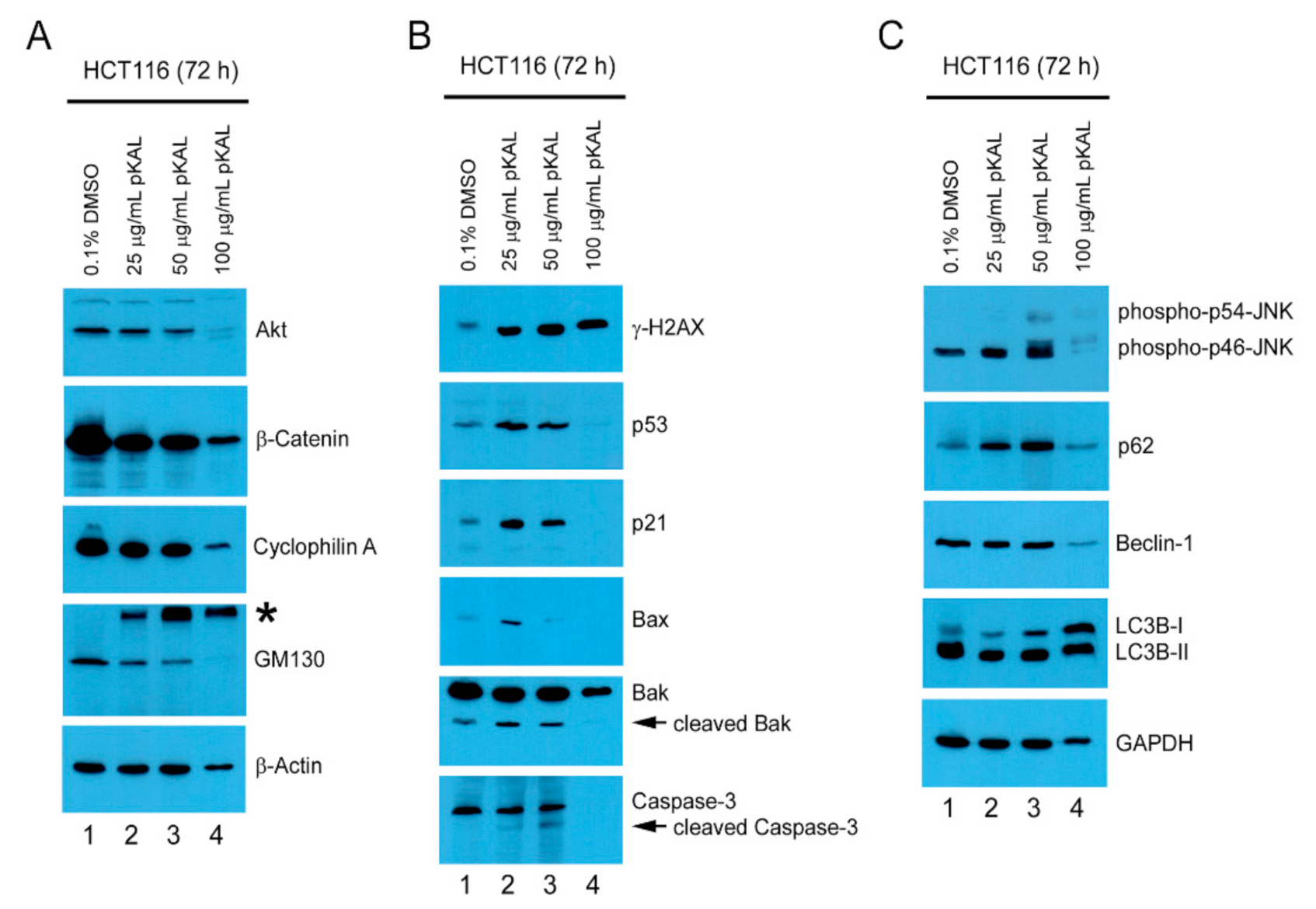
2.4. pKAL-Induced Cell Death Mechanisms in p53 Wild-Type HCT116 Cells
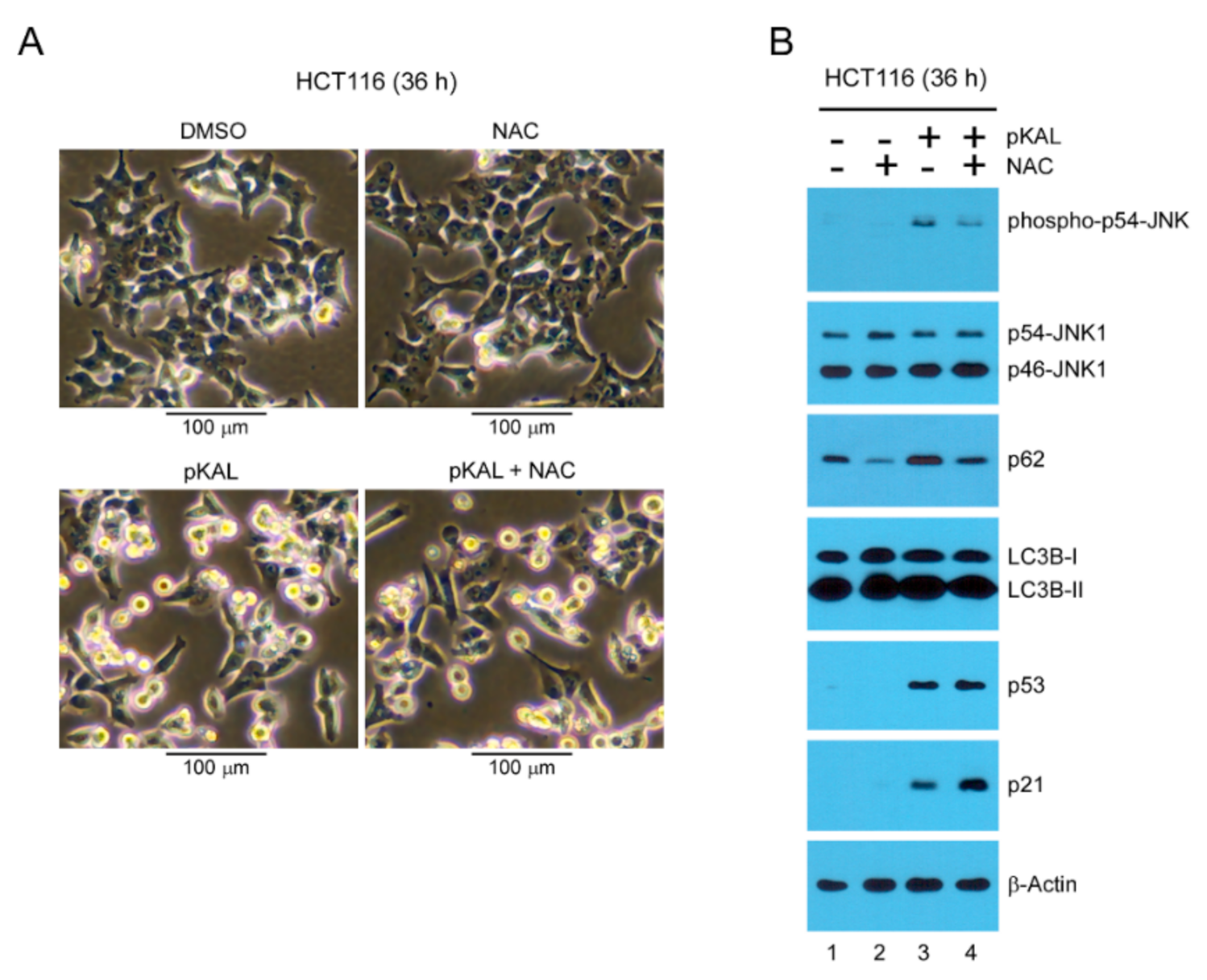
2.5. Effect of the ROS Inhibitor NAC on Morphological Changes and Protein Levels Altered by pKAL
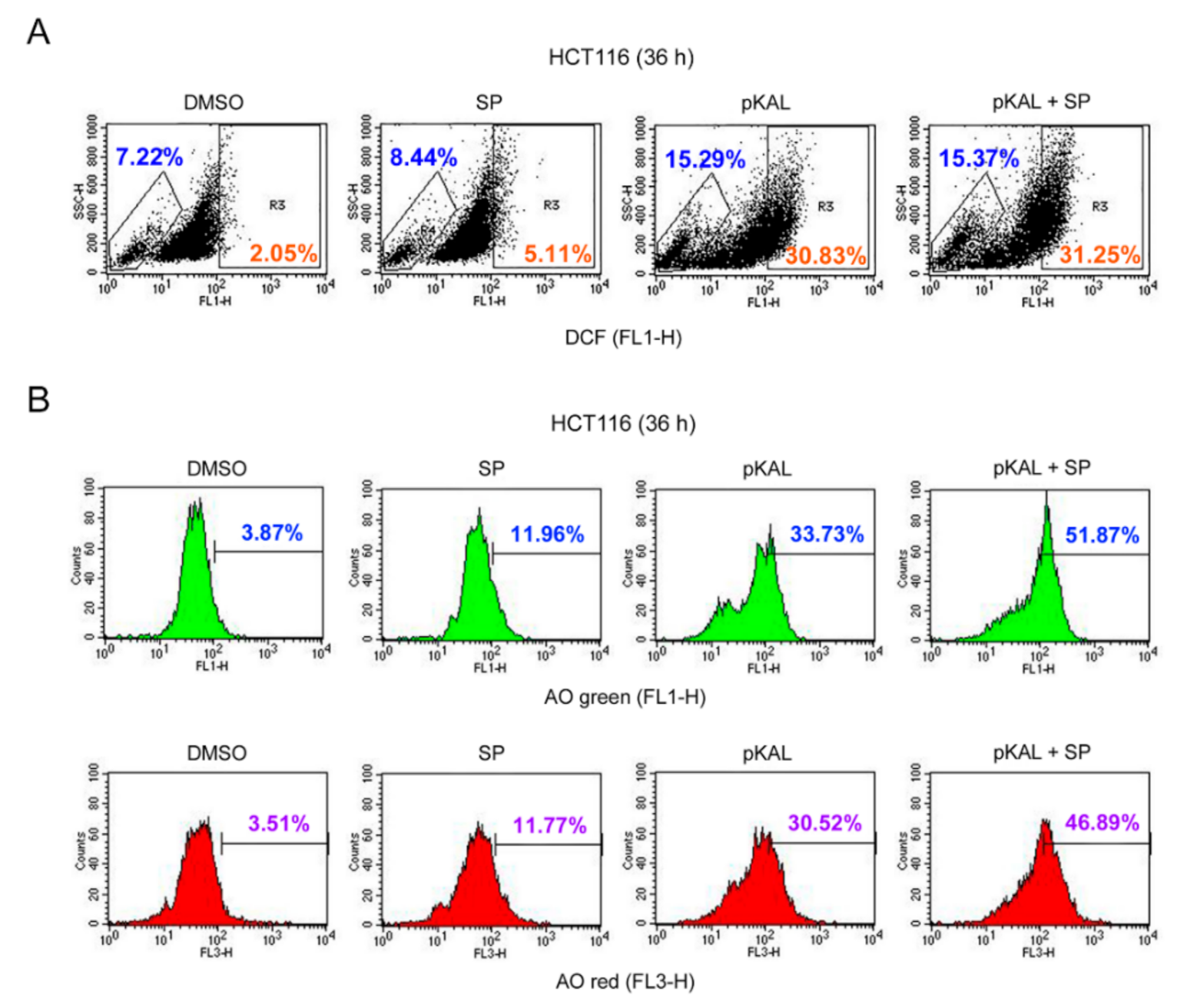
2.6. Effect of the JNK Inhibitor SP600125 on the Regulation of ROS, DNA Conformational Change, and Acidic Vesicles Induced by pKAL
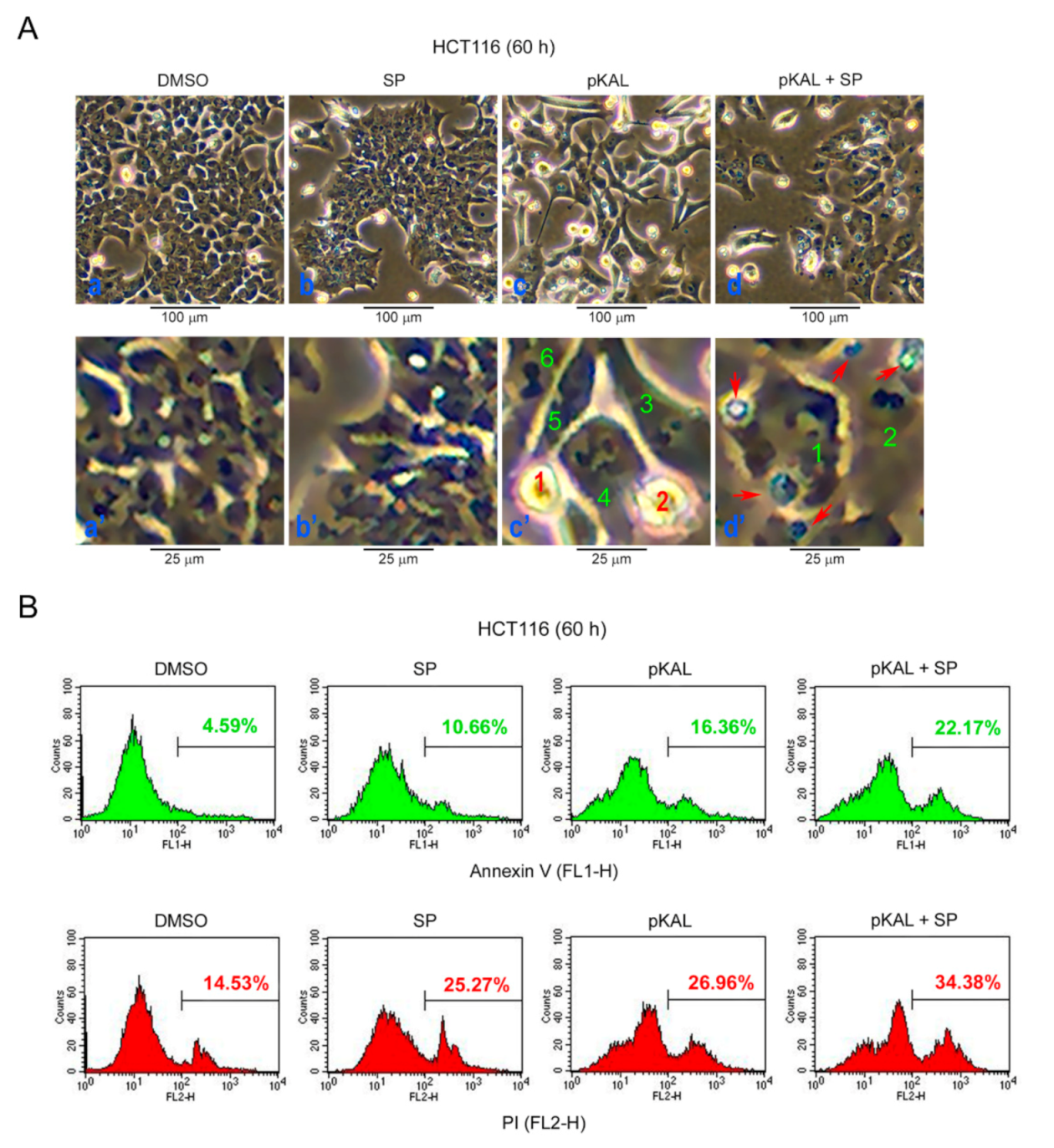
2.7. Effect of SP600125 on the Regulation of Morphological CHANGES, Apoptosis, and PI UpTake Caused by pKAL
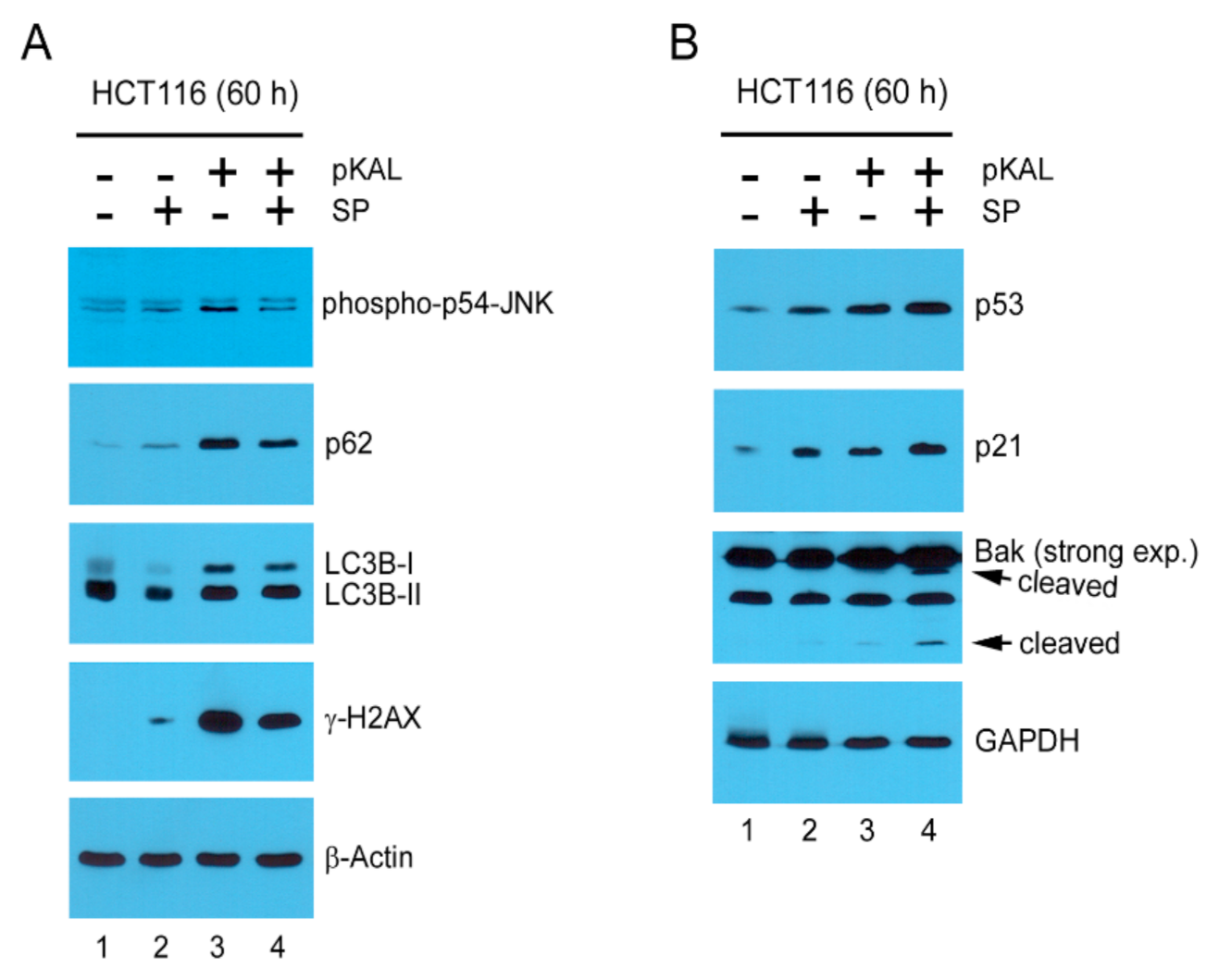
2.8. Mechanisms Related to the Enhancement of pKAL-Induced Cell Death by SP600125
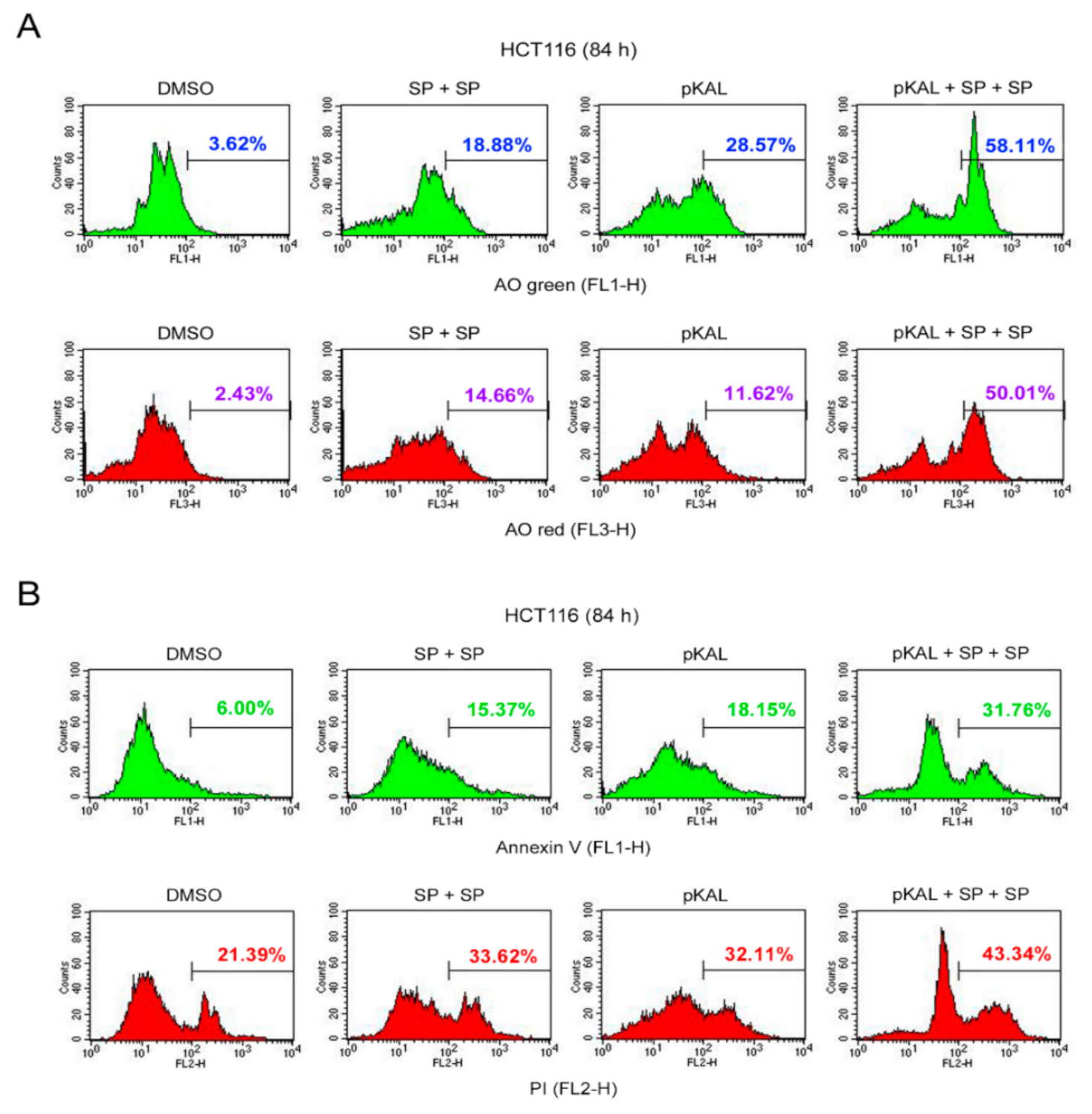
2.9. Effect of Twice Sequential Treatments of SP600125 on the Regulation of DNA Conformational Change, Acidic Vesicles, Apoptosis, and PI Uptake Caused by pKAL
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. pKAL Components
4.3. Cell Culture
4.4. Phase-Contrast Light Microscopy
4.5. Flow Cytometric Analysis of Annexin V/PI-Stained Cells
4.6. Flow Cytometric Analysis of DCF-Stained Cells
4.7. Flow Cytometric Analysis of AO-Stained Cells
4.8. DNA Content Analysis Using Flow Cytometry
4.9. Western Blot Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| pKAL | Polyphenols isolated from Korean Artemisia annua L. |
| JNK | c-Jun N-terminal kinase |
| g-H2AX | Phosphorylation of H2AX at Ser-139 |
| ROS | Reactive oxygen species |
| PI | Propidium iodide |
| DCF-DA | 2’,7’-dichlorofluorescein diacetate |
| AO | Acridine orange |
| SSC-H | Side scatter pulse height |
| Akt | Protein kinases B |
| GM130 | Golgi matrix protein 130 |
| MAPK | Mitogen-activated protein kinase |
| ERK | Extracellular regulated kinase |
| LC3B | Microtubule-associated protein 1 light chain 3B |
| ATM | Ataxia-telangiectasia mutated |
| ATR | Ataxia-telangiectasia and Rad3-related |
| DNA-PK | DNA-dependent protein kinase |
| PS | Phosphatidylserine |
| PI3K | Phosphatidylinositol 3-kinase |
| Nox2 | NADPH oxidase 2 |
| CML | Chronic myelogenous leukemia |
| PB1 | Phox1 and Bem1p |
| TNF | Tumor necrosis factor |
| TRAF6 | TNF receptor-associated factor 6 |
| LIR | LC3-interacting region |
| KIR | Keap1-interacting region |
| UBA | Ubiquitin-associated |
| FBSNC | Fetal bovine serumNitrocellulose |
| RT | Room temperature |
| PBS | Phosphate-buffered saline |
| BSA | Bovine serum albumin |
| DMSO | Dimethyl sulfoxide |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| HRP | Horseradish-peroxidase |
References
- Lin, S.S.; Li, Y.Y.; Zamyatnin, A.A.; Werner, J.; Bazhin, A.V. Reactive oxygen species and colorectal cancer. J. Cell. Physiol. 2018, 233, 5119–5132. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.N.; Almoyad, M.; Huq, F. Polyphenols in Colorectal Cancer: Current State of Knowledge including Clinical Trials and Molecular Mechanism of Action. Biomed. Res. Int. 2018, 2018, 4154185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval-Acuna, C.; Ferreira, J.; Speisky, H. Polyphenols and mitochondria: An update on their increasingly emerging ROS-scavenging independent actions. Arch. Biochem. Biophys. 2014, 559, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Mileo, A.M.; Miccadei, S. Polyphenols as Modulator of Oxidative Stress in Cancer Disease: New Therapeutic Strategies. Oxid. Med. Cell. Longev. 2016, 2016, 6475624. [Google Scholar] [CrossRef] [Green Version]
- Afshari, K.; Haddadi, N.S.; Haj-Mirzaian, A.; Farzaei, M.H.; Rohani, M.M.; Akramian, F.; Naseri, R.; Sureda, A.; Ghanaatian, N.; Abdolghaffari, A.H. Natural flavonoids for the prevention of colon cancer: A comprehensive review of preclinical and clinical studies. J. Cell. Physiol. 2019, 234, 21519–21546. [Google Scholar] [CrossRef]
- Cicenas, J.; Zalyte, E.; Rimkus, A.; Dapkus, D.; Noreika, R.; Urbonavicius, S. JNK, p38, ERK, and SGK1 Inhibitors in Cancer. Cancers 2018, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Sabapathy, K.; Hochedlinger, K.; Nam, S.Y.; Bauer, A.; Karin, M.; Wagner, E.F. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell 2004, 15, 713–725. [Google Scholar] [CrossRef]
- Shaukat, Z.; Liu, D.; Hussain, R.; Khan, M.; Gregory, S.L. The Role of JNK Signalling in Responses to Oxidative DNA Damage. Curr. Drug Targets 2016, 17, 154–163. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar]
- Wu, X.P.; Xiong, M.; Xu, C.S.; Duan, L.N.; Dong, Y.Q.; Luo, Y.; Niu, T.H.; Lu, C.R. Resveratrol induces apoptosis of human chronic myelogenous leukemia cells in vitro through p38 and JNK-regulated H2AX phosphorylation. Acta Pharmacol. Sin. 2015, 36, 353–361. [Google Scholar] [CrossRef]
- Vuoso, D.C.; D’Angelo, S.; Ferraro, R.; Caserta, S.; Guido, S.; Cammarota, M.; Porcelli, M.; Cacciapuoti, G. Annurca apple polyphenol extract promotes mesenchymal-to-epithelial transition and inhibits migration in triple-negative breast cancer cells through ROS/JNK signaling. Sci. Rep. 2020, 10, 15921. [Google Scholar] [CrossRef] [PubMed]
- Viiri, J.; Hyttinen, J.M.T.; Ryhanen, T.; Rilla, K.; Paimela, T.; Kuusisto, E.; Siitonen, A.; Urtti, A.; Salminen, A.; Kaarniranta, K. p62/sequestosome 1 as a regulator of proteasome inhibitor-induced autophagy in human retinal pigment epithelial cells. Mol. Vis. 2010, 16, 1399–1414. [Google Scholar] [PubMed]
- Tiwari, R.V.; Parajuli, P.; Sylvester, P.W. gamma-Tocotrienol-induced autophagy in malignant mammary cancer cells. Exp. Biol. Med. (Maywood) 2014, 239, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; Czompa, A.; Reboul, C.; Csepanyi, E.; Czegledi, A.; Bak, I.; Balla, G.; Balla, J.; Tosaki, A.; Lekli, I. The Cellular Autophagy Markers Beclin-1 and LC3B-II are Increased During Reperfusion in Fibrillated Mouse Hearts. Curr. Pharm. Des. 2013, 19, 6912–6918. [Google Scholar] [CrossRef] [PubMed]
- Nihira, K.; Miki, Y.; Ono, K.; Suzuki, T.; Sasano, H. An inhibition of p62/SQSTM1 caused autophagic cell death of several human carcinoma cells. Cancer Sci. 2014, 105, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Vegliante, R.; Desideri, E.; Di Leo, L.; Ciriolo, M.R. Dehydroepiandrosterone triggers autophagic cell death in human hepatoma cell line HepG2 via JNK-mediated p62/SQSTM1 expression. Carcinogenesis 2016, 37, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Bai, H.M.; Chen, L.A.; Li, B.; Lu, Y.C. Reduced expression of LC3B-II and Beclin 1 in glioblastoma multiforme indicates a down-regulated autophagic capacity that relates to the progression of astrocytic tumors. J. Clin. Neurosci. 2010, 17, 1515–1519. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.H.; Wu, W.D.; Fu, B.S.; Shi, L.; Wang, X.; Kuca, K. JNK signaling in cancer cell survival. Med. Res. Rev. 2019, 39, 2082–2104. [Google Scholar] [CrossRef]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef]
- Firestone, G.L.; Sundar, S.N. Anticancer activities of artemisinin and its bioactive derivatives. Expert Rev. Mol. Med. 2009, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.S.; Li, J.; Wang, Z.; Mi, C.; Ma, J.; Piao, L.X.; Xu, G.H.; Li, X.; Jin, X. Artemisinin inhibits inflammatory response via regulating NF-kappaB and MAPK signaling pathways. Immunopharmacol. Immunotoxicol. 2017, 39, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.C.; Lee, S.H.; Lee, M.; Kim, H.J.; Oak, M.H.; Lee, I.S.; Kang, B.Y. Enhanced IL-12p40 production in LPS-stimulated macrophages by inhibiting JNK activation by artemisinin. Arch. Pharm. Res. 2012, 35, 1961–1968. [Google Scholar] [CrossRef]
- Jung, E.J.; Lee, W.S.; Paramanantham, A.; Kim, H.J.; Shin, S.C.; Kim, G.S.; Jung, J.M.; Ryu, C.H.; Hong, S.C.; Chung, K.H.; et al. p53 Enhances Artemisia annua L. Polyphenols-Induced Cell Death Through Upregulation of p53-Dependent Targets and Cleavage of PARP1 and Lamin A/C in HCT116 Colorectal Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9315. [Google Scholar] [CrossRef]
- Crowley, L.C.; Scott, A.P.; Marfell, B.J.; Boughaba, J.A.; Chojnowski, G.; Waterhouse, N.J. Measuring Cell Death by Propidium Iodide Uptake and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, 087163. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, 087288. [Google Scholar] [CrossRef] [PubMed]
- Rieger, A.M.; Nelson, K.L.; Konowalchuk, J.D.; Barreda, D.R. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J. Vis. Exp. 2011, 50, 2597. [Google Scholar] [CrossRef]
- Thome, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajstura, M.; Halicka, H.D.; Pryjma, J.; Darzynkiewicz, Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete “sub-G1” peaks on DNA content histograms. Cytom. Part A 2007, 71, 125–131. [Google Scholar] [CrossRef]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Healy, E.; Dempsey, M.; Lally, C.; Ryan, M.P. Apoptosis and necrosis: Mechanisms of cell death induced by cyclosporine A in a renal proximal tubular cell line. Kidney Int. 1998, 54, 1955–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogunwobi, O.O.; Mahmood, F.; Akingboye, A. Biomarkers in Colorectal Cancer: Current Research and Future Prospects. Int. J. Mol. Sci. 2020, 21, 5311. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Rodriguez-Mateos, A.; Corona, G.; Oruna-Concha, M.J.; Spencer, J.P.E. Polyphenols and Human Health: Prevention of Disease and Mechanisms of Action. Nutrients 2010, 2, 1106–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mileo, A.M.; Nistico, P.; Miccadei, S. Polyphenols: Immunomodulatory and Therapeutic Implication in Colorectal Cancer. Front. Immunol. 2019, 10, 729. [Google Scholar] [CrossRef]
- Tatapudy, S.; Aloisio, F.; Barber, D.; Nystul, T. Cell fate decisions: Emerging roles for metabolic signals and cell morphology. EMBO Rep. 2017, 18, 2105–2118. [Google Scholar] [CrossRef]
- Alexandrova, A.Y.; Kopnin, P.B.; Vasiliev, J.M.; Kopnin, B.P. ROS up-regulation mediates Ras-induced changes of cell morphology and motility. Exp. Cell Res. 2006, 312, 2066–2073. [Google Scholar] [CrossRef]
- Diederich, M.; Cerella, C. Non-canonical programmed cell death mechanisms triggered by natural compounds. Semin. Cancer Biol. 2016, 40–41, 4–34. [Google Scholar] [CrossRef]
- Henry, C.M.; Hollville, E.; Martin, S.J. Measuring apoptosis by microscopy and flow cytometry. Methods 2013, 61, 90–97. [Google Scholar] [CrossRef]
- Borges, G.A.; Elias, S.T.; Amorim, B.; De Lima, C.L.; Coletta, R.D.; Castilho, R.M.; Squarize, C.H.; Guerra, E.N.S. Curcumin downregulates the PI3K-AKT-mTOR pathway and inhibits growth and progression in head and neck cancer cells. Phytother. Res. 2020, 34, 3311–3324. [Google Scholar] [CrossRef]
- Shi, W.; Tang, Y.; Zhi, Y.; Li, Z.; Yu, S.; Jiang, J.; Zhu, J.; Li, J.; Wang, F.; Su, L.; et al. Akt inhibition-dependent downregulation of the Wnt/beta-Catenin Signaling pathway contributes to antimony-induced neurotoxicity. Sci. Total Environ. 2020, 737, 140252. [Google Scholar] [CrossRef]
- Christofferson, D.E.; Yuan, J. Cyclophilin A release as a biomarker of necrotic cell death. Cell Death Differ. 2010, 17, 1942–1943. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.Y.; Yuan, W.; Cao, M.F.; Chen, R.; Wu, X.L.; Yan, J.C. Cyclophilin A Protects Cardiomyocytes against Hypoxia/Reoxygenation-Induced Apoptosis via the AKT/Nox2 Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 2717986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreft, M.E.; Di Giandomenico, D.; Beznoussenko, G.V.; Resnik, N.; Mironov, A.A.; Jezernik, K. Golgi apparatus fragmentation as a mechanism responsible for uniform delivery of uroplakins to the apical plasma membrane of uroepithelial cells. Biol. Cell 2010, 102, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N. Emerging New Roles of GM130, a cis-Golgi Matrix Protein, in Higher Order Cell Functions. J. Pharmacol. Sci. 2010, 112, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Azqueta, A.; Collins, A. Polyphenols and DNA Damage: A Mixed Blessing. Nutrients 2016, 8, 785. [Google Scholar] [CrossRef]
- Bekeschus, S.; Schutz, C.S.; Niessner, F.; Wende, K.; Weltmann, K.D.; Gelbrich, N.; von Woedtke, T.; Schmidt, A.; Stope, M.B. Elevated H2AX Phosphorylation Observed with kINPen Plasma Treatment Is Not Caused by ROS-Mediated DNA Damage but Is the Consequence of Apoptosis. Oxid. Med. Cell. Longev. 2019, 2019, 8535163. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.; Reale, M.; Ullah, H.; Sureda, A.; Tejada, S.; Wang, Y.; Zhang, Z.J.; Xiao, J. Anti-cancer effects of polyphenols via targeting p53 signaling pathway: Updates and future directions. Biotechnol. Adv. 2020, 38, 107385. [Google Scholar] [CrossRef]
- Zhou, Y.L.; Ho, W.S. Combination of liquiritin, isoliquiritin and isoliquirigenin induce apoptotic cell death through upregulating p53 and p21 in the A549 non-small cell lung cancer cells. Oncol. Rep. 2014, 31, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A.; Sooro, M.A.; Zhang, P.H. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Sohn, E.J.; Yoon, S.; Won, G.; Kim, C.G.; Jung, J.H.; Kim, S.H. Activation of JNK and IRE1 is critically involved in tanshinone I-induced p62 dependent autophagy in malignant pleural mesothelioma cells: Implication of p62 UBA domain. Oncotarget 2017, 8, 25032–25045. [Google Scholar] [PubMed]
- Ferreira, J.F.S.; Luthria, D.L.; Sasaki, T.; Heyerick, A. Flavonoids from Artemisia annua L. as Antioxidants and Their Potential Synergism with Artemisinin against Malaria and Cancer. Molecules 2010, 15, 3135–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skowyra, M.; Gallego, M.G.; Segovia, F.; Almajano, M.P. Antioxidant Properties of Artemisia annua Extracts in Model Food Emulsions. Antioxidants (Basel) 2014, 3, 116–128. [Google Scholar] [PubMed] [Green Version]
- Song, Y.; Desta, K.T.; Kim, G.S.; Lee, S.J.; Lee, W.S.; Kim, Y.H.; Jin, J.S.; Abd El-Aty, A.M.; Shin, H.C.; Shim, J.H.; et al. Polyphenolic profile and antioxidant effects of various parts of Artemisia annua L. Biomed. Chromatogr. 2016, 30, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Krigerts, J.; Salmina, K.; Selga, T.; Sorokins, H.; Freivalds, T. Differential staining of peripheral nuclear chromatin with Acridine orange implies an A-form epichromatin conformation of the DNA. Nucleus 2018, 9, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, E.J.; Paramanantham, A.; Kim, H.J.; Shin, S.C.; Kim, G.S.; Jung, J.-M.; Ryu, C.H.; Hong, S.C.; Chung, K.H.; Kim, C.W.; et al. Artemisia annua L. Polyphenol-Induced Cell Death Is ROS-Independently Enhanced by Inhibition of JNK in HCT116 Colorectal Cancer Cells. Int. J. Mol. Sci. 2021, 22, 1366. https://doi.org/10.3390/ijms22031366
Jung EJ, Paramanantham A, Kim HJ, Shin SC, Kim GS, Jung J-M, Ryu CH, Hong SC, Chung KH, Kim CW, et al. Artemisia annua L. Polyphenol-Induced Cell Death Is ROS-Independently Enhanced by Inhibition of JNK in HCT116 Colorectal Cancer Cells. International Journal of Molecular Sciences. 2021; 22(3):1366. https://doi.org/10.3390/ijms22031366
Chicago/Turabian StyleJung, Eun Joo, Anjugam Paramanantham, Hye Jung Kim, Sung Chul Shin, Gon Sup Kim, Jin-Myung Jung, Chung Ho Ryu, Soon Chan Hong, Ky Hyun Chung, Choong Won Kim, and et al. 2021. "Artemisia annua L. Polyphenol-Induced Cell Death Is ROS-Independently Enhanced by Inhibition of JNK in HCT116 Colorectal Cancer Cells" International Journal of Molecular Sciences 22, no. 3: 1366. https://doi.org/10.3390/ijms22031366
APA StyleJung, E. J., Paramanantham, A., Kim, H. J., Shin, S. C., Kim, G. S., Jung, J. -M., Ryu, C. H., Hong, S. C., Chung, K. H., Kim, C. W., & Lee, W. S. (2021). Artemisia annua L. Polyphenol-Induced Cell Death Is ROS-Independently Enhanced by Inhibition of JNK in HCT116 Colorectal Cancer Cells. International Journal of Molecular Sciences, 22(3), 1366. https://doi.org/10.3390/ijms22031366