
Flexibility and Adaptation of Cancer Cells in a Heterogenous Metabolic Microenvironment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
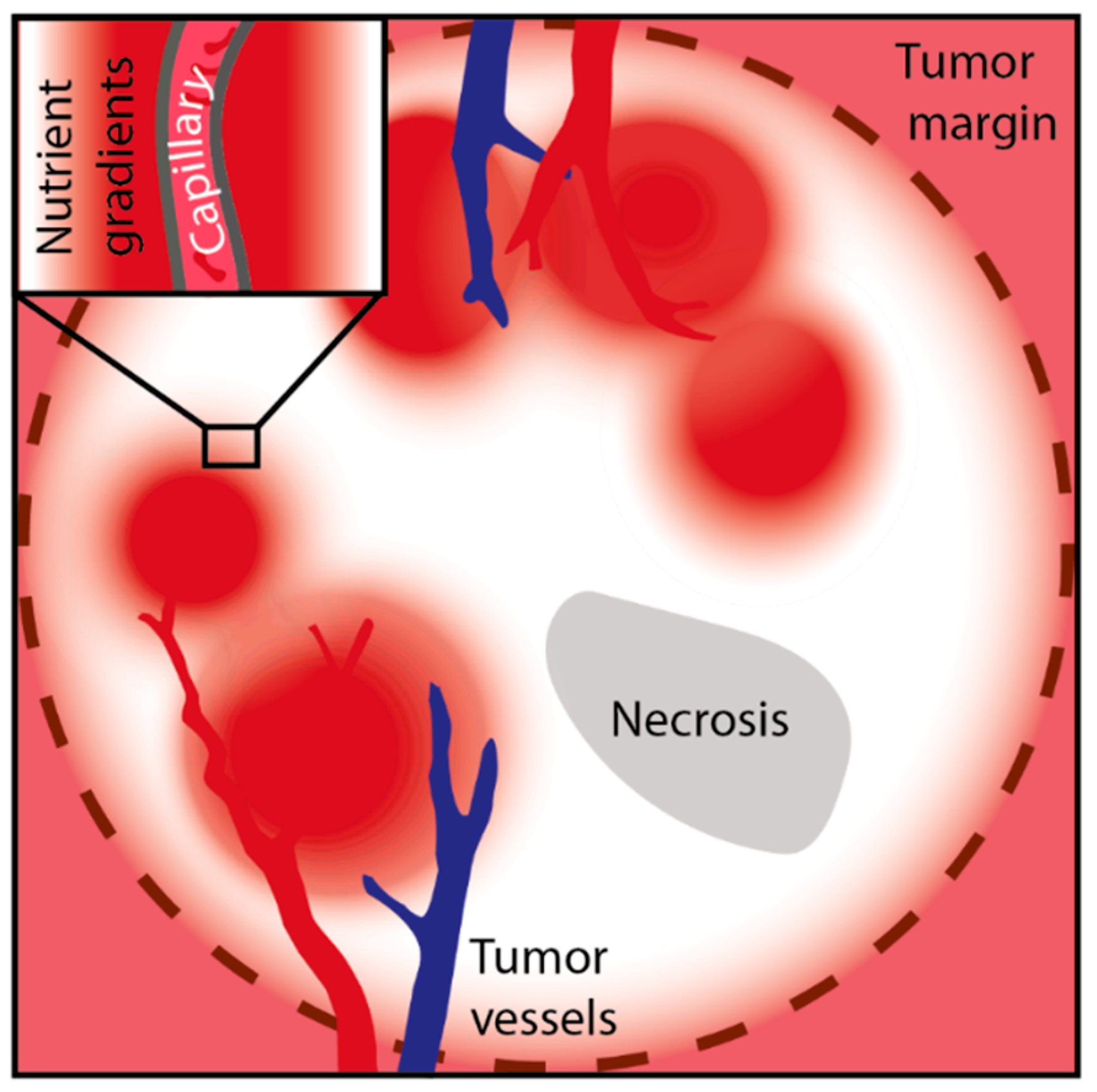
2. Blood Perfusion and the Metabolic Microenvironment
3. Availability of Nutrients in Solid Cancers
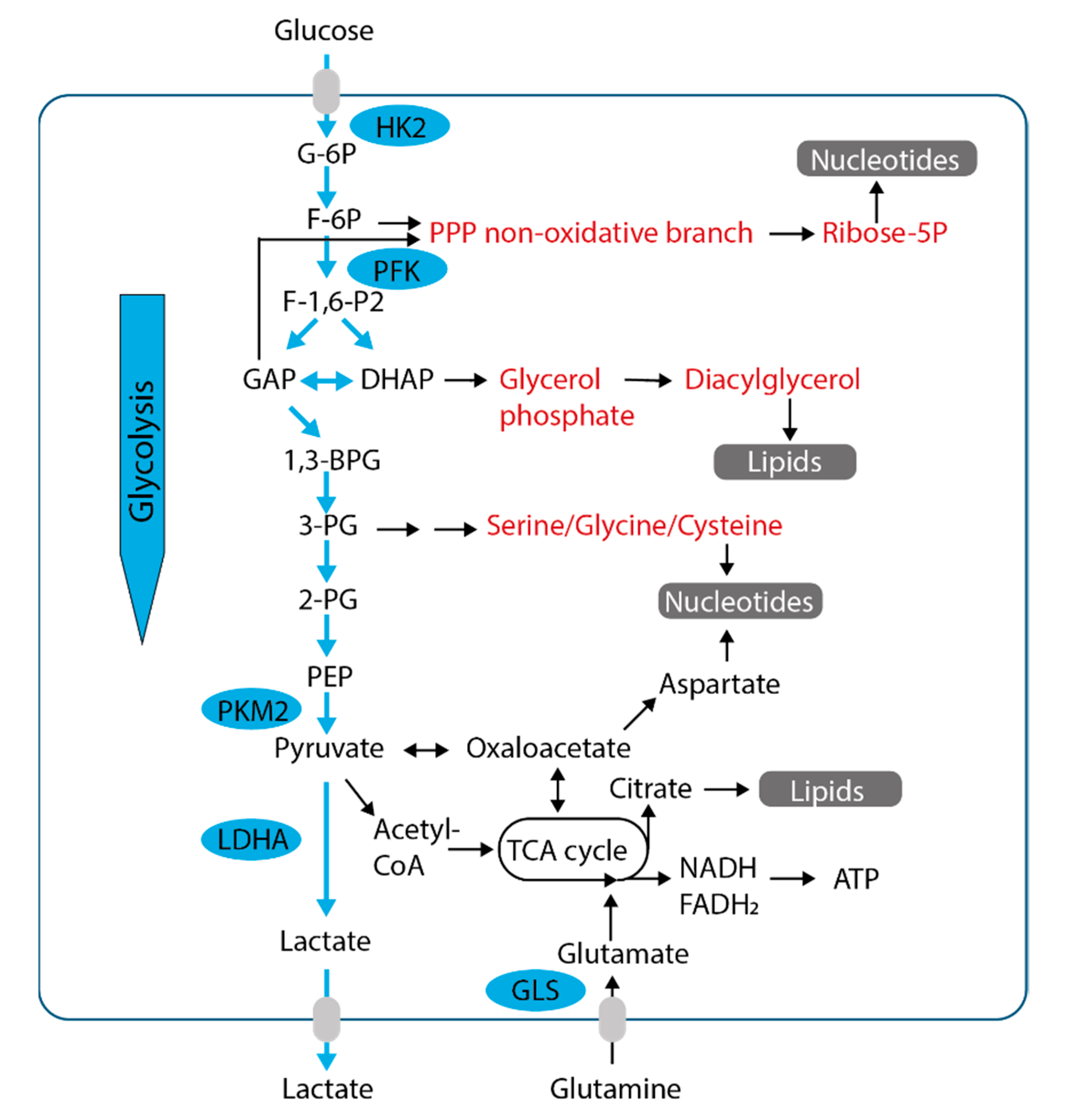
4. Glucose Metabolism and Oxidative Phosphorylation in Cancer—Partners in Crime
5. The TCA Cycle Is a Metabolic Hub with Major Importance for Metabolic Adaptations
6. Glutamine as Anabolic Precursor
7. Lactate and Pyruvate as Alternative Fuels and Carbon Sources
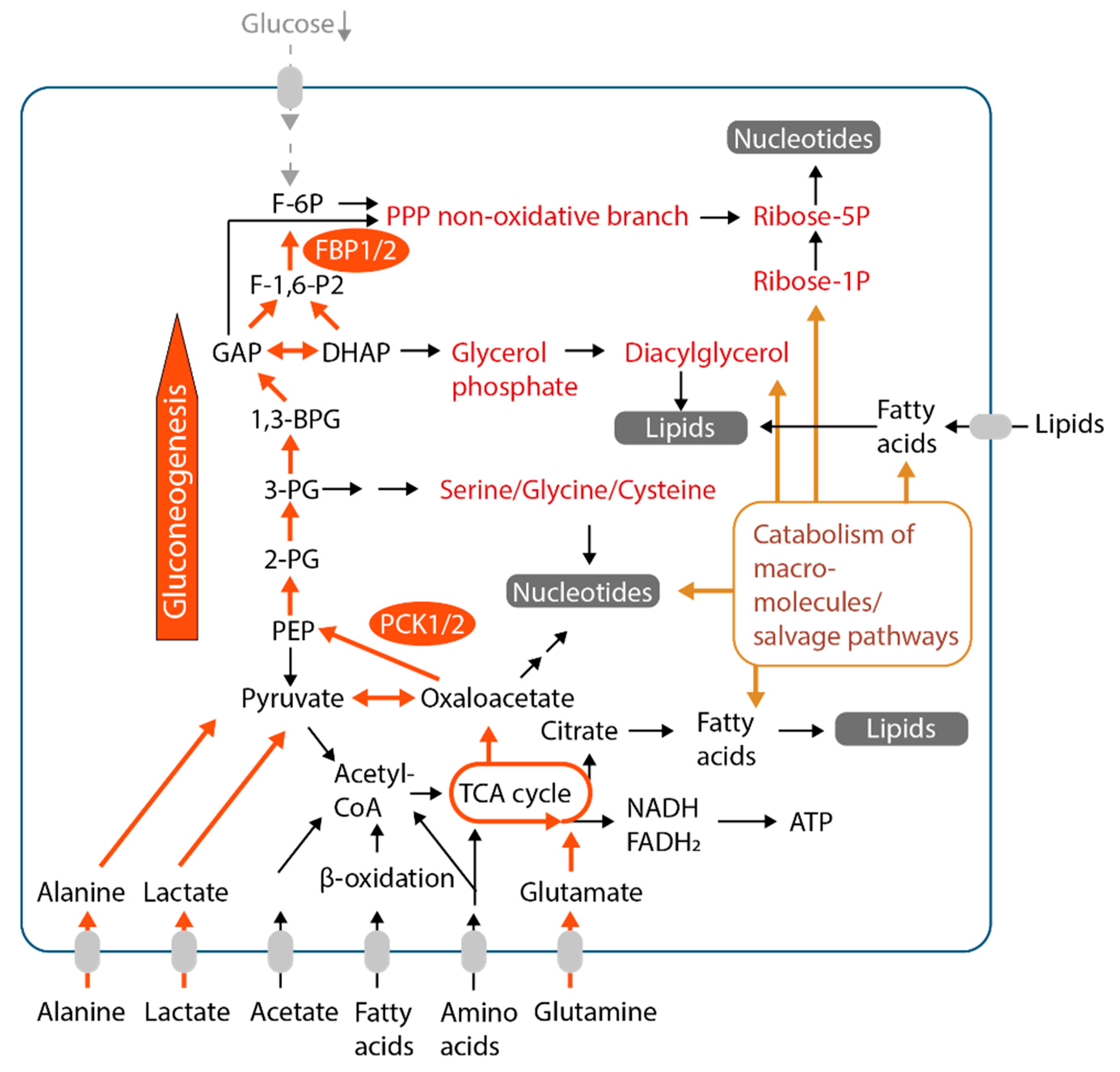
8. Gluconeogenesis Enhances Metabolic Flexibility and Anabolism Under Glucose Deprivation
9. Fatty Acids as Fuels and Constituents of Structural Lipids
10. Autophagy and Macromolecule Degradation Provide Metabolic Intermediates in Glucose-Starved Cancer Cells
11. Metabolic Alterations in Immune Cells under Glucose Starvation
12. Cell Signaling under Starvation—Orchestration of the Adaptive Response
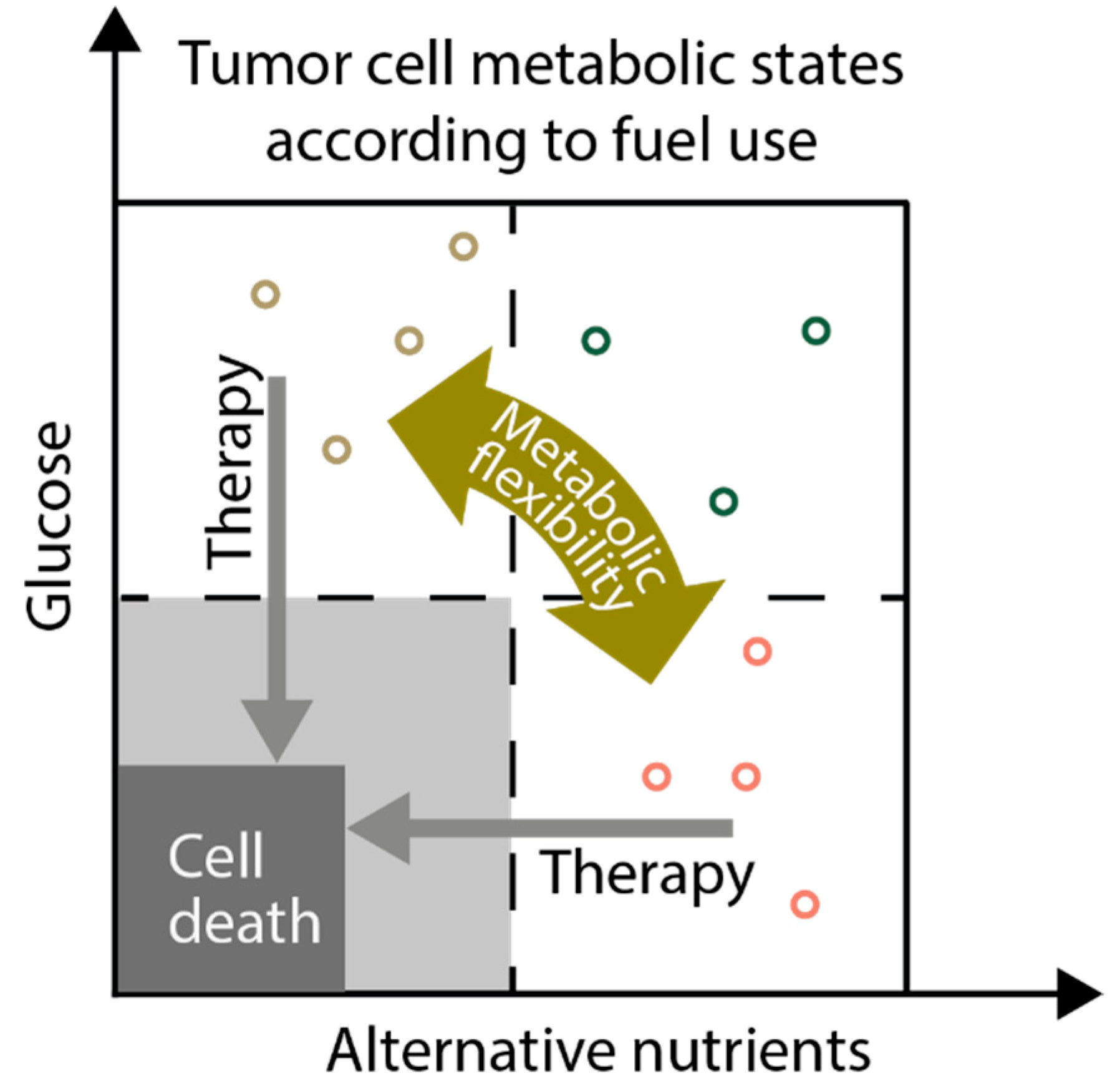
13. Targeting Metabolic Flexibility as Anticancer Strategy
14. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garcia-Canaveras, J.C.; Chen, L.; Rabinowitz, J.D. The Tumor Metabolic Microenvironment: Lessons from Lactate. Cancer Res. 2019, 79, 3155–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2019, 21, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; De Berardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell. Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer Invasion and the Microenvironment: Plasticity and Reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of Cancer Cell Metabolism. Nat. Rev. Cancer. 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Palm, W.; Thompson, C.B. Nutrient Acquisition Strategies of Mammalian Cells. Nature 2017, 546, 234–242. [Google Scholar] [CrossRef]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The Impact of O2 Availability on Human Cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Fendt, S.M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer. Discov. 2020, 10, 1797–1807. [Google Scholar] [CrossRef]
- Kreuzaler, P.; Panina, Y.; Segal, J.; Yuneva, M. Adapt and Conquer: Metabolic Flexibility in Cancer Growth, Invasion and Evasion. Mol. Metab. 2020, 33, 83–101. [Google Scholar] [CrossRef]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in Cancer Cells Repurposing of a Starvation-Induced Metabolic Pathway? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, C. Gluconeogenesis in Cancer: Function and Regulation of PEPCK, FBPase, and G6Pase. Trends Cancer. 2019, 5, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Otto, A.M. Metabolic Constants and Plasticity of Cancer Cells in a Limiting Glucose and Glutamine microenvironment—A Pyruvate Perspective. Front. Oncol. 2020, 10, 596197. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Venneti, S. Glutamine Metabolism in Brain Tumors. Cancers (Basel) 2019, 11, 1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph Protects Metastasizing Melanoma Cells from Ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Psallidas, I.; Kalomenidis, I.; Porcel, J.M.; Robinson, B.W.; Stathopoulos, G.T. Malignant Pleural Effusion: From Bench to Bedside. Eur. Respir. Rev. 2016, 25, 189–198. [Google Scholar] [CrossRef]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-Induced CD36 Expression Drives Ovarian Cancer Progression and Metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes Promote Ovarian Cancer Metastasis and Provide Energy for Rapid Tumor Growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.L. Hypoxia a Key Regulatory Factor in Tumour Growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Vaupel, P. Tumor Microenvironmental Physiology and its Implications for Radiation Oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling Hypoxia and Free Radicals Regulate Angiogenesis and Radiotherapy Response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb. Perspect. Med. 2016, 6, a027094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Cao, G.; Sun, X.; Lee, J.; Rubin, D.L.; Napel, S.; Kurian, A.W.; Daniel, B.L.; Li, R. Intratumoral Spatial Heterogeneity at Perfusion MR Imaging Predicts Recurrence-Free Survival in Locally Advanced Breast Cancer Treated with Neoadjuvant Chemotherapy. Radiology 2018, 288, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Park, C.M.; Park, S.J.; Yoon, J.H.; Hahn, S.; Goo, J.M. Tumor Heterogeneity in Lung Cancer: Assessment with Dynamic Contrast-Enhanced MR Imaging. Radiology 2016, 280, 940–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell. Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose Deprivation Contributes to the Development of KRAS Pathway Mutations in Tumor Cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative Metabolome Profiling of Colon and Stomach Cancer Microenvironment by Capillary Electrophoresis Time-of-Flight Mass Spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef] [Green Version]
- Duarte, I.F.; Rocha, C.M.; Barros, A.S.; Gil, A.M.; Goodfellow, B.J.; Carreira, I.M.; Bernardo, J.; Gomes, A.; Sousa, V.; Carvalho, L. Can Nuclear Magnetic Resonance (NMR) Spectroscopy Reveal Different Metabolic Signatures for Lung Tumours? Virchows Arch. 2010, 457, 715–725. [Google Scholar] [CrossRef]
- Rocha, C.M.; Barros, A.S.; Gil, A.M.; Goodfellow, B.J.; Humpfer, E.; Spraul, M.; Carreira, I.M.; Melo, J.B.; Bernardo, J.; Gomes, A.; et al. Metabolic Profiling of Human Lung Cancer Tissue by 1H High Resolution Magic Angle Spinning (HRMAS) NMR Spectroscopy. J. Proteome. Res. 2010, 9, 319–332. [Google Scholar] [CrossRef]
- Rocha, C.M.; Barros, A.S.; Goodfellow, B.J.; Carreira, I.M.; Gomes, A.; Sousa, V.; Bernardo, J.; Carvalho, L.; Gil, A.M.; Duarte, I.F. NMR Metabolomics of Human Lung Tumours Reveals Distinct Metabolic Signatures for Adenocarcinoma and Squamous Cell Carcinoma. Carcinogenesis 2015, 36, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of Microenvironmental Metabolites in Murine Cancers Reveals Determinants of Tumor Nutrient Availability. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- De Heer, E.C.; Jalving, M.; Harris, A.L. HIFs, Angiogenesis, and Metabolism: Elusive Enemies in Breast Cancer. J. Clin. Investig. 2020, 130, 5074–5087. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Bartman, C.R.; Jankowski, C.S.R.; Zhang, Z.; Xu, X.; Xing, X.; Wang, L.; Lu, W.; Hui, S.; Rabinowitz, J.D. The Source of Glycolytic Intermediates in Mammalian Tissues. Cell. Metab. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Favaro, E.; Harris, A.L. Glycogen Metabolism in Cancer. Biochem. Pharmacol. 2014, 92, 3–11. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic Determinants of Cancer Cell Sensitivity to Glucose Limitation and Biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Galons, J.P.; Fantini, J.; Vion-Dury, J.; Cozzone, P.J.; Canioni, P. Metabolic Changes in Undifferentiated and Differentiated Human Colon Adenocarcinoma Cells Studied by Multinuclear Magnetic Resonance Spectroscopy. Biochimie 1989, 71, 949–961. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting Lactate-Fueled Respiration Selectively Kills Hypoxic Tumor Cells in Mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Feron, O. Cancer Cell Metabolism and Mitochondria: Nutrient Plasticity for TCA Cycle Fueling. Biochim. Biophys. Acta Rev. Cancer. 2017, 1868, 7–15. [Google Scholar] [CrossRef]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell. 2016, 36, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Glycolysis and Gluconeogenesis. In Biochemistry; W. H. Freeman: New York, NY, USA, 2011. [Google Scholar]
- Muir, A.; Danai, L.V.; Gui, D.Y.; Waingarten, C.Y.; Lewis, C.A.; Vander Heiden, M.G. Environmental Cystine Drives Glutamine Anaplerosis and Sensitizes Cancer Cells to Glutaminase Inhibition. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P.; Wilson, M.C. The Monocarboxylate Transporter Family--Role and Regulation. IUBMB Life 2012, 64, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Javaeed, A.; Ghauri, S.K. MCT4 has a Potential to be used as a Prognostic Biomarker a Systematic Review and Meta-Analysis. Oncol. Rev. 2019, 13, 403. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking Lactate Export by Inhibiting the Myc Target MCT1 Disables Glycolysis and Glutathione Synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [Green Version]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate Transporters in Cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Certo, M.; Tsai, C.H.; Pucino, V.; Ho, P.C.; Mauro, C. Lactate Modulation of Immune Responses in Inflammatory Versus Tumour Microenvironments. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N. Navigating Metabolism, 1st ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Kurhanewicz, J.; Vigneron, D.B.; Ardenkjaer-Larsen, J.H.; Bankson, J.A.; Brindle, K.; Cunningham, C.H.; Gallagher, F.A.; Keshari, K.R.; Kjaer, A.; Laustsen, C.; et al. Hyperpolarized (13)C MRI: Path to Clinical Translation in Oncology. Neoplasia 2019, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Gammon, S.; Zacharias, N.M.; Liu, T.; Salzillo, T.; Xi, Y.; Wang, J.; Bhattacharya, P.; Piwnica-Worms, D. Hyperpolarized [1-(13)C]Pyruvate-to-[1-(13)C]Lactate Conversion is Rate-Limited by Monocarboxylate Transporter-1 in the Plasma Membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 22378–22389. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Abu-Remaileh, M.; Kanarek, N.; Freinkman, E.; Gao, X.; Louissaint, A., Jr.; Lewis, C.A.; Sabatini, D.M. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 2017, 169, 258–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate Dependence of Tumors. Cell 2014, 159, 1591–1602. [Google Scholar] [CrossRef] [Green Version]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth Under Metabolic Stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Bulusu, V.; Tumanov, S.; Michalopoulou, E.; van den Broek, N.J.; MacKay, G.; Nixon, C.; Dhayade, S.; Schug, Z.T.; Vande Voorde, J.; Blyth, K.; et al. Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell. Rep. 2017, 18, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Sellers, K.; Fox, M.P.; Bousamra, M., II; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate Carboxylase is Critical for Non-Small-Cell Lung Cancer Proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K.; Elia, I.; Buescher, J.M.; Orth, M.F.; Davidson, S.M.; et al. Breast Cancer-Derived Lung Metastases show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell. Rep. 2016, 17, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Vriezen, N.; Romein, B.; Luyben, K.C.; van Dijken, J.P. Effects of Glutamine Supply on Growth and Metabolism of Mammalian Cells in Chemostat Culture. Biotechnol. Bioeng. 1997, 54, 272–286. [Google Scholar] [CrossRef]
- Chen, Y.J.; Mahieu, N.G.; Huang, X.; Singh, M.; Crawford, P.A.; Johnson, S.L.; Gross, R.W.; Schaefer, J.; Patti, G.J. Lactate Metabolism is Associated with Mammalian Mitochondria. Nat. Chem. Biol. 2016, 12, 937–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakimi, P.; Johnson, M.T.; Yang, J.; Lepage, D.F.; Conlon, R.A.; Kalhan, S.C.; Reshef, L.; Tilghman, S.M.; Hanson, R.W. Phosphoenolpyruvate Carboxykinase and the Critical Role of Cataplerosis in the Control of Hepatic Metabolism. Nutr. Metab. (Lond.) 2005, 2, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, R.W.; Reshef, L. Glyceroneogenesis Revisited. Biochimie 2003, 85, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kalhan, S.C.; Hanson, R.W. What is the Metabolic Role of Phosphoenolpyruvate Carboxykinase? J. Biol. Chem. 2009, 284, 27025–27029. [Google Scholar] [CrossRef] [Green Version]
- Beale, E.G.; Harvey, B.J.; Forest, C. PCK1 and PCK2 as Candidate Diabetes and Obesity Genes. Cell Biochem. Biophys. 2007, 48, 89–95. [Google Scholar] [CrossRef]
- Leithner, K.; Hrzenjak, A.; Trotzmuller, M.; Moustafa, T.; Kofeler, H.C.; Wohlkoenig, C.; Stacher, E.; Lindenmann, J.; Harris, A.L.; Olschewski, A.; et al. PCK2 Activation Mediates an Adaptive Response to Glucose Depletion in Lung Cancer. Oncogene 2015, 34, 1044–1050. [Google Scholar] [CrossRef]
- Mendez-Lucas, A.; Hyrossova, P.; Novellasdemunt, L.; Vinals, F.; Perales, J.C. Mitochondrial Phosphoenolpyruvate Carboxykinase (PEPCK-M) is a Pro-Survival, Endoplasmic Reticulum (ER) Stress Response Gene Involved in Tumor Cell Adaptation to Nutrient Availability. J. Biol. Chem. 2014, 289, 22090–22102. [Google Scholar] [CrossRef] [Green Version]
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.C.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choiniere, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell 2015, 60, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Lee, H.J.; Wu, X.; Huo, L.; Kim, S.J.; Xu, L.; Wang, Y.; He, J.; Bollu, L.R.; Gao, G.; et al. Gain of Glucose-Independent Growth upon Metastasis of Breast Cancer Cells to the Brain. Cancer Res. 2015, 75, 554–565. [Google Scholar] [CrossRef] [Green Version]
- Montal, E.D.; Dewi, R.; Bhalla, K.; Ou, L.; Hwang, B.J.; Ropell, A.E.; Gordon, C.; Liu, W.J.; DeBerardinis, R.J.; Sudderth, J.; et al. PEPCK Coordinates the Regulation of Central Carbon Metabolism to Promote Cancer Cell Growth. Mol. Cell 2015, 60, 571–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Li, J.; Fan, T.W.M.; Hou, S.X. Glycolytic Reprogramming through PCK2 Regulates Tumor Initiation of Prostate Cancer Cells. Oncotarget 2017, 8, 83602–83618. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.Y.; Jiang, S.S.; Shan, Y.S.; Hung, W.C.; Chen, M.H.; Lin, H.Y.; Chen, Y.L.; Tsai, H.J.; Chen, L.T. Mitochondrial Phosphoenolpyruvate Carboxykinase (PEPCK-M) Regulates the Cell Metabolism of Pancreatic Neuroendocrine Tumors (pNET) and De-Sensitizes pNET to mTOR Inhibitors. Oncotarget 2017, 8, 103613–103625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, M.; Dorfman, R.G.; Pan, Y.; Tang, D.; Xu, L.; Zhao, Z.; Zhou, Q.; Zhou, L.; Wang, Y.; et al. SIRT2 Promotes the Migration and Invasion of Gastric Cancer through RAS/ERK/JNK/MMP-9 Pathway by Increasing PEPCK1-Related Metabolism. Neoplasia 2018, 20, 745–756. [Google Scholar] [CrossRef]
- Leithner, K.; Triebl, A.; Trotzmuller, M.; Hinteregger, B.; Leko, P.; Wieser, B.I.; Grasmann, G.; Bertsch, A.L.; Zullig, T.; Stacher, E.; et al. The Glycerol Backbone of Phospholipids Derives from Noncarbohydrate Precursors in Starved Lung Cancer Cells. Proc. Natl. Acad. Sci. USA 2018, 115, 6225–6230. [Google Scholar] [CrossRef] [Green Version]
- Montal, E.D.; Bhalla, K.; Dewi, R.E.; Ruiz, C.F.; Haley, J.A.; Ropell, A.E.; Gordon, C.; Haley, J.D.; Girnun, G.D. Inhibition of Phosphoenolpyruvate Carboxykinase Blocks Lactate Utilization and Impairs Tumor Growth in Colorectal Cancer. Cancer. Metab. 2019, 7. [Google Scholar] [CrossRef]
- Hodakoski, C.; Hopkins, B.D.; Zhang, G.; Su, T.; Cheng, Z.; Morris, R.; Rhee, K.Y.; Goncalves, M.D.; Cantley, L.C. Rac-Mediated Macropinocytosis of Extracellular Protein Promotes Glucose Independence in Non-Small Cell Lung Cancer. Cancers (Basel) 2019, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Felici, J.; Hyroššová, P.; Aragó, M.; Rodríguez-Arévalo, S.; García-Rovés, P.M.; Escolano, C.; Perales, J.C. Phosphoenolpyruvate from Glycolysis and PEPCK Regulate Cancer Cell Fate by Altering Cytosolic Ca(2). Cells 2019, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Hussain, R.; Shaukat, Z.; Khan, M.; Saint, R.; Gregory, S.L. Phosphoenolpyruvate Carboxykinase Maintains Glycolysis-Driven Growth in Drosophila Tumors. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.X.; Jin, L.; Sun, S.J.; Liu, P.; Feng, X.; Cheng, Z.L.; Liu, W.R.; Guan, K.L.; Shi, Y.H.; Yuan, H.X.; et al. Metabolic Reprogramming by PCK1 Promotes TCA Cataplerosis, Oxidative Stress and Apoptosis in Liver Cancer Cells and Suppresses Hepatocellular Carcinoma. Oncogene 2018, 37, 1637–1653. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, Y.; Wang, C.; Sun, Z.; Li, L.; Cheng, S.; Zhou, W. Overexpression of PCK1 Gene Antagonizes Hepatocellular Carcinoma through the Activation of Gluconeogenesis and Suppression of Glycolysis Pathways. Cell. Physiol. Biochem. 2018, 47, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.L.; Chen, H.Z.; Yang, P.B.; Li, Y.P.; Zhang, F.N.; Zhang, J.Y.; Wang, W.J.; Zhao, W.X.; Zhang, S.; Chen, Q.T.; et al. Nur77 Suppresses Hepatocellular Carcinoma Via Switching Glucose Metabolism Toward Gluconeogenesis through Attenuating Phosphoenolpyruvate Carboxykinase Sumoylation. Nat. Commun. 2017, 8, 14420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuo, L.; Xiang, J.; Pan, X.; Gao, Q.; Zhang, G.; Yang, Y.; Liang, L.; Xia, J.; Wang, K.; Tang, N. PCK1 Downregulation Promotes TXNRD1 Expression and Hepatoma Cell Growth Via the Nrf2/Keap1 Pathway. Front. Oncol. 2018, 8, 611. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, Z.; Xia, Y.; Shao, F.; Xia, W.; Wei, Y.; Li, X.; Qian, X.; Lee, J.H.; Du, L.; et al. The Gluconeogenic Enzyme PCK1 Phosphorylates INSIG1/2 for Lipogenesis. Nature 2020, 580, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Keshet, R.; Lee, J.S.; Adler, L.; Iraqi, M.; Ariav, Y.; Lim, L.Q.J.; Lerner, S.; Rabinovich, S.; Oren, R.; Katzir, R.; et al. Targeting Purine Synthesis in ASS1-Expressing Tumors Enhances the Response to Immune Checkpoint Inhibitors. Nature Cancer 2020, 1, 894–908. [Google Scholar] [CrossRef]
- Sun, Y.; Gao, J.; Jing, Z.; Zhao, Y.; Sun, Y.; Zhao, X. PURα Promotes the Transcriptional Activation of PCK2 in Oesophageal Squamous Cell Carcinoma Cells. Genes (Basel) 2020, 11, 1301. [Google Scholar] [CrossRef]
- Carroll, P.A.; Diolaiti, D.; McFerrin, L.; Gu, H.; Djukovic, D.; Du, J.; Cheng, P.F.; Anderson, S.; Ulrich, M.; Hurley, J.B.; et al. Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis. Cancer Cell. 2015, 27, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Smolle, E.; Leko, P.; Stacher-Priehse, E.; Brcic, L.; El-Heliebi, A.; Hofmann, L.; Quehenberger, F.; Hrzenjak, A.; Popper, H.H.; Olschewski, H.; et al. Distribution and Prognostic Significance of Gluconeogenesis and Glycolysis in Lung Cancer. Mol. Oncol. 2020, 14, 2853–2867. [Google Scholar] [CrossRef]
- Semakova, J.; Hyroššová, P.; Méndez-Lucas, A.; Cutz, E.; Bermudez, J.; Burgess, S.; Alcántara, S.; Perales, J.C. PEPCK-C Reexpression in the Liver Counters Neonatal Hypoglycemia in Pck1 (del/del) Mice, Unmasking Role in Non-Gluconeogenic Tissues. J. Physiol. Biochem. 2017, 73, 89–98. [Google Scholar] [CrossRef]
- Abulizi, A.; Cardone, R.L.; Stark, R.; Lewandowski, S.L.; Zhao, X.; Hillion, J.; Ma, L.; Sehgal, R.; Alves, T.C.; Thomas, C.; et al. Multi-Tissue Acceleration of the Mitochondrial Phosphoenolpyruvate Cycle Improves Whole-Body Metabolic Health. Cell Metab. 2020, 32, 751–766.e11. [Google Scholar] [CrossRef]
- Arago, M.; Moreno-Felici, J.; Abas, S.; Rodriguez-Arevalo, S.; Hyrossova, P.; Figueras, A.; Vinals, F.; Perez, B.; Loza, M.I.; Brea, J.; et al. Pharmacology and Preclinical Validation of a Novel Anticancer Compound Targeting PEPCK-M. Biomed. Pharmacother. 2020, 121, 109601. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig, T.; Turrado, C.; Benhamú, B.; Aguilar, H.; Relat, J.; Ortega-Gutiérrez, S.; Casals, G.; Marrero, P.F.; Urruticoechea, A.; Haro, D.; et al. Novel Inhibitors of Fatty Acid Synthase with Anticancer Activity. Clin. Cancer Res. 2009, 15, 7608–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-Transformed Cells Support Growth by Scavenging Unsaturated Fatty Acids from Lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [Green Version]
- Rohrig, F.; Schulze, A. The Multifaceted Roles of Fatty Acid Synthesis in Cancer. Nat. Rev. Cancer. 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of De Novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. eBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [Green Version]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte Lipolysis Links Obesity to Breast Cancer Growth: Adipocyte-Derived Fatty Acids Drive Breast Cancer Cell Proliferation and Migration. Cancer Metab. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Santi, A.; Caselli, A.; Ranaldi, F.; Paoli, P.; Mugnaioni, C.; Michelucci, E.; Cirri, P. Cancer Associated Fibroblasts Transfer Lipids and Proteins to Cancer Cells through Cargo Vesicles Supporting Tumor Growth. Biochim. Biophys. Acta 2015, 1853, 3211–3223. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.; Cheng, Z.; Yang, X.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of Lipid Metabolism in Cancer-Associated Fibroblasts Potentiates Migration of Colorectal Cancer Cells. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting Metastasis-Initiating Cells through the Fatty Acid Receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Lucas, A.; Lin, W.; Driscoll, P.C.; Legrave, N.; Novellasdemunt, L.; Xie, C.; Charles, M.; Wilson, Z.; Jones, N.P.; Rayport, S.; et al. Identifying Strategies to Target the Metabolic Flexibility of Tumours. Nat. Metab. 2020, 2, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Zechner, R. Biochemistry and Pathophysiology of Intravascular and Intracellular Lipolysis. Genes Dev. 2013, 27, 459–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozeveld, C.N.; Johnson, K.M.; Zhang, L.; Razidlo, G.L. KRAS Controls Pancreatic Cancer Cell Lipid Metabolism and Invasive Potential through the Lipase HSL. Cancer Res. 2020, 80, 4932–4945. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Xie, H.; Heier, C.; Huang, J.; Zheng, Q.; Eichmann, T.O.; Schoiswohl, G.; Ni, J.; Zechner, R.; Ni, S.; et al. Enhanced Monoacylglycerol Lipolysis by ABHD6 Promotes NSCLC Pathogenesis. eBioMedicine 2020, 53, 102696. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty Acid Uptake and Lipid Storage Induced by HIF-1alpha Contribute to Cell Growth and Survival After Hypoxia-Reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, D.; Tumanov, S.; Qiu, B.; Michalopoulou, E.; Spata, M.; Azzam, A.; Xie, H.; Simon, M.C.; Kamphorst, J.J. Triglycerides Promote Lipid Homeostasis during Hypoxic Stress by Balancing Fatty Acid Saturation. Cell Rep. 2018, 24, 2596–2605.e5. [Google Scholar] [CrossRef] [Green Version]
- Blunsom, N.J.; Cockcroft, S. Phosphatidylinositol Synthesis at the Endoplasmic Reticulum. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158471. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Fatty acid metabolism. In Biochemistry; W. H. Freeman: New York, NY, USA, 2011. [Google Scholar]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of Protein is an Amino Acid Supply Route in Ras-Transformed Cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Nguyen, T.T.; Ravi, A.; Kubiniok, P.; Finicle, B.T.; Jayashankar, V.; Malacrida, L.; Hou, J.; Robertson, J.; Gao, D.; et al. PTEN Deficiency and AMPK Activation Promote Nutrient Scavenging and Anabolism in Prostate Cancer Cells. Cancer Discov. 2018, 8, 866–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finicle, B.T.; Jayashankar, V.; Edinger, A.L. Nutrient Scavenging in Cancer. Nat. Rev. Cancer 2018, 18, 619–633. [Google Scholar] [CrossRef] [PubMed]
- King, B.; Araki, J.; Palm, W.; Thompson, C.B. Yap/Taz Promote the Scavenging of Extracellular Nutrients through Macropinocytosis. Genes Dev. 2020, 34, 1345–1358. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent Insights into the Function of Autophagy in Cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer. 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Zhou, Y.; Yao, J.; Qiao, C.; Ni, T.; Guo, R.; Guo, Q.; Lu, N. Lactate Promotes PGE2 Synthesis and Gluconeogenesis in Monocytes to Benefit the Growth of Inflammation-Associated Colorectal Tumor. Oncotarget 2015, 6, 16198–16214. [Google Scholar] [CrossRef] [Green Version]
- Püschel, F.; Favaro, F.; Redondo-Pedraza, J.; Lucendo, E.; Iurlaro, R.; Marchetti, S.; Majem, B.; Eldering, E.; Nadal, E.; Ricci, J.E.; et al. Starvation and Antimetabolic Therapy Promote Cytokine Release and Recruitment of Immune Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 9932–9941. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Ji, T.; Zhang, H.; Dong, W.; Chen, X.; Xu, P.; Chen, D.; Liang, X.; Yin, X.; Liu, Y.; et al. A Pck1-Directed Glycogen Metabolic Program Regulates Formation and Maintenance of Memory CD8(+) T Cells. Nat. Cell Biol. 2018, 20, 21–27. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell Death Induced by Endoplasmic Reticulum Stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational Control is Required for the Unfolded Protein Response and in Vivo Glucose Homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Wang, C.; Huang, Z.; Du, Y.; Cheng, Y.; Chen, S.; Guo, F. ATF4 Regulates Lipid Metabolism and Thermogenesis. Cell Res. 2010, 20, 174–184. [Google Scholar] [CrossRef]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 Pathway is Critical for Tumour Cell Survival and Proliferation in Response to Nutrient Deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Koo, S.H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB Coactivator TORC2 is a Key Regulator of Fasting Glucose Metabolism. Nature 2005, 437, 1109–1111. [Google Scholar] [CrossRef]
- Harris, T.E.; Lawrence, J.C., Jr. TOR Signaling. Sci. STKE 2003, 2003, re15. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Orozco, J.M.; Krawczyk, P.A.; Scaria, S.M.; Cangelosi, A.L.; Chan, S.H.; Kunchok, T.; Lewis, C.A.; Sabatini, D.M. Dihydroxyacetone Phosphate Signals Glucose Availability to mTORC1. Nat. Metab. 2020, 2, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Senoo, H.; Wai, M.; Matsubayashi, H.T.; Sesaki, H.; Iijima, M. Hetero-Oligomerization of Rho and Ras GTPases Connects GPCR Activation to mTORC2-AKT Signaling. Cell. Rep. 2020, 33, 108427. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K Pathway in Cancer: Are we Making Headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Gremke, N.; Polo, P.; Dort, A.; Schneikert, J.; Elmshäuser, S.; Brehm, C.; Klingmüller, U.; Schmitt, A.; Reinhardt, H.C.; Timofeev, O.; et al. MTOR-Mediated Cancer Drug Resistance Suppresses Autophagy and Generates a Druggable Metabolic Vulnerability. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B. New Antimetabolites in Cancer Chemotherapy and their Clinical Impact. Br. J. Cancer 1998, 78, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Meena, A.S.; Bhat, M.K. Targeting Metabolic Flexibility by Simultaneously Inhibiting Respiratory Complex I and Lactate Generation Retards Melanoma Progression. Oncotarget 2015, 6, 37281–37299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, A.; Nambu, T.; Ebara, S.; Hasegawa, Y.; Toyoshima, K.; Tsuchiya, Y.; Tomita, D.; Fujimoto, J.; Kurasawa, O.; Takahara, C.; et al. Inhibition of GCN2 Sensitizes ASNS-Low Cancer Cells to Asparaginase by Disrupting the Amino Acid Response. Proc. Natl. Acad. Sci. USA 2018, 115, E7776–E7785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurelac, I.; Iommarini, L.; Vatrinet, R.; Amato, L.B.; De Luise, M.; Leone, G.; Girolimetti, G.; Umesh Ganesh, N.; Bridgeman, V.L.; Ombrato, L.; et al. Inducing Cancer Indolence by Targeting Mitochondrial Complex I is Potentiated by Blocking Macrophage-Mediated Adaptive Responses. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Schlappack, O.K.; Zimmermann, A.; Hill, R.P. Glucose Starvation and Acidosis: Effect on Experimental Metastatic Potential, DNA Content and MTX Resistance of Murine Tumour Cells. Br. J. Cancer 1991, 64, 663–670. [Google Scholar] [CrossRef] [Green Version]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic Deletion of Malic Enzyme 2 Confers Collateral Lethality in Pancreatic Cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grasmann, G.; Mondal, A.; Leithner, K. Flexibility and Adaptation of Cancer Cells in a Heterogenous Metabolic Microenvironment. Int. J. Mol. Sci. 2021, 22, 1476. https://doi.org/10.3390/ijms22031476
Grasmann G, Mondal A, Leithner K. Flexibility and Adaptation of Cancer Cells in a Heterogenous Metabolic Microenvironment. International Journal of Molecular Sciences. 2021; 22(3):1476. https://doi.org/10.3390/ijms22031476
Chicago/Turabian StyleGrasmann, Gabriele, Ayusi Mondal, and Katharina Leithner. 2021. "Flexibility and Adaptation of Cancer Cells in a Heterogenous Metabolic Microenvironment" International Journal of Molecular Sciences 22, no. 3: 1476. https://doi.org/10.3390/ijms22031476
APA StyleGrasmann, G., Mondal, A., & Leithner, K. (2021). Flexibility and Adaptation of Cancer Cells in a Heterogenous Metabolic Microenvironment. International Journal of Molecular Sciences, 22(3), 1476. https://doi.org/10.3390/ijms22031476