
Does C-C Motif Chemokine Ligand 2 (CCL2) Link Obesity to a Pro-Inflammatory State?


Abstract
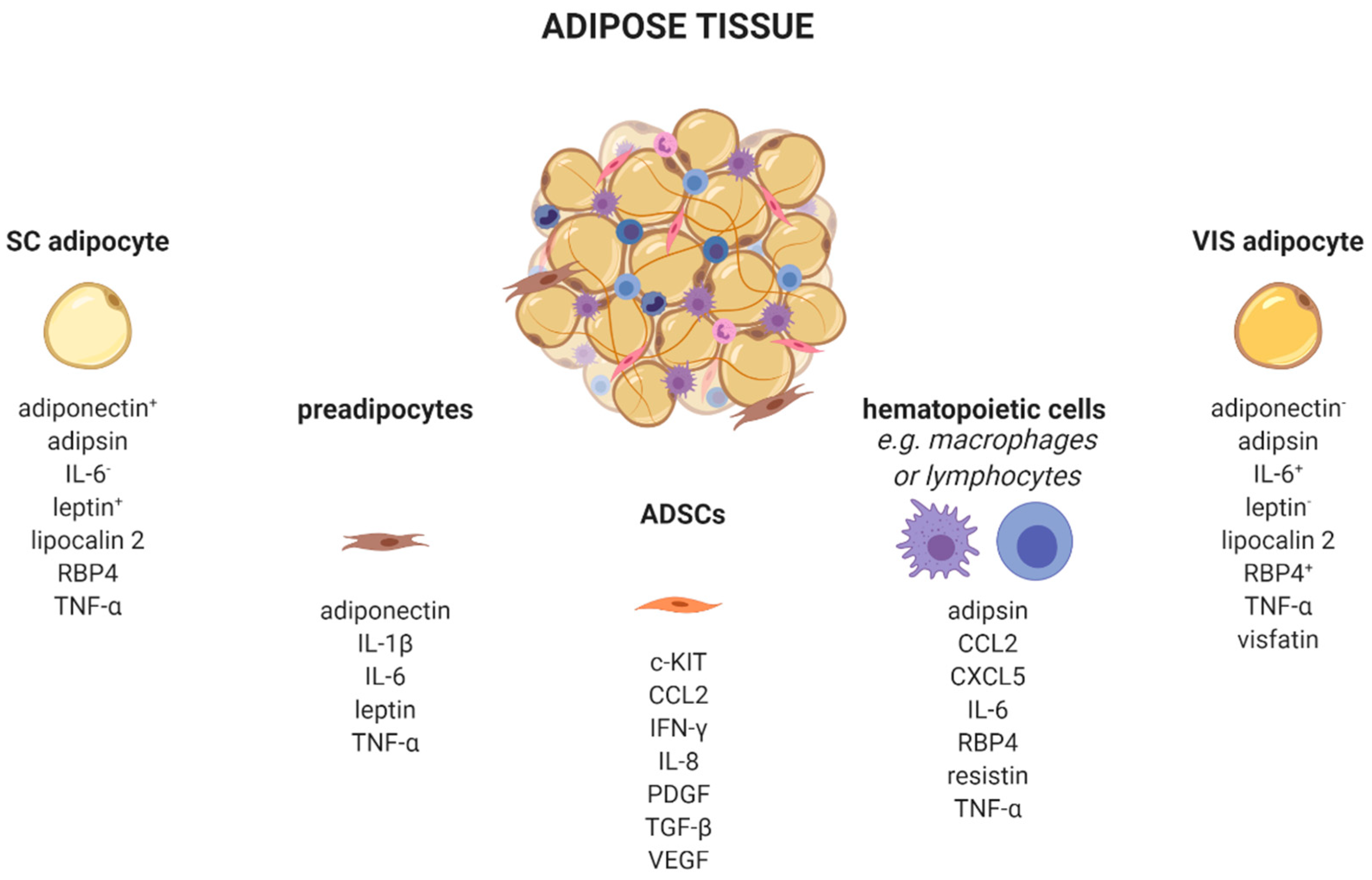
:1. Introduction
2. C-C Motif Chemokine Ligand 2
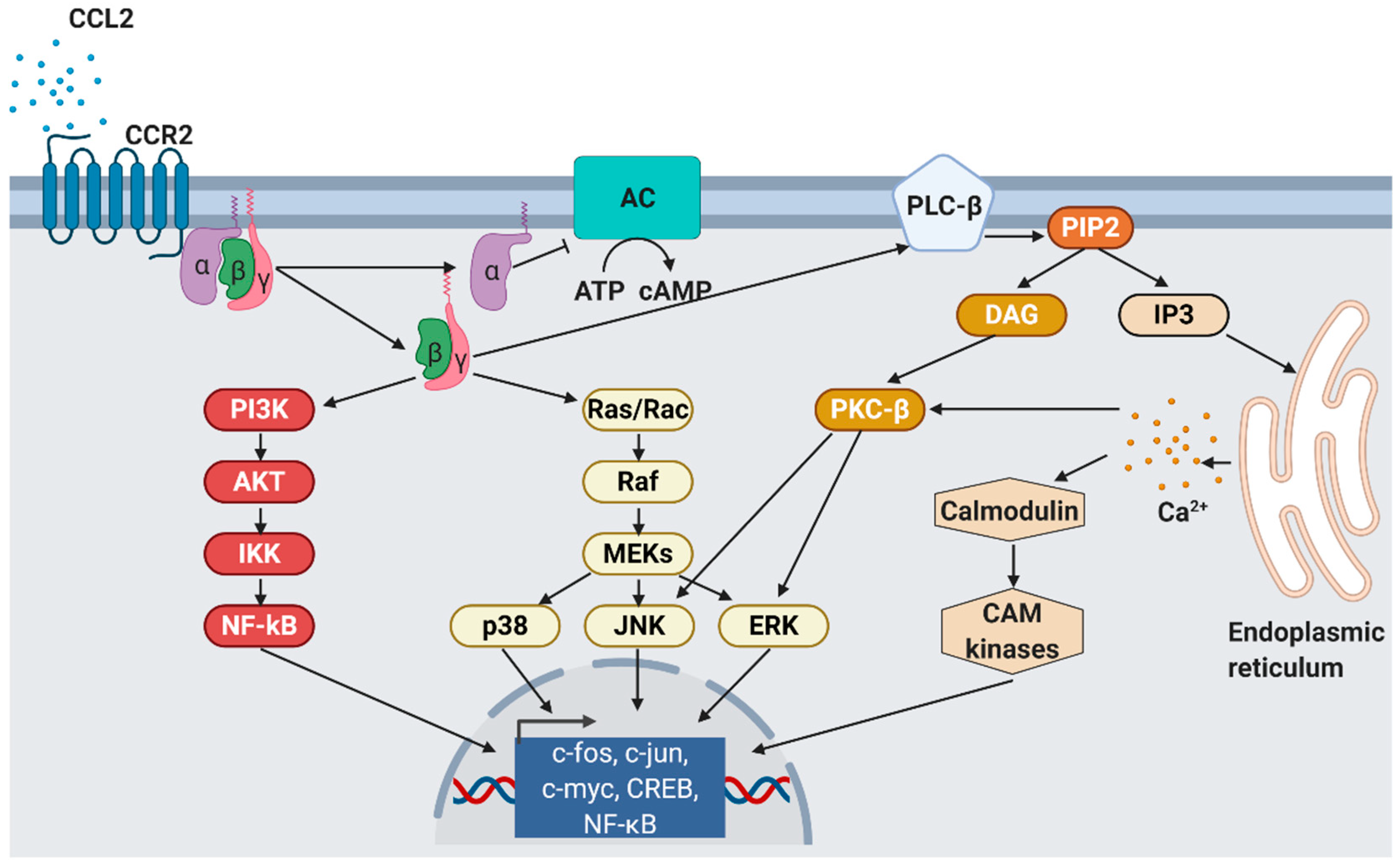
2.1. Structure, Sources and Signaling
2.2. Animal Models
2.3. Human Mutations in the CCL2 Pathway and Associated Diseases
2.4. Effects of Antibody Administration Affecting CCL2/CCR2 Signaling
2.5. CCL2 in Obesity and Obesity Related Diseases
2.5.1. CCL2 Reflects a Pro-Inflammatory State
2.5.2. CCL2 and Insulin Resistance
2.5.3. CCL2 and Cardiovascular Diseases
2.6. CCL2 as Drug Target
3. Summary and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| AdRiKO | Adipose-specific Rictor knockout |
| ADSCs | Adipose-derived stem cells |
| ASCVD | Atherosclerotic cardiovascular disease |
| AT | Adipose tissue |
| BMI | Body mass index |
| CaM | Calmodulin |
| cAMP | Cyclic adenosine monophosphate |
| CCL2 | C-C motif chemokine ligand 2 |
| CCR | CC chemokine receptor |
| c-kit | KIT proto-oncogene, receptor tyrosine kinase |
| CNS | Central nervous system |
| CRP | C-reactive protein |
| CXCL | C-X-C motif chemokine ligand |
| DAG | Diacylglycerol |
| DN | Diabetic nephropathy |
| ERK | Extracellular signal-regulated kinase |
| FAE | Femoral artery excision |
| FOXK1 | Forkhead box K1 |
| GDP | Guanosine diphosphate |
| GLUT4 | Glucose transporter 4 |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GPCR | G-protein coupled receptor |
| GTP | Guanosine triphosphate |
| HFD | High-fat diet |
| HIV | Human immunodeficiency virus |
| IFN-γ | Interferon gamma |
| IKK | Inhibitor of nuclear factor-κB kinase |
| IL | Interleukin |
| IP3 | Inositol 1,4,5-trisphosphate |
| IRF | Interferon regulatory factor |
| IκB | Inhibitor of NF-κB |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| LDL | Low density lipoprotein |
| LPS | Lipopolysaccharide |
| MAP | Mitogen-activated protein |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MCPIP1 | MCP-1-induced protein |
| M-CSF | Macrophage colony-stimulating factor |
| MDSC | Myeloid-derived suppressor cells |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NCD | Noncommunicable diseases |
| NF-κB | Nuclear factor-κB |
| NTD | Neural tube defect |
| PAI1 | Plasminogen activator inhibitor-1 |
| PDGF | Platelet-derived growth factor |
| PI3K | Phosphatidylinositol 3-kinase |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PKC-β | Protein kinase C-β |
| PP2A | Protein phosphatase 2A |
| RANTES | Regulated upon activation, normal T cell expressed and presumably secreted |
| RBP4 | Retinol binding protein |
| RETN | Resistin |
| RHEB | Ras homolog enriched in brain |
| sc | Subcutaneous |
| SNP | Single nucleotide polymorphism |
| snRNA-seq | Single-nucleus RNA sequencing |
| STAT | Signal transducers and activators of transcription |
| TAMs | Tumor-associated macrophages |
| TGFβ | Transforming growth factor beta |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor alpha |
| TRIF | TIR-domain containing adapter-inducing interferon β |
| VEGF | Vascular-endothelial growth factor |
| vis | Visceral |
| WAT | White adipose tissue |
References
- Obregon, M.-J. Adipose tissues and thyroid hormones. Front. Physiol. 2014, 5, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Lehr, S.; Hartwig, S.; Lamers, D.; Famulla, S.; Müller, S.; Hanisch, F.-G.; Cuvelier, C.; Ruige, J.; Eckardt, K.; Ouwens, D.M.; et al. Identification and validation of novel adipokines released from primary human adipocytes. Mol. Cell. Proteomics 2012, 11, M111.010504. [Google Scholar] [CrossRef] [Green Version]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Arner, P. Editorial: Visfatin—A True Or False Trail To Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2006, 91, 28–30. [Google Scholar] [CrossRef] [Green Version]
- Funahashi, T.; Nakamura, T.; Shimomura, I.; Maeda, K.; Kuriyama, H.; Takahashi, M.; Arita, Y.; Kihara, S.; Matsuzawa, Y. 3. Role of adipocytokines on the pathogenesis of atherosclerosis in visceral obesity. Intern. Med. 1999, 38, 202–206. [Google Scholar] [CrossRef] [Green Version]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Joseph, F. Adipose Tissue and Adipokines: The Association with and Application of Adipokines in Obesity. Scientifica 2014, 2014, 28592. [Google Scholar] [CrossRef]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef] [Green Version]
- Ebert, T.; Gebhardt, C.; Scholz, M.; Wohland, T.; Schleinitz, D.; Fasshauer, M.; Blüher, M.; Stumvoll, M.; Kovacs, P.; Tönjes, A. Relationship between 12 adipocytokines and distinct components of the metabolic syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 1015–1023. [Google Scholar] [CrossRef]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Flehmig, G.; Scholz, M.; Klöting, N.; Fasshauer, M.; Tönjes, A.; Stumvoll, M.; Youn, B.S.; Blüher, M. Identification of adipokine clusters related to parameters of fat mass, insulin sensitivity and inflammation. PLoS ONE 2014, 9, e99785. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Blüher, M.; Stumvoll, M. Adipokines in gestational diabetes. Lancet Diabetes Endocrinol. 2014, 2, 488–499. [Google Scholar] [CrossRef]
- Klöting, N.; Fasshauer, M.; Dietrich, A.; Kovacs, P.; Schön, M.R.; Kern, M.; Stumvoll, M.; Blüher, M. Insulin-sensitive obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E506–E515. [Google Scholar] [CrossRef]
- Ebert, T.; Roth, I.; Richter, J.; Tönjes, A.; Kralisch, S.; Lossner, U.; Kratzsch, J.; Blüher, M.; Stumvoll, M.; Fasshauer, M. Different associations of adipokines in lean and healthy adults. Horm. Metab. Res. 2014, 46, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Tönjes, A.; Fasshauer, M.; Kratzsch, J.; Stumvoll, M.; Blüher, M. Adipokine pattern in subjects with impaired fasting glucose and impaired glucose tolerance in comparison to normal glucose tolerance and diabetes. PLoS ONE 2010, 5, e13911. [Google Scholar] [CrossRef] [Green Version]
- Ahima, R.S. Revisiting leptin’s role in obesity and weight loss. J. Clin. Investig. 2008, 118, 2380–2383. [Google Scholar] [CrossRef] [Green Version]
- Nigro, E.; Scudiero, O.; Monaco, M.L.; Palmieri, A.; Mazzarella, G.; Costagliola, C.; Bianco, A.; Daniele, A. New insight into adiponectin role in obesity and obesity-related diseases. Biomed Res. Int. 2014, 2014, 658913. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Esteve Ràfols, M. Adipose tissue: Cell heterogeneity and functional diversity. Endocrinol. Nutr. 2014, 61, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, P. The role of adipokines in chronic inflammation. Immunol. Targets Ther. 2016, 5, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Dong, H.; Balaz, M.; Slyper, M.; Drokhlyansky, E.; Colleluori, G.; Giordano, A.; Kovanicova, Z.; Stefanicka, P.; Balazova, L.; et al. snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature 2020, 587, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Vidal, H. Gene expression in visceral and subcutaneous adipose tissues. Ann. Med. 2001, 33, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Fried, S.K.; Bunkin, D.A.; Greenberg, A.S. Omental and Subcutaneous Adipose Tissues of Obese Subjects Release Interleukin-6: Depot Difference and Regulation by Glucocorticoid 1. J. Clin. Endocrinol. Metab. 1998, 83, 847–850. [Google Scholar] [CrossRef]
- Yang, R.-Z.; Lee, M.-J.; Hu, H.; Pray, J.; Wu, H.-B.; Hansen, B.C.; Shuldiner, A.R.; Fried, S.K.; McLenithan, J.C.; Gong, D.-W. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: Possible role in modulating insulin action. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1253–E1261. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, Y.; He, W.; Liu, S.; Li, H.; Li, L. Visfatin is regulated by interleukin-6 and affected by the PPAR-γ pathway in BeWo cells. Mol. Med. Rep. 2019, 19, 400–406. [Google Scholar] [CrossRef]
- Klöting, N.; Graham, T.E.; Berndt, J.; Kralisch, S.; Kovacs, P.; Wason, C.J.; Fasshauer, M.; Schön, M.R.; Stumvoll, M.; Blüher, M.; et al. Serum Retinol-Binding Protein Is More Highly Expressed in Visceral than in Subcutaneous Adipose Tissue and Is a Marker of Intra-abdominal Fat Mass. Cell Metab. 2007, 6, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Lo, J.C.; Ljubicic, S.; Leibiger, B.; Kern, M.; Leibiger, I.B.; Moede, T.; Kelly, M.E.; Chatterjee Bhowmick, D.; Murano, I.; Cohen, P.; et al. Adipsin Is an Adipokine that Improves β Cell Function in Diabetes. Cell 2014, 158, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Auguet, T.; Quintero, Y.; Terra, X.; Martínez, S.; Lucas, A.; Pellitero, S.; Aguilar, C.; Hernández, M.; Del Castillo, D.; Richart, C. Upregulation of Lipocalin 2 in Adipose Tissues of Severely Obese Women: Positive Relationship With Proinflammatory Cytokines. Obesity 2011, 19, 2295–2300. [Google Scholar] [CrossRef]
- Winkler, G.; Kiss, S.; Keszthelyi, L.; Sápi, Z.; Ory, I.; Salamon, F.; Kovács, M.; Vargha, P.; Szekeres, O.; Speer, G.; et al. Expression of tumor necrosis factor (TNF)-alpha protein in the subcutaneous and visceral adipose tissue in correlation with adipocyte cell volume, serum TNF-alpha, soluble serum TNF-receptor-2 concentrations and C-peptide level. Eur. J. Endocrinol. 2003, 149, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.Y.; Sharma, R.; Gase, G.; Ussar, S.; Li, Y.; Welch, L.; Berryman, D.E.; Kispert, A.; Blüher, M.; Kahn, C.R. Tbx15 Defines a Glycolytic Subpopulation and White Adipocyte Heterogeneity. Diabetes 2017, 66, 2822–2829. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Luong, Q.; Sharma, R.; Dreyfuss, J.M.; Ussar, S.; Kahn, C.R. Developmental and functional heterogeneity of white adipocytes within a single fat depot. EMBO J. 2019, 38, e99291. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.K.; Dankel, S.N.; Rastegarpanah, B.; Cai, W.; Xue, R.; Crovella, M.; Tseng, Y.; Kahn, C.R.; Kasif, S. Single-cell transcriptional networks in differentiating preadipocytes suggest drivers associated with tissue heterogeneity. Nat. Commun. 2020, 11, 2117. [Google Scholar] [CrossRef] [PubMed]
- Preisner, F.; Leimer, U.; Sandmann, S.; Zoernig, I.; Germann, G.; Koellensperger, E. Impact of Human Adipose Tissue-Derived Stem Cells on Malignant Melanoma Cells in An In Vitro Co-culture Model. Stem Cell Rev. Rep. 2018, 14, 125–140. [Google Scholar] [CrossRef]
- Salha, S.; Gehmert, S.; Brébant, V.; Anker, A.; Loibl, M.; Prantl, L.; Gehmert, S. PDGF regulated migration of mesenchymal stem cells towards malignancy acts via the PI3K signaling pathway. Clin. Hemorheol. Microcirc. 2019, 70, 543–551. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Qian, C. c-Kit-Positive Adipose Tissue-Derived Mesenchymal Stem Cells Promote the Growth and Angiogenesis of Breast Cancer. Biomed Res. Int. 2017, 2017, 7407168. [Google Scholar] [CrossRef]
- Zimmerlin, L.; Park, T.S.; Zambidis, E.T.; Donnenberg, V.S.; Donnenberg, A.D. Mesenchymal stem cell secretome and regenerative therapy after cancer. Biochimie 2013, 95, 2235–2245. [Google Scholar] [CrossRef]
- Guo, W.; Wong, S.; Xie, W.; Lei, T.; Luo, Z. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 576–586. [Google Scholar] [CrossRef] [Green Version]
- Renovato-Martins, M.; Moreira-Nunes, C.; Atella, G.C.; Barja-Fidalgo, C.; Moraes, J.A. de Obese Adipose Tissue Secretion Induces Inflammation in Preadipocytes: Role of Toll-Like Receptor-4. Nutrients 2020, 12, 2828. [Google Scholar] [CrossRef]
- La Cava, A.; Matarese, G. The weight of leptin in immunity. Nat. Rev. Immunol. 2004, 4, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Rutledge, B.J.; Gu, L.; Fiorillo, J.; Lukacs, N.W.; Kunkel, S.L.; North, R.; Gerard, C.; Rollins, B.J. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 1998, 187, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.I.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Rouault, C.; Marcelin, G.; Adriouch, S.; Rose, C.; Genser, L.; Ambrosini, M.; Bichet, J.-C.; Zhang, Y.; Marquet, F.; Aron-Wisnewsky, J.; et al. Senescence-associated β-galactosidase in subcutaneous adipose tissue associates with altered glycaemic status and truncal fat in severe obesity. Diabetologia 2021, 64, 240–254. [Google Scholar] [PubMed]
- Miller, M.C.; Mayo, K.H. Chemokines from a Structural Perspective. Int. J. Mol. Sci. 2017, 18, 2088. [Google Scholar] [CrossRef] [Green Version]
- Rollins, B.J. Chemokines. Blood 1997, 90, 909–928. [Google Scholar] [CrossRef]
- Clore, G.M.; Gronenborn, A.M. Three-dimensional structures of α and β chemokines. FASEB J. 1995, 9, 57–62. [Google Scholar] [CrossRef]
- Alexander, J.M.; Nelson, C.A.; Van Berkel, V.; Lau, E.K.; Studts, J.M.; Brett, T.J.; Speck, S.H.; Handel, T.M.; Virgin, H.W.; Fremont, D.H. Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell 2002, 111, 343–356. [Google Scholar] [CrossRef] [Green Version]
- Cochran, B.H.; Reffel, A.C.; Stiles, C.D. Molecular cloning of gene sequences regulated by platelet-derived growth factor. Cell 1983, 33, 939–947. [Google Scholar] [CrossRef]
- Rollins, B.J.; Morrison, E.D.; Stiles, C.D. Cloning and expression of JE, a gene inducible by platelet-derived growth factor and whose product has cytokine-like properties. Proc. Natl. Acad. Sci. USA 1988, 85, 3738–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrabian, M.; Sparkes, R.S.; Mohandas, T.; Fogelman, A.M.; Lusis, A.J. Localization of monocyte chemotactic protein-1 gene (SCYA2) to human chromosome 17q11.2-q21.1. Genomics 1991, 9, 200–203. [Google Scholar] [CrossRef]
- Barna, B.P.; Pettay, J.; Barnett, G.H.; Zhou, P.; Iwasaki, K.; Estes, M.L. Regulation of monocyte chemoattractant protein-1 expression in adult human non-neoplastic astrocytes is sensitive to tumor necrosis factor (TNF) or antibody to the 55-kDa TNF receptor. J. Neuroimmunol. 1994, 50, 101–107. [Google Scholar] [CrossRef]
- Cushing, S.D.; Berliner, J.A.; Valente, A.J.; Territo, M.C.; Navab, M.; Parhami, F.; Gerrity, R.; Schwartz, C.J.; Fogelman, A.M. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5134–5138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, Z.; Strieter, R.M.; Neild, G.H.; Thompson, R.C.; Kunkel, S.L.; Westwick, J. IL-1 receptor antagonist inhibits monocyte chemotactic peptide 1 generation by human mesangial cells. Kidney Int. 1992, 42, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Standiford, T.J.; Kunkel, S.L.; Phan, S.H.; Rollins, B.J.; Strieter, R.M. Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J. Biol. Chem. 1991, 266, 9912–9918. [Google Scholar] [CrossRef]
- Yoshimura, T.; Yuhki, N.; Moore, S.K.; Appella, E.; Lerman, M.I.; Leonard, E.J. Human monocyte chemoattractant protein-1 (MCP-1). Full-length cDNA cloning, expression in mitogen-stimulated blood mononuclear leukocytes, and sequence similarity to mouse competence gene JE. FEBS Lett. 1989, 244, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, T.; Robinson, E.A.; Tanaka, S.; Appella, E.; Kuratsu, J.I.; Leonard, E.J. Purification and amino acid analysis of two human glioma-derived monocyte chemoattractants. J. Exp. Med. 1989, 169, 1449–1459. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, C.C.; Romero, I.A.; Cancello, R.; Camoin, L.; Strosberg, A.D. Chemokines control fat accumulation and leptin secretion by cultured human adipocytes. Mol. Cell. Endocrinol. 2001, 175, 81–92. [Google Scholar] [CrossRef]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef] [Green Version]
- Tsaur, I.; Noack, A.; Makarevic, J.; Oppermann, E.; Waaga-Gasser, A.M.; Gasser, M.; Borgmann, H.; Huesch, T.; Gust, K.M.; Reiter, M.; et al. CCL2 Chemokine as a Potential Biomarker for Prostate Cancer: A Pilot Study. Cancer Res. Treat. 2014, 47, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Lubowicka, E.; Przylipiak, A.; Zajkowska, M.; Piskór, B.M.; Malinowski, P.; Fiedorowicz, W.; Ławicki, S. Plasma Chemokine CCL2 and Its Receptor CCR2 Concentrations as Diagnostic Biomarkers for Breast Cancer Patients. Biomed Res. Int. 2018, 2018, 2124390. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Aljada, A.; Ghanim, H.; Saadeh, R.; Dandona, P. Insulin Inhibits NFκB and MCP-1 Expression in Human Aortic Endothelial Cells. J. Clin. Endocrinol. Metab. 2001, 86, 450–453. [Google Scholar] [CrossRef] [Green Version]
- Nakatsumi, H.; Matsumoto, M.; Nakayama, K.I. Noncanonical Pathway for Regulation of CCL2 Expression by an mTORC1-FOXK1 Axis Promotes Recruitment of Tumor-Associated Macrophages. Cell Rep. 2017, 21, 2471–2486. [Google Scholar] [CrossRef] [Green Version]
- Callewaere, C.; Banisadr, G.; Rostène, W.; Parsadaniantz, S.M. Chemokines and chemokine receptors in the brain: Implication in neuroendocrine regulation. J. Mol. Endocrinol. 2007, 38, 355–363. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interf. Cytokine Res. 2009, 29, 313–325. [Google Scholar] [CrossRef]
- Oettgen, H.; Broide, D.H. Introduction to mechanisms of allergic disease. In Allergy; Elsevier: Amsterdam, The Netherlands, 2012; pp. 1–32. [Google Scholar]
- Charo, I.F.; Myers, S.J.; Herman, A.; Franci, C.; Connolly, A.J.; Coughlin, S.R. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc. Natl. Acad. Sci. USA 1994, 91, 2752–2756. [Google Scholar] [CrossRef] [Green Version]
- Bartoli, C.; Civatte, M.; Pellissier, J.; Figarella-Branger, D. CCR2A and CCR2B, the two isoforms of the monocytes chemoattractant protein-1 receptor are up-regulated and expressed by different cell subsets in idiopathic inflammatory myopathies. Acta Neuropathol. 2001, 102, 385–392. [Google Scholar] [CrossRef]
- Jen, C.H.; Leary, J.A. A competitive binding study of chemokine, sulfated receptor, and glycosaminoglycan interactions by nano-electrospray ionization mass spectrometry. Anal. Biochem. 2010, 407, 134–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc, R.S.; Kallenbach, J.G.; Bachman, J.F.; Mitchell, A.; Paris, N.D.; Chakkalakal, J.V. Inhibition of inflammatory CCR2 signaling promotes aged muscle regeneration and strength recovery after injury. Nat. Commun. 2020, 11, 4167. [Google Scholar] [CrossRef] [PubMed]
- Sarafi, M.N.; Garcia-Zepeda, E.A.; MacLean, J.A.; Charo, I.F.; Luster, A.D. Murine Monocyte Chemoattractant Protein (MCP)-5: A Novel CC Chemokine That Is a Structural and Functional Homologue of Human MCP-1. J. Exp. Med. 1997, 185, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, S.; Cho, J. Role of chemokine CCL2 and its receptor CCR2 in neurodegenerative diseases. Arch. Pharm. Res. 2013, 36, 1039–1050. [Google Scholar] [CrossRef]
- Nomiyama, H.; Hieshima, K.; Nakayama, T.; Sakaguchi, T.; Fujisawa, R.; Tanase, S.; Nishiura, H.; Matsuno, K.; Takamori, H.; Tabira, Y.; et al. Human CC chemokine liver-expressed chemokine/CCL16 is a functional ligand for CCR1, CCR2 and CCR5, and constitutively expressed by hepatocytes. Int. Immunol. 2001, 13, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Sozzani, S.; Zhou, D.; Locati, M.; Rieppi, M.; Proost, P.; Magazin, M.; Vita, N.; van Damme, J.; Mantovani, A. Receptors and transduction pathways for monocyte chemotactic protein-2 and monocyte chemotactic protein-3. Similarities and differences with MCP-1. J. Immunol. 1994, 152, 3615–3622. [Google Scholar]
- Mellado, M.; Rodríguez-Frade, J.M.; Aragay, A.; del Real, G.; Martín, A.M.; Vila-Coro, A.J.; Serrano, A.; Mayor, F.; Martínez-A, C. The chemokine monocyte chemotactic protein 1 triggers Janus kinase 2 activation and tyrosine phosphorylation of the CCR2B receptor. J. Immunol. 1998, 161, 805–813. [Google Scholar]
- Cambien, B.; Pomeranz, M.; Millet, M.A.; Rossi, B.; Schmid-Alliana, A. Signal transduction involved in MCP-1-mediated monocytic transendothelial migration. Blood 2001, 97, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Wain, J.H.; Kirby, J.A.; Ali, S. Leucocyte chemotaxis: Examination of mitogen-activated protein kinase and phosphoinositide 3-kinase activation by Monocyte Chemoattractant Proteins-1, -2, -3 and -4. Clin. Exp. Immunol. 2002, 127, 436–444. [Google Scholar] [CrossRef]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 receptor-mediated calcium signaling and its role in autophagy in cancer. Front. Oncol. 2017, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Racioppi, L.; Noeldner, P.K.; Lin, F.; Arvai, S.; Means, A.R. Calcium/calmodulin-dependent protein kinase kinase 2 regulates macrophage-mediated inflammatory responses. J. Biol. Chem. 2012, 287, 11579–11591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, F.; Eckard, L.; Chen, S.; Nutt, L.K. Metabolic regulation of apoptosis via Ca+/Calmodulin Kinase II (CaMKII). BMC Proc. 2012, 6, P36. [Google Scholar]
- Sriraman, V.; Modi, S.R.; Bodenburg, Y.; Denner, L.A.; Urban, R.J. Identification of ERK and JNK as signaling mediators on protein kinase C activation in cultured granulosa cells. Mol. Cell. Endocrinol. 2008, 294, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.; Miltz, W.; Mir, A.K.; Wiessner, C. Targeting monocyte chemoattractant protein-1 signalling in disease. Expert Opin. Ther. Targets 2003, 7, 35–48. [Google Scholar] [CrossRef]
- UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [Green Version]
- Kurihara, T.; Warr, G.; Loy, J.; Bravo, R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 1997, 186, 1757–1762. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell 1998, 2, 275–281. [Google Scholar] [CrossRef]
- Combadiere, C.; Ahuja, S.K.; Murphy, P.M. Cloning and functional expression of a human eosinophil CC chemokine receptor. J. Biol. Chem. 1995, 270, 16491–16494. [Google Scholar] [CrossRef] [Green Version]
- Franci, C.; Proost, P.; Charo, I.F. Monocyte chemoattractant protein-3, but not monocyte chemoattractant protein-2, is a functional ligand of the human monocyte chemoattractant protein-1 receptor. J. Immunol. 1995, 154, 6511–6517. [Google Scholar]
- Ford, L.B.; Cerovic, V.; Milling, S.W.F.; Graham, G.J.; Hansell, C.A.H.; Nibbs, R.J.B. Characterization of Conventional and Atypical Receptors for the Chemokine CCL2 on Mouse Leukocytes. J. Immunol. 2014, 193, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Goncalves, R.; Mosser, D.M. The Isolation and Characterization of Murine Macrophages. Curr. Protoc. Immunol. 2008, 83, 14.1.1–14.1.14. [Google Scholar] [CrossRef]
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael, L.H.; Rollins, B.J.; Entman, M.L.; Frangogiannis, N.G. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005, 96, 881–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, W.I.; Motomura, Y.; Wang, H.; El-Sharkawy, R.T.; Verdu, E.F.; Verma-Gandhu, M.; Rollins, B.J.; Collins, S.M. Critical role of MCP-1 in the pathogenesis of experimental colitis in the context of immune and enterochromaffin cells. Am. J. Physiol. Liver Physiol. 2006, 291, G803–G811. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H.; Schwarting, A.; Kinoshita, K.; Lan, H.Y.; Rollins, B.J.; Kelley, V.R. Monocyte chemoattractant protein-1 promotes macrophage-mediated tubular injury, but not glomerular injury, in nephrotoxic serum nephritis. J. Clin. Investig. 1999, 103, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shireman, P.K.; Contreras-Shannon, V.; Ochoa, O.; Karia, B.P.; Michalek, J.E.; McManus, L.M. MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J. Leukoc. Biol. 2007, 81, 775–785. [Google Scholar] [CrossRef]
- Contreras-Shannon, V.; Ochoa, O.; Reyes-Reyna, S.M.; Sun, D.; Michalek, J.E.; Kuziel, W.A.; McManus, L.M.; Shireman, P.K. Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2−/− mice following ischemic injury. Am. J. Physiol. Cell Physiol. 2007, 292, 953–967. [Google Scholar] [CrossRef]
- Jensen, L.E.; Etheredge, A.J.; Brown, K.S.; Mitchell, L.E.; Whitehead, A.S. Maternal genotype for the monocyte chemoattractant protein 1 A(-2518)G promoter polymorphism is associated with the risk of spina bifida in offspring. Am. J. Med. Genet. Part A 2006, 140, 1114–1118. [Google Scholar] [CrossRef]
- Lu, Z.Y.; Jensen, L.E.; Huang, Y.; Kealey, C.; Blair, I.A.; Whitehead, A.S. The up-regulation of monocyte chemoattractant protein-1 (MCP-1) in Ea.hy 926 endothelial cells under long-term low folate stress is mediated by the p38 MAPK pathway. Atherosclerosis 2009, 205, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Hammons, A.L.; Summers, C.M.; Woodside, J.V.; McNulty, H.; Strain, J.J.; Young, I.S.; Murray, L.; Boreham, C.A.; Scott, J.M.; Mitchell, L.E.; et al. Folate/homocysteine phenotypes and MTHFR 677C>T genotypes are associated with serum levels of monocyte chemoattractant protein-1. Clin. Immunol. 2009, 133, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Sung, F.L.; Siow, Y.L.; Wang, G.; Lynn, E.G.; Karmin, O. Homocysteine stimulates the expression of monocyte chemoattractant protein-1 in endothelial cells leading to enhanced monocyte chemotaxis. Mol. Cell. Biochem. 2001, 216, 121–128. [Google Scholar] [CrossRef]
- Modi, W.S.; Goedert, J.J.; Strathdee, S.; Buchbinder, S.; Detels, R.; Donfield, S.; O’Brien, S.J.; Winkler, C. MCP-1-MCP-3-Eotaxin gene cluster influences HIV-1 transmission. Aids 2003, 17, 2357–2365. [Google Scholar] [CrossRef]
- Palmieri, O.; Latiano, A.; Salvatori, E.; Valvano, M.R.; Bossa, F.; Latiano, T.; Corritore, G.; Di Mauro, L.; Andriulli, A.; Annesec, V. The A2518G polymorphism of monocyte chemoattractant protein-1 is associated with crohn’s disease. Am. J. Gastroenterol. 2010, 105, 1586–1594. [Google Scholar] [CrossRef]
- Li, K.S.; Wang, B.Y.; Liu, S.Y.; Yao, S.P.; Guo, L.; Mao, D.W. The combination of polymorphisms within MCP-1 and IL-1β associated with ulcerative colitis. Int. J. Immunogenet. 2009, 36, 135–139. [Google Scholar] [CrossRef]
- Angeles-Martínez, J.; Posadas-Sánchez, R.; Álvarez-León, E.; Villarreal-Molina, T.; Cardoso-Saldaña, G.; Fragoso, J.M.; Juárez-Rojas, J.G.; Medina-Urrutia, A.; Posadas-Romero, C.; Vargas-Alarcón, G. Monocyte chemoattractant protein-1 gene (MCP-1) polymorphisms are associated with risk of premature coronary artery disease in Mexican patients from the Genetics of Atherosclerotic Disease (GEA) study. Immunol. Lett. 2015, 167, 125–130. [Google Scholar] [CrossRef]
- González-Escribano, M.F.; Torres, B.; Aguilar, F.; Rodríguez, R.; García, A.; Valenzuela, Á.; Núñez-Roldán, A. MCP-1 promoter polymorphism in Spanish patients with rheumatoid arthritis. Hum. Immunol. 2003, 64, 741–744. [Google Scholar] [CrossRef]
- He, J.; Chen, Y.; Lin, Y.; Zhang, W.; Cai, Y.; Chen, F.; Liao, Q.; Yin, Z.; Wang, Y.; Tao, S.; et al. Association study of MCP-1 promoter polymorphisms with the susceptibility and progression of sepsis. PLoS ONE 2017, 12, e0176781. [Google Scholar] [CrossRef]
- Sang, G.-Y.; Meng, C.-R.; Hao, Y.-F.; Dai, J.-H. Monocyte chemoattractant protein-1 (MCP-1)-2518 A/G polymorphism and lupus nephritis risk: A PRISMA-compliant meta-analysis. Medicine 2017, 96, e9401. [Google Scholar] [CrossRef]
- Yang, Z.; Guo, Z.; Dong, J.; Sheng, S.; Wang, Y.; Yu, L.; Wang, H.; Tang, L. miR-374a Regulates Inflammatory Response in Diabetic Nephropathy by Targeting MCP-1 Expression. Front. Pharmacol. 2018, 9, 900. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neurooncol. 2011, 104, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Smart, C.; Hu, Q.; Cheng, N. Continuous Delivery of Neutralizing Antibodies Elevate CCL2 Levels in Mice Bearing MCF10CA1d Breast Tumor Xenografts. Transl. Oncol. 2017, 10, 734–743. [Google Scholar] [CrossRef]
- Loberg, R.D.; Ying, C.; Craig, M.; Day, L.L.; Sargent, E.; Neeley, C.; Wojno, K.; Snyder, L.A.; Yan, L.; Pienta, K.J. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007, 67, 9417–9424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, K.Y.; Han, J.; Zhang, X.; Hsu, S.H.; He, S.; Wani, N.A.; Barajas, J.M.; Snyder, L.A.; Frankel, W.L.; Caligiuri, M.A.; et al. Blocking the CCL2-CCR2 axis using CCL2-neutralizing antibody is an effective therapy for hepatocellular cancer in a mouse model. Mol. Cancer Ther. 2017, 16, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haringman, J.J.; Gerlag, D.M.; Smeets, T.J.M.; Baeten, D.; Van Den Bosch, F.; Bresnihan, B.; Breedveld, F.C.; Dinant, H.J.; Legay, F.; Gram, H.; et al. A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Vergunst, C.E.; Gerlag, D.M.; Lopatinskaya, L.; Klareskog, L.; Smith, M.D.; van den Bosch, F.; Dinant, H.J.; Lee, Y.; Wyant, T.; Jacobson, E.W.; et al. Modulation of CCR2 in rheumatoid arthritis: A double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2008, 58, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, T.; Richelsen, B.; Bruun, J.M. Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int. J. Obes. 2005, 29, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Dahlman, I.; Kaaman, M.; Olsson, T.; Tan, G.D.; Bickerton, A.S.T.; Wåhlén, K.; Andersson, J.; Nordström, E.A.; Blomqvist, L.; Sjögren, A.; et al. A unique role of monocyte chemoattractant protein 1 among chemokines in adipose tissue of obese subjects. J. Clin. Endocrinol. Metab. 2005, 90, 5834–5840. [Google Scholar] [CrossRef] [Green Version]
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.L.; et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 2005, 54, 2277–2286. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Lee, I.S.; Choue, R. Obesity, inflammation and diet. Pediatr. Gastroenterol. Hepatol. Nutr. 2013, 16, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Charles A Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. Principles of innate and adaptive immunity. In Immunobiology: The Immune System in Health and Disease; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Rajala, M.W.; Scherer, P.E. Minireview: The adipocyte—At the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 2003, 144, 3765–3773. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M. The inflammatory process of adipose tissue. Pediatr. Endocrinol. Rev. 2008, 6, 24–31. [Google Scholar] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [PubMed] [Green Version]
- Surmi, B.K.; Hasty, A.H. Macrophage infiltration into adipose tissue: Initiation, propagation and remodeling. Future Lipidol. 2008, 3, 545–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braune, J.; Lindhorst, A.; Fröba, J.; Hobusch, C.; Kovacs, P.; Blüher, M.; Eilers, J.; Bechmann, I.; Gericke, M. Multinucleated Giant Cells in Adipose Tissue Are Specialized in Adipocyte Degradation. Diabetes 2021, 70, 538–548. [Google Scholar] [CrossRef]
- Massier, L.; Chakaroun, R.; Tabei, S.; Crane, A.; Didt, K.D.; Fallmann, J.; Von Bergen, M.; Haange, S.B.; Heyne, H.; Stumvoll, M.; et al. Adipose tissue derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut 2020, 69, 1796–1806. [Google Scholar] [CrossRef]
- Rolle-Kampczyk, U.; Gebauer, S.; Haange, S.-B.; Schubert, K.; Kern, M.; Moulla, Y.; Dietrich, A.; Schön, M.R.; Klöting, N.; von Bergen, M.; et al. Accumulation of distinct persistent organic pollutants is associated with adipose tissue inflammation. Sci. Total Environ. 2020, 748, 142458. [Google Scholar] [CrossRef]
- Cullberg, K.B.; Larsen, J.Ø.; Pedersen, S.B.; Richelsen, B. Effects of LPS and dietary free fatty acids on MCP-1 in 3T3-L1 adipocytes and macrophages in vitro. Nutr. Diabetes 2014, 4, e113. [Google Scholar] [CrossRef]
- Ahmad, R.; Al-Roub, A.; Kochumon, S.; Akther, N.; Thomas, R.; Kumari, M.; Koshy, M.S.; Tiss, A.; Hannun, Y.A.; Tuomilehto, J.; et al. The Synergy between Palmitate and TNF-α for CCL2 Production Is Dependent on the TRIF/IRF3 Pathway: Implications for Metabolic Inflammation. J. Immunol. 2018, 200, 3599–3611. [Google Scholar] [CrossRef]
- Arner, E.; Mejhert, N.; Kulyté, A.; Balwierz, P.J.; Pachkov, M.; Cormont, M.; Lorente-Cebrián, S.; Ehrlund, A.; Laurencikiene, J.; Hedén, P.; et al. Adipose tissue MicroRNAs as regulators of CCL2 production in human obesity. Diabetes 2012, 61, 1986–1993. [Google Scholar] [CrossRef] [Green Version]
- Andrei, A.M.; Berbecaru-Iovan, A.; Din-Anghel, F.R.I.; Stanciulescu, C.E.; Berbecaru-Iovan, S.; Banita, I.M.; Pisoschi, C.G. Interplay between Hypoxia, Inflammation and Adipocyte Remodeling in the Metabolic Syndrome. In Hypoxia and Human Diseases; InTech: London, UK, 2017. [Google Scholar]
- Uchiyama, T.; Itaya-Hironaka, A.; Yamauchi, A.; Makino, M.; Sakuramoto-Tsuchida, S.; Shobatake, R.; Ota, H.; Takeda, M.; Ohbayashi, C.; Takasawa, S. Intermittent Hypoxia Up-Regulates CCL2, RETN, and TNFα mRNAs in Adipocytes via Down-regulation of miR-452. Int. J. Mol. Sci. 2019, 20, 1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ojeda, F.J.; Méndez-Gutiérrez, A.; Aguilera, C.M.; Plaza-Díaz, J. Extracellular matrix remodeling of adipose tissue in obesity and metabolic diseases. Int. J. Mol. Sci. 2019, 20, 4888. [Google Scholar] [CrossRef] [Green Version]
- Henegar, C.; Tordjman, J.; Achard, V.; Lacasa, D.; Cremer, I.; Guerre-Millo, M.; Poitou, C.; Basdevant, A.; Stich, V.; Viguerie, N.; et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008, 9, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Divoux, A.; Tordjman, J.; Lacasa, D.; Veyrie, N.; Hugol, D.; Aissat, A.; Basdevant, A.; Guerre-Millo, M.; Poitou, C.; Zucker, J.D.; et al. Fibrosis in human adipose tissue: Composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 2010, 59, 2817–2825. [Google Scholar] [CrossRef] [Green Version]
- Marcelin, G.; Silveira, A.L.M.; Martins, L.B.; Ferreira, A.V.M.; Clément, K. Deciphering the cellular interplays underlying obesity-induced adipose tissue fibrosis. J. Clin. Investig. 2019, 129, 4032–4040. [Google Scholar] [CrossRef] [PubMed]
- Lassen, P.B.; Charlotte, F.; Liu, Y.; Bedossa, P.; Le Naour, G.; Tordjman, J.; Poitou, C.; Bouillot, J.L.; Genser, L.; Zucker, J.D.; et al. The fat score, a fibrosis score of adipose tissue: Predicting weight-loss outcome after gastric bypass. J. Clin. Endocrinol. Metab. 2017, 102, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Pessentheiner, A.R.; Ducasa, G.M.; Gordts, P.L.S.M. Proteoglycans in Obesity-Associated Metabolic Dysfunction and Meta-Inflammation. Front. Immunol. 2020, 11, 769. [Google Scholar] [CrossRef]
- Proudfoot, A.E.I.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.C.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [Green Version]
- Gschwandtner, M.; Piccinini, A.M.; Gerlza, T.; Adage, T.; Kungl, A.J. Interfering with the CCL2-glycosaminoglycan axis as a potential approach to modulate neuroinflammation. Neurosci. Lett. 2016, 626, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Unamuno, X.; Gómez-Ambrosi, J.; Ramírez, B.; Rodríguez, A.; Becerril, S.; Valentí, V.; Moncada, R.; Silva, C.; Salvador, J.; Frühbeck, G.; et al. NLRP3 inflammasome blockade reduces adipose tissue inflammation and extracellular matrix remodeling. Cell. Mol. Immunol. 2019. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Chisanga, D.; Blume, J.; Gloury, R.; Britt, K.; Henstridge, D.C.; Zhan, Y.; Torres, S.V.; Liene, S.; Collins, N.; et al. Sex-specific adipose tissue imprinting of regulatory T cells. Nature 2020, 579, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Kuziel, G.; Thompson, V.; D’amato, J.V.; Arendt, L.M. Stromal CCL2 signaling promotes mammary tumor fibrosis through recruitment of myeloid-lineage cells. Cancers 2020, 12, 2083. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Huh, J.Y.; Sohn, J.H.; Choe, S.S.; Lee, Y.S.; Lim, C.Y.; Jo, A.; Park, S.B.; Han, W.; Kim, J.B. Lipid-Overloaded Enlarged Adipocytes Provoke Insulin Resistance Independent of Inflammation. Mol. Cell. Biol. 2015, 35, 1686–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, Y.; Nakae, J.; Watanabe, N.; Kikuchi, T.; Tateya, S.; Tamori, Y.; Kaneko, M.; Abe, T.; Onodera, M.; Itoh, H. Colonic Pro-inflammatory Macrophages Cause Insulin Resistance in an Intestinal Ccl2/Ccr2-Dependent Manner. Cell Metab. 2016, 24, 295–310. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Kang, J.S.; Kim, H.M.; Lee, E.S.; Lee, J.-H.; Chung, C.H.; Lee, E.Y. CCR2 knockout ameliorates obesity-induced kidney injury through inhibiting oxidative stress and ER stress. PLoS ONE 2019, 14, e0222352. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, S.U.; Cohen, J.L.; Vangala, P.; Tencerova, M.; Nicoloro, S.M.; Yawe, J.C.; Shen, Y.; Czech, M.P.; Aouadi, M. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014, 19, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef]
- Dusi, V.; Ghidoni, A.; Ravera, A.; De Ferrari, G.M.; Calvillo, L. Chemokines and Heart Disease: A Network Connecting Cardiovascular Biology to Immune and Autonomic Nervous Systems. Mediators Inflamm. 2016, 2016, 5902947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Economou, E.; Tousoulis, D.; Katinioti, A.; Stefanadis, C.; Trikas, A.; Pitsavos, C.; Tentolouris, C.; Toutouza, M.G.; Toutouzas, P. Chemokines in patients with ischaemic heart disease and the effect of coronary angioplasty. Int. J. Cardiol. 2001, 80, 55–60. [Google Scholar] [CrossRef]
- De Lemos, J.A.; Morrow, D.A.; Sabatine, M.S.; Murphy, S.A.; Gibson, C.M.; Antman, E.M.; McCabe, C.H.; Cannon, C.P.; Braunwald, E. Association between plasma levels of monocyte chemoattractant protein-1 and long-term clinical outcomes in patients with acute coronary syndromes. Circulation 2003, 107, 690–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, D.; Su, D.; Li, X.; Li, Z.; Wang, Y.; Qiu, J.; Lin, P.; Zhang, Y.; Guo, P.; Xia, M.; et al. Serum levels of monocyte chemoattractant protein-1 and all-cause and cardiovascular mortality among patients with coronary artery disease. PLoS ONE 2015, 10, e0120633. [Google Scholar] [CrossRef]
- Bucova, M.; Lietava, J.; Penz, P.; Mrazek, F.; Petrkova, J.; Bernadic, M.; Petrek, M. Association of MCP-1 -2518 A/G single nucleotide polymorphism with the serum level of CRP in Slovak patients with ischemic heart disease, angina pectoris, and hypertension. Mediators Inflamm. 2009, 2009, 390951. [Google Scholar] [CrossRef] [Green Version]
- Guzmán-Ornelas, M.-O.; Petri, M.H.; Vázquez-Del Mercado, M.; Chavarría-Ávila, E.; Corona-Meraz, F.-I.; Ruíz-Quezada, S.-L.; Madrigal-Ruíz, P.-M.; Castro-Albarrán, J.; Sandoval-García, F.; Navarro-Hernández, R.-E. CCL2 Serum Levels and Adiposity Are Associated with the Polymorphic Phenotypes -2518A on CCL2 and 64ILE on CCR2 in a Mexican Population with Insulin Resistance. J. Diabetes Res. 2016, 2016, 5675739. [Google Scholar] [CrossRef]
- Koper-Lenkiewicz, O.M.; Kamińska, J.; Lisowska, A.; Milewska, A.; Hirnle, T.; Dymicka-Piekarska, V. Factors Associated with RANTES Concentration in Cardiovascular Disease Patients. Biomed Res. Int. 2019, 2019, 3026453. [Google Scholar]
- Semple, B.D.; Bye, N.; Rancan, M.; Ziebell, J.M.; Morganti-Kossmann, M.C. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): Evidence from severe TBI patients and CCL2−/− mice. J. Cereb. Blood Flow Metab. 2010, 30, 769–782. [Google Scholar] [CrossRef]
- Fabene, P.F.; Bramanti, P.; Constantin, G. The emerging role for chemokines in epilepsy. J. Neuroimmunol. 2010, 224, 22–27. [Google Scholar] [CrossRef]
- Shen, Z.; Kuang, S.; Zhang, M.; Huang, X.; Chen, J.; Guan, M.; Qin, W.; Xu, H.H.K.; Lin, Z. Inhibition of CCL2 by bindarit alleviates diabetes-associated periodontitis by suppressing inflammatory monocyte infiltration and altering macrophage properties. Cell. Mol. Immunol. 2020. online ahead of print. [Google Scholar] [CrossRef]
- Wolf, S.; Johnson, S.; Perwitasari, O.; Mahalingam, S.; Tripp, R.A. Targeting the pro-inflammatory factor CCL2 (MCP-1) with Bindarit for influenza A (H7N9) treatment. Clin. Transl. Immunol. 2017, 6, e135. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Knight, D.A.; A Snyder, L.; Smyth, M.J.; Stewart, T.J. A role for CCL2 in both tumor progression and immunosurveillance. Oncoimmunology 2013, 2, e25474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Li, Y.; Yan, H.; Zhang, E.; Huang, X.; Chen, Q.; Chen, J.; Qu, J.; Liu, Y.; He, J.; et al. CCL2 promotes macrophages-associated chemoresistance via MCPIP1 dual catalytic activities in multiple myeloma. Cell Death Dis. 2019, 10, 781. [Google Scholar] [CrossRef]
- Auvynet, C.; Baudesson De Chanville, C.; Hermand, P.; Dorgham, K.; Piesse, C.; Pouchy, C.; Carlier, L.; Poupel, L.; Barthélémy, S.; Felouzis, V.; et al. ECL1i, d(LGTFLKC), a novel, small peptide that specifically inhibits CCL2-dependent migration. FASEB J. 2016, 30, 2370–2381. [Google Scholar] [PubMed] [Green Version]
- Monickaraj, F.; Oruganti, S.R.; McGuire, P.; Das, A. A potential novel therapeutic target in diabetic retinopathy: A chemokine receptor (CCR2/CCR5) inhibitor reduces retinal vascular leakage in an animal model. Graefe’s Arch. Clin. Exp. Ophthalmol. 2021, 259, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic | Dose | Therapeutic Target/Disease | Effect | Literature |
|---|---|---|---|---|
| anti-human CCL2 Ab (ABN912) | 0.3/1.0/3.0/10.0 mg/kg | rheumatoid arthritis | no benefits compared to placebo; dose-related CCL2 increase | [114] |
| anti-human CCL2 Ab (CNTO888) | 2 mg/kg, twice a week | prostate cancer | 47% reduced tumor burden | [112] |
| anti-human CCL2 Ab (MAB279) | 0.3 mg/kg/day over 4 weeks | breast tumor xenograft | no effects of tumor growth or angiogenesis and on macrophage recruitment | [111] |
| anti-mouse CCL2 Ab | 2 mg/kg/dose, twice a week | mouse and human glioma xenografts | life prolonged modestly | [110] |
| anti-mouse CCL2 Ab (C1142) | 2 mg/kg, twice a week | hepatitis and hepatocellular carcinoma | suppressed liver inflammation and damage; reduced carcinoma incidence; reduced inflammatory markers | [113] |
| anti-mouse CCL2 Ab + temozolomide | 2 mg/kg/dose, twice a week +800 µg in 100 µL PBS | mouse and human glioma xenografts | significantly prolonged life | [110] |
| Bindarit | 50 mg/kg, daily | diabetes-associated periodontitis | suppressed inflammation; reduced monocyte infiltration; reduced alveolar bone loss; increased epithelial thickness | [150] |
| Bindarit | 70 mg/kg, twice daily | H7N9 virus influenza | no anti-inflammatory effects | [151] |
| ECL1i, d(LGTFLKC) | 90 µg, twice | peritonitis | limits monocyte and macrophage recruitment | [154] |
| TAK-779 | 30 mg/kg, daily | diabetic retinopathy | reduces retinal vascular permeability | [155] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dommel, S.; Blüher, M. Does C-C Motif Chemokine Ligand 2 (CCL2) Link Obesity to a Pro-Inflammatory State? Int. J. Mol. Sci. 2021, 22, 1500. https://doi.org/10.3390/ijms22031500
Dommel S, Blüher M. Does C-C Motif Chemokine Ligand 2 (CCL2) Link Obesity to a Pro-Inflammatory State? International Journal of Molecular Sciences. 2021; 22(3):1500. https://doi.org/10.3390/ijms22031500
Chicago/Turabian StyleDommel, Sebastian, and Matthias Blüher. 2021. "Does C-C Motif Chemokine Ligand 2 (CCL2) Link Obesity to a Pro-Inflammatory State?" International Journal of Molecular Sciences 22, no. 3: 1500. https://doi.org/10.3390/ijms22031500
APA StyleDommel, S., & Blüher, M. (2021). Does C-C Motif Chemokine Ligand 2 (CCL2) Link Obesity to a Pro-Inflammatory State? International Journal of Molecular Sciences, 22(3), 1500. https://doi.org/10.3390/ijms22031500