
Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties


Abstract
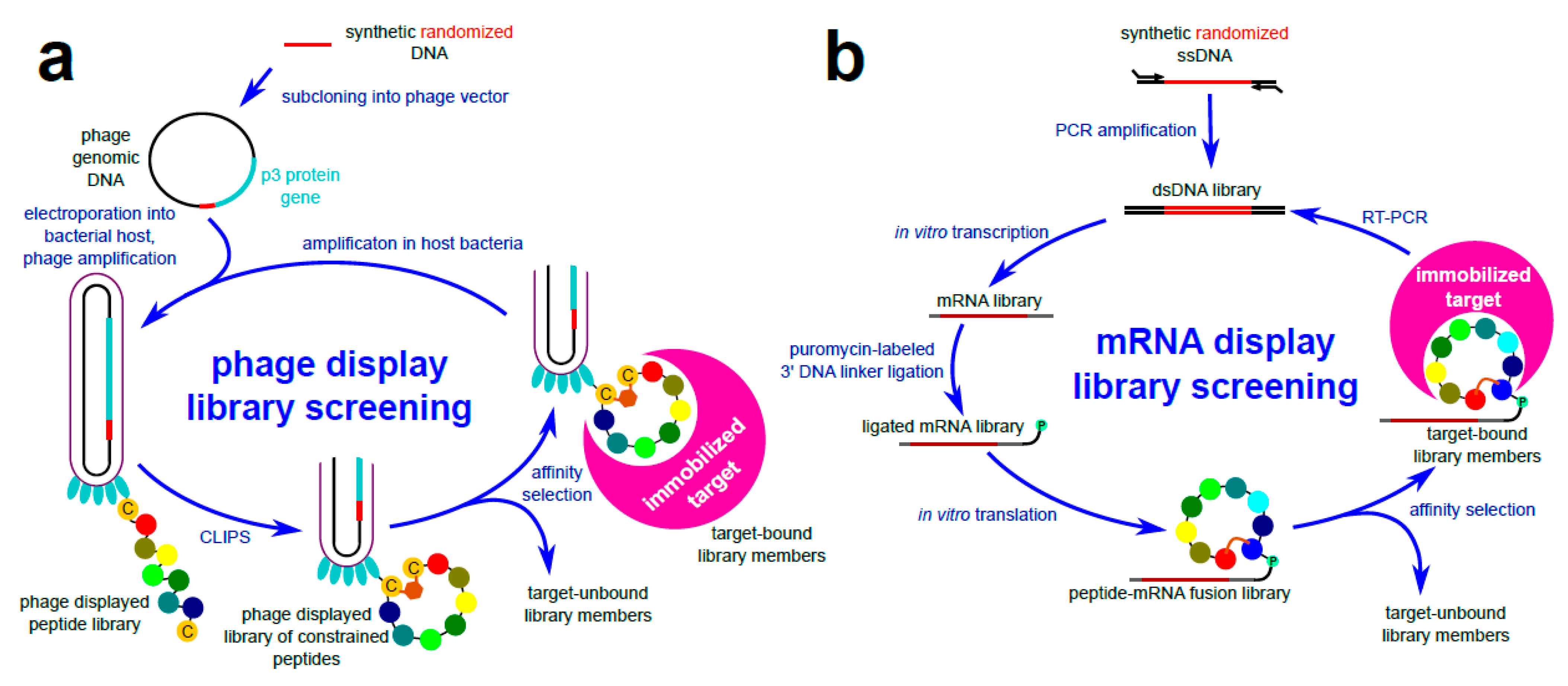
:1. Introduction
2. Properties of Conformationally Constrained Peptides
3. Constrainment Strategies and Chemistries
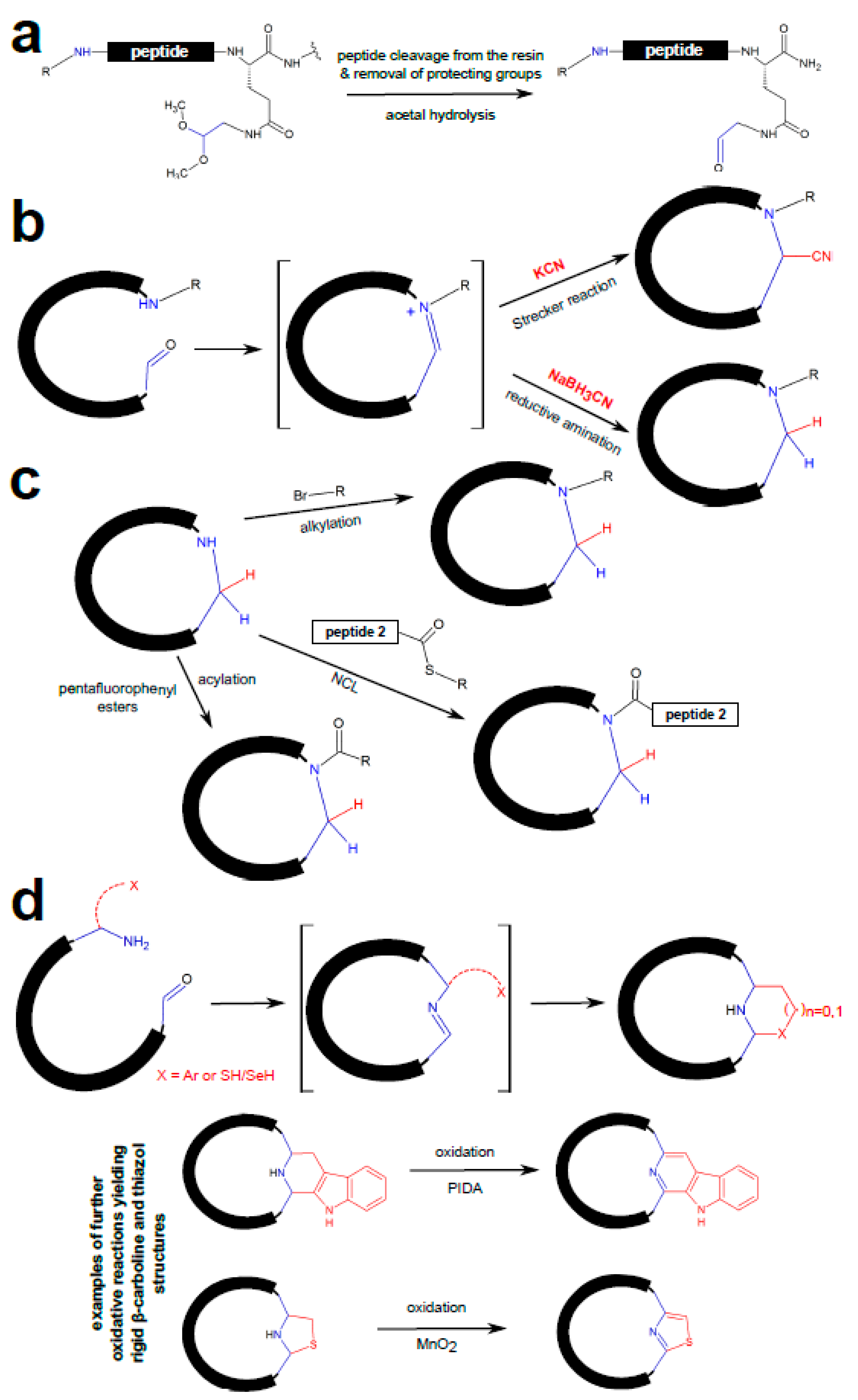
3.1. Chemical Peptide Ligation and Bridging
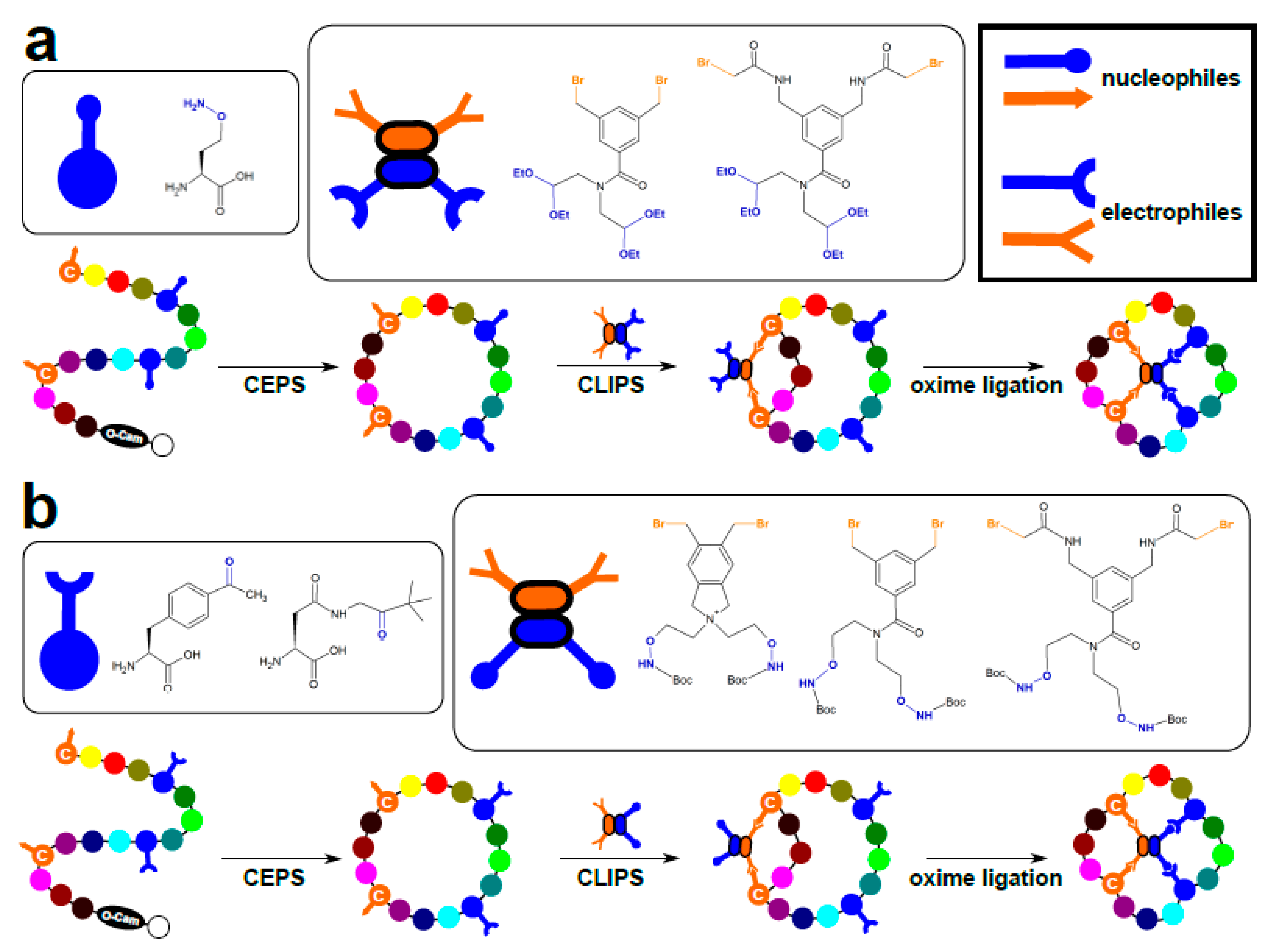
3.2. Chemical Ligation of Peptides onto Scaffolds (CLIPS)
3.3. Enzymatic Peptide Cyclization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | N-Terminal Recognition Sequence | C-Terminal Recognition Sequence (Cleavage Site Denoted with Caron Sign) | Ring Sizes (Residues) | Advantages | Disadvantages |
|---|---|---|---|---|---|
| sortase A | (G)n | LPXTˇG | ≥16 | commercially available, recombinant, high ligation yield | LPXT(G)n footprint, reaction reversible, competing oligomerization, moderate catalytic efficiency (0.1–1 molar eq. required) |
| butelase 1 | X1X2, where X1 is not P, D, or E, and X2 is L, I, V or C | (N/D)ˇHV | ≥10 | broad substrate specificity (thus minimal footprint), high ligation yield, high catalytic efficiency (~0.005 molar eq. required), reaction irreversible | only accessible through isolation, very few residues tolerated in position X2 |
| peptiligases [omniligase] | any dipeptide lacking P | essentially any tetrapeptide without C-terminal P; requires activated C-terminal carboxy group (ester) | ≥13 | very broad substrate specificity (hence no footprint), extraordinary catalytic efficiency (only ~0.0003 molar eq. required), reaction irreversible, high yield, [tolerates co-solvents and chaotropic agents, commercially available, compatible with non-peptidic backbone moieties] | requires activated C-terminal carboxy group (ester) |
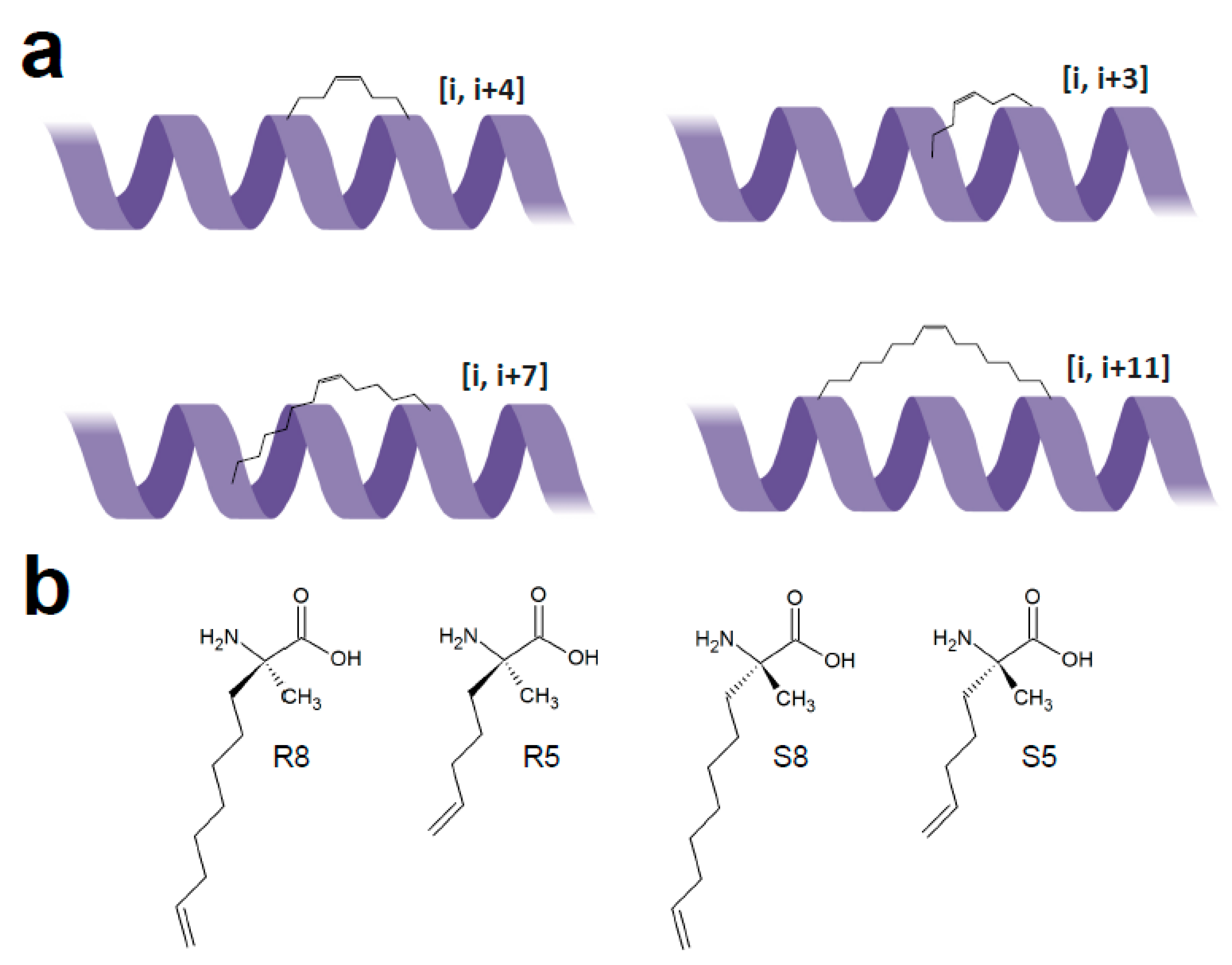
3.4. Peptide Stapling
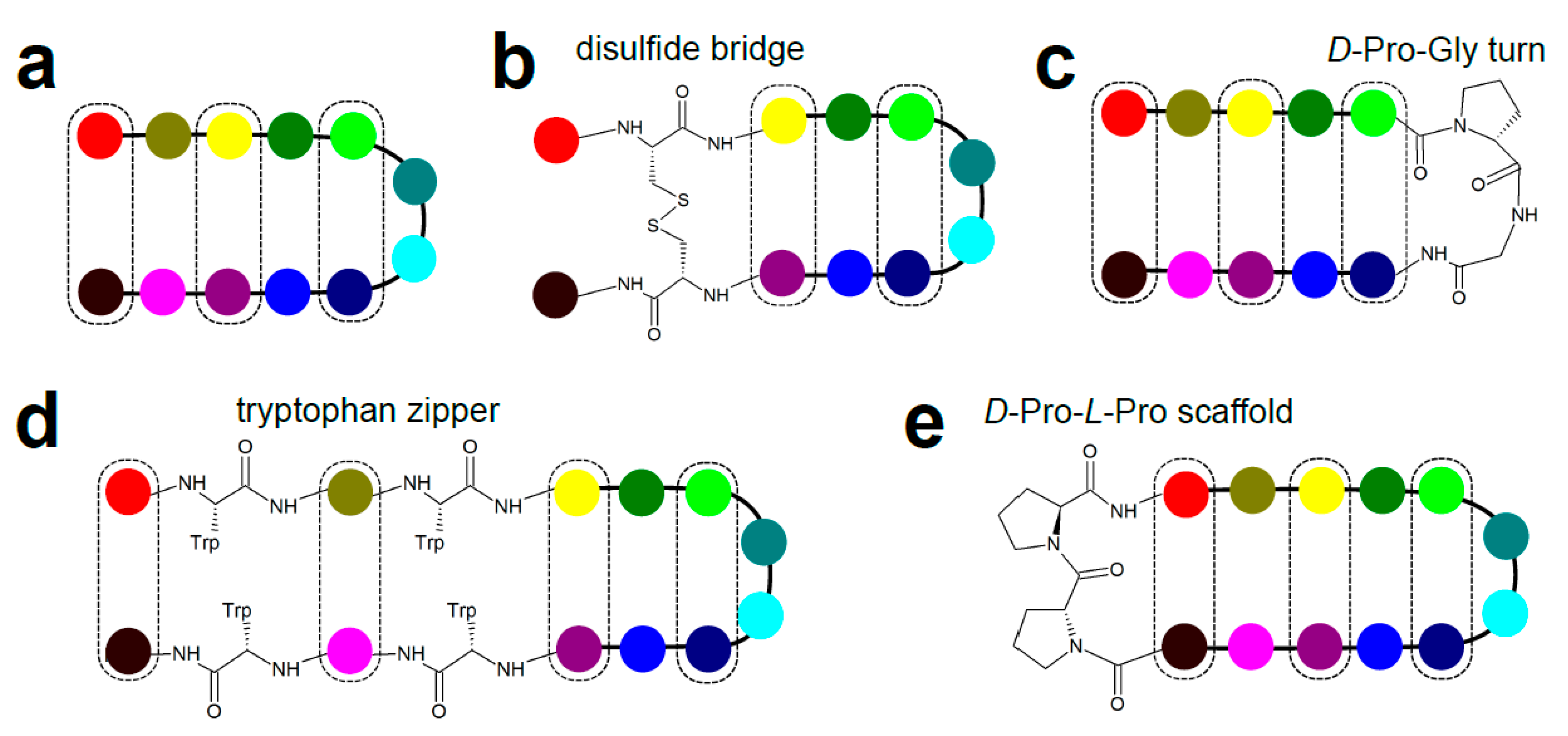
3.5. β-Hairpins and Hairpin Loops
3.6. In Vitro Molecular Evolution
4. Applications of Constrained Peptides
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Empting, M. An introduction to cyclic peptides. In Cyclic Peptides: From Bioorganic Synthesis to Applications; Koehnke, J., Naismith, J., van der Donk, W.A., Eds.; Royal Society of Chemistry: Croydon, UK, 2017; Volume 6, pp. 1–14. [Google Scholar]
- Fang, Z.; Song, Y.; Zhan, P.; Zhang, Q.; Liu, X. Conformational restriction: An effective tactic in ‘follow-on’-based drug discovery. Future Med. Chem. 2014, 6, 885–901. [Google Scholar] [CrossRef]
- Rhodes, C.A.; Pei, D. Bicyclic peptides as next-generation therapeutics. Chemistry 2017, 23, 12690–12703. [Google Scholar] [CrossRef]
- Passioura, T.; Katoh, T.; Goto, Y.; Suga, H. Selection-based discovery of drug-like macrocyclic peptides. Annu. Rev. Biochem. 2014, 83, 727–752. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Atmaj, J.; Van Oosterwijk, N.; Groves, M.R.; Domling, A. Stapled peptides inhibitors: A new window for target drug discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef]
- Robinson, J.A. Beta-hairpin peptidomimetics: Design, structures and biological activities. Acc. Chem. Res. 2008, 41, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, G.R.; Starovasnik, M.A.; Reynolds, M.E.; Lowman, H.B. A novel family of hairpin peptides that inhibit IgE activity by binding to the high-affinity IgE receptor. Biochemistry 2001, 40, 9828–9835. [Google Scholar] [CrossRef]
- Skelton, N.J.; Russell, S.; de Sauvage, F.; Cochran, A.G. Amino acid determinants of beta-hairpin conformation in erythropoeitin receptor agonist peptides derived from a phage display library. J. Mol. Biol. 2002, 316, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Katchalski-Katzir, E.; Kasher, R.; Balass, M.; Scherf, T.; Harel, M.; Fridkin, M.; Sussman, J.L.; Fuchs, S. Design and synthesis of peptides that bind alpha-bungarotoxin with high affinity and mimic the three-dimensional structure of the binding-site of acetylcholine receptor. Biophys. Chem. 2003, 100, 293–305. [Google Scholar] [CrossRef]
- DeLano, W.L.; Ultsch, M.H.; de Vos, A.M.; Wells, J.A. Convergent solutions to binding at a protein-protein interface. Science 2000, 287, 1279–1283. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Huang, L.; Shen, B. Rational cyclization-based minimization of entropy penalty upon the binding of Nrf2-derived linear peptides to Keap1: A new strategy to improve therapeutic peptide activity against sepsis. Biophys. Chem. 2019, 244, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Driggers, E.M.; Hale, S.P.; Lee, J.; Terrett, N.K. The exploration of macrocycles for drug discovery—An underexploited structural class. Nat. Rev. Drug Discov. 2008, 7, 608–624. [Google Scholar] [CrossRef]
- Tyndall, J.D.; Fairlie, D.P. Conformational homogeneity in molecular recognition by proteolytic enzymes. J. Mol. Recognit. 1999, 12, 363–370. [Google Scholar] [CrossRef]
- Timmerman, P.; Beld, J.; Puijk, W.C.; Meloen, R.H. Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem 2005, 6, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Bertoldo, D.; Khan, M.M.; Dessen, P.; Held, W.; Huelsken, J.; Heinis, C. Phage selection of peptide macrocycles against beta-catenin to interfere with Wnt signaling. Chem. Med. Chem. 2016, 11, 834–839. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, P.; Qian, Z.; Habir, N.A.; Pei, D. Direct Ras inhibitors identified from a structurally rigidified bicyclic peptide library. Tetrahedron 2014, 70, 7714–7720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adaligil, E.; Patil, K.; Rodenstein, M.; Kumar, K. Discovery of peptide antibiotics composed of D-amino acids. ACS Chem. Biol. 2019, 14, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- ‘t Hart, P.; Wood, T.M.; Tehrani, K.; van Harten, R.M.; Sleszynska, M.; Rentero Rebollo, I.; Hendrickx, A.P.A.; Willems, R.J.L.; Breukink, E.; Martin, N.I. De novo identification of lipid II binding lipopeptides with antibacterial activity against vancomycin-resistant bacteria. Chem. Sci. 2017, 8, 7991–7997. [Google Scholar] [CrossRef] [Green Version]
- Richelle, G.J.J.; Schmidt, M.; Ippel, H.; Hackeng, T.M.; van Maarseveen, J.H.; Nuijens, T.; Timmerman, P. A one-pot “triple-C” multicyclization methodology for the synthesis of highly constrained isomerically pure tetracyclic peptides. Chembiochem 2018, 19, 1934–1938. [Google Scholar] [CrossRef] [PubMed]
- Trinh, T.B.; Upadhyaya, P.; Qian, Z.; Pei, D. Discovery of a direct Ras inhibitor by screening a combinatorial library of cell-permeable bicyclic peptides. ACS Comb. Sci. 2016, 18, 75–85. [Google Scholar] [CrossRef]
- Rhodes, C.A.; Dougherty, P.G.; Cooper, J.K.; Qian, Z.; Lindert, S.; Wang, Q.E.; Pei, D. Cell-permeable bicyclic peptidyl inhibitors against NEMO-IkappaB kinase interaction directly from a combinatorial library. J. Am. Chem. Soc. 2018, 140, 12102–12110. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-penetrating peptides: Design strategies beyond primary structure and amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Q.; Moellering, R.E.; Hilinski, G.J.; Kim, Y.-W.; Grossmann, T.N.; Yehab, J.T.-H.; Verdine, G.L. Towards understanding cell penetration by stapled peptides. Med. Chem. Comm. 2015, 6, 111–119. [Google Scholar] [CrossRef]
- Bird, G.H.; Mazzola, E.; Opoku-Nsiah, K.; Lammert, M.A.; Godes, M.; Neuberg, D.S.; Walensky, L.D. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol. 2016, 12, 845–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasan, R.; Dias, R.L.; Moehle, K.; Zerbe, O.; Vrijbloed, J.W.; Obrecht, D.; Robinson, J.A. Using a beta-hairpin to mimic an alpha-helix: Cyclic peptidomimetic inhibitors of the p53-HDM2 protein-protein interaction. Angew. Chem. Int. Ed. Engl. 2004, 43, 2109–2112. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.T.; Shortridge, M.D.; Varani, G. A small cyclic beta-hairpin peptide mimics the Rbfox2 RNA recognition motif and binds to the precursor miRNA-20b. Chembiochem 2019, 20, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Shortridge, M.D.; Walker, M.J.; Pavelitz, T.; Chen, Y.; Yang, W.; Varani, G. A macrocyclic peptide ligand binds the oncogenic microRNA-21 precursor and suppresses dicer processing. ACS Chem. Biol. 2017, 12, 1611–1620. [Google Scholar] [CrossRef]
- Bratkovič, T.; Lunder, M.; Popovič, T.; Kreft, S.; Turk, B.; Štrukelj, B.; Urleb, U. Affinity selection to papain yields potent peptide inhibitors of cathepsins L., B., H., and K. Biochem. Biophys. Res. Commun. 2005, 332, 897–903. [Google Scholar] [CrossRef]
- Bratkovič, T.; Berlec, A.; Popovič, T.; Lunder, M.; Kreft, S.; Urleb, U.; Štrukelj, B. Engineered staphylococcal protein A’s IgG-binding domain with cathepsin L inhibitory activity. Biochem. Biophys. Res. Commun. 2006, 349, 449–453. [Google Scholar] [CrossRef]
- Lu, Z.; Murray, K.S.; Van Cleave, V.; LaVallie, E.R.; Stahl, M.L.; McCoy, J.M. Expression of thioredoxin random peptide libraries on the Escherichia coli cell surface as functional fusions to flagellin: A system designed for exploring protein-protein interactions. Biotechnology 1995, 13, 366–372. [Google Scholar] [CrossRef]
- Borghouts, C.; Kunz, C.; Groner, B. Peptide aptamer libraries. Comb. Chem. High Throughput Screen 2008, 11, 135–145. [Google Scholar]
- Reverdatto, S.; Burz, D.S.; Shekhtman, A. Peptide aptamers: Development and applications. Curr. Top. Med. Chem. 2015, 15, 1082–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Škrlec, K.; Štrukelj, B.; Berlec, A. Non-immunoglobulin scaffolds: A focus on their targets. Trends Biotechnol. 2015, 33, 408–418. [Google Scholar] [CrossRef]
- Reguera, L.; Rivera, D.G. Multicomponent reaction toolbox for peptide macrocyclization and stapling. Chem. Rev. 2019, 119, 9836–9860. [Google Scholar] [CrossRef] [PubMed]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Li, Z.; Shao, S.; Ren, X.; Sun, J.; Guo, Z.; Wang, S.; Song, M.M.; Chang, C.A.; Xue, M. Construction of a sequenceable protein mimetic peptide library with a true 3D diversifiable chemical space. J. Am. Chem. Soc. 2018, 140, 14552–14556. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Q.; Wong, C.T.T.; Li, X. Chemoselective peptide cyclization and bicyclization directly on unprotected peptides. J. Am. Chem. Soc. 2019, 141, 12274–12279. [Google Scholar] [CrossRef]
- Thapa, P.; Zhang, R.Y.; Menon, V.; Bingham, J.P. Native chemical ligation: A boon to peptide chemistry. Molecules 2014, 19, 14461–14483. [Google Scholar] [CrossRef] [Green Version]
- Malins, L.R.; de Gruyter, J.N.; Robbins, K.J.; Scola, P.M.; Eastgate, M.D.; Ghadiri, M.R.; Baran, P.S. Peptide macrocyclization inspired by non-ribosomal imine natural products. J. Am. Chem. Soc. 2017, 139, 5233–5241. [Google Scholar] [CrossRef]
- Bionda, N.; Cryan, A.L.; Fasan, R. Bioinspired strategy for the ribosomal synthesis of thioether-bridged macrocyclic peptides in bacteria. ACS Chem. Biol. 2014, 9, 2008–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bionda, N.; Fasan, R. Ribosomal synthesis of natural-product-like bicyclic peptides in Escherichia coli. Chembiochem 2015, 16, 2011–2016. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.P.; Abel-Santos, E.; Wall, M.; Wahnon, D.C.; Benkovic, S.J. Production of cyclic peptides and proteins in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 13638–13643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Morales-Sanfrutos, J.; Angelini, A.; Cutting, B.; Heinis, C. Structurally diverse cyclisation linkers impose different backbone conformations in bicyclic peptides. Chembiochem 2012, 13, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Sindlinger, J.; Schwarzer, D.; Koch, P.; Boeckler, F.M. The symmetric tetravalent sulfhydryl-specific linker NATBA facilitates a combinatorial “tool kit” strategy for phage display-based selection of functionalized bicyclic peptides. ACS Omega 2018, 3, 12361–12368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, M.R.; Yu, H.; Wickware, J.M.; Lin, Y.S.; Derda, R. Light-responsive bicyclic peptides. Org. Biomol. Chem 2018, 16, 7588–7594. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Spokoyny, A.M.; Zhang, C.; Simon, M.D.; Yu, H.; Lin, Y.S.; Pentelute, B.L. Convergent diversity-oriented side-chain macrocyclization scan for unprotected polypeptides. Org. Biomol. Chem. 2014, 12, 566–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spokoyny, A.M.; Zou, Y.; Ling, J.J.; Yu, H.; Lin, Y.S.; Pentelute, B.L. A perfluoroaryl-cysteine SNAr chemistry approach to unprotected peptide stapling. J. Am. Chem. Soc. 2013, 135, 5946–5949. [Google Scholar] [CrossRef] [Green Version]
- Antos, J.M.; Popp, M.W.; Ernst, R.; Chew, G.L.; Spooner, E.; Ploegh, H.L. A straight path to circular proteins. J. Biol. Chem. 2009, 284, 16028–16036. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Guo, X.; Guo, Z. Sortase A-catalyzed peptide cyclization for the synthesis of macrocyclic peptides and glycopeptides. Chem. Commun. 2011, 47, 9218–9220. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.K.; Wang, S.; Qiu, Y.; Hemu, X.; Lian, Y.; Tam, J.P. Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 2014, 10, 732–738. [Google Scholar] [CrossRef]
- Nguyen, G.K.; Hemu, X.; Quek, J.P.; Tam, J.P. Butelase-mediated macrocyclization of D-amino-acid-containing peptides. Angew Chem. Int. Ed. Engl. 2016, 55, 12802–12806. [Google Scholar] [CrossRef] [PubMed]
- Braisted, A.C.; Judice, J.K.; Wells, J.A. Synthesis of proteins by subtiligase. Methods Enzymol. 1997, 289, 298–313. [Google Scholar]
- Toplak, A.; Nuijens, T.; Quaedflieg, P.J.L.M.; Wu, B.; Janssen, D.B. Peptiligase, an enzyme for efficient chemoenzymatic peptide synthesis and cyclization in water. Adv. Synth. Catal. 2016, 358, 2140–2147. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Toplak, A.; Quaedflieg, P.; van Maarseveen, J.H.; Nuijens, T. Enzyme-catalyzed peptide cyclization. Drug Discov. Today Technol. 2017, 26, 11–16. [Google Scholar] [CrossRef]
- Schmidt, M.; Toplak, A.; Quaedflieg, P.J.L.M.; Ippel, H.; Richelle, G.J.J.; Hackeng, T.M.; van Maarseveen, J.H.; Nuijens, T. Omniligase-1: A powerful tool for peptide head-to-tail cyclization. Adv. Synth. Catal. 2017, 359, 2050–2055. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Huang, Y.H.; Texeira de Oliveira, E.F.; Toplak, A.; Wijma, H.J.; Janssen, D.B.; van Maarseveen, J.H.; Craik, D.J.; Nuijens, T. Efficient enzymatic cyclization of disulfide-rich peptides by using peptide ligases. Chembiochem 2019, 20, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Streefkerk, D.E.; Schmidt, M.; Ippel, J.H.; Hackeng, T.M.; Nuijens, T.; Timmerman, P.; van Maarseveen, J.H. Synthesis of constrained tetracyclic peptides by consecutive CEPS, CLIPS, and oxime ligation. Org. Lett. 2019, 21, 2095–2100. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, H.E.; Grubbs, R.H. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew. Chem. Int. Ed. Engl. 1998, 37, 3281–3284. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Shepherd, N.E.; Hoang, H.N.; Abbenante, G.; Fairlie, D.P. Single turn peptide alpha helices with exceptional stability in water. J. Am. Chem. Soc. 2005, 127, 2974–2983. [Google Scholar] [CrossRef]
- Scrima, M.; Le Chevalier-Isaad, A.; Rovero, P.; Papini, A.M.; Chorev, M.; D’Ursi, A.M. CuI-catalyzed azide-alkyne intramolecular i-to-(i+4) side-chain-to-side-chain cyclization promotes the formation of helix-like secondary structures. Eur. J. Org. Chem. 2010, 3, 446–457. [Google Scholar] [CrossRef]
- Madden, M.M.; Rivera Vera, C.I.; Song, W.; Lin, Q. Facile synthesis of stapled, structurally reinforced peptide helices via a photoinduced intramolecular 1,3-dipolar cycloaddition reaction. Chem. Commun. 2009, 37, 5588–5590. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.Y.; King, D.S.; Chmielewski, J.; Singh, S.; Schultz, P.G. General approach to the synthesis of short alpha-helical peptides. J. Am. Chem. Soc. 1991, 113, 9391–9392. [Google Scholar] [CrossRef]
- Brunel, F.M.; Dawson, P.E. Synthesis of constrained helical peptides by thioether ligation: Application to analogs of gp41. Chem. Commun. 2005, 20, 2552–2554. [Google Scholar] [CrossRef] [PubMed]
- Assem, N.; Ferreira, D.J.; Wolan, D.W.; Dawson, P.E. Acetone-linked peptides: A convergent approach for peptide macrocyclization and labeling. Angew. Chem. Int. Ed. Engl. 2015, 54, 8665–8668. [Google Scholar] [CrossRef] [Green Version]
- Hilinski, G.J.; Kim, Y.W.; Hong, J.; Kutchukian, P.S.; Crenshaw, C.M.; Berkovitch, S.S.; Chang, A.; Ham, S.; Verdine, G.L. Stitched alpha-helical peptides via bis ring-closing metathesis. J. Am. Chem. Soc. 2014, 136, 12314–12322. [Google Scholar] [CrossRef]
- Serrano, J.C.; Sipthorp, J.; Xu, W.; Itzhaki, L.S.; Ley, S.V. A new methodology for incorporating chiral linkers into stapled peptides. Chembiochem 2017, 18, 1066–1071. [Google Scholar] [CrossRef]
- Hu, K.; Geng, H.; Zhang, Q.; Liu, Q.; Xie, M.; Sun, C.; Li, W.; Lin, H.; Jiang, F.; Wang, T.; et al. An in-tether chiral center modulates the helicity, cell permeability, and target binding affinity of a peptide. Angew. Chem. Int. Ed. Engl. 2016, 55, 8013–8017. [Google Scholar] [CrossRef]
- Jiang, Y.; Hu, K.; Shi, X.; Tang, Q.; Wang, Z.; Ye, X.; Li, Z. Switching substitution groups on the in-tether chiral centre influences backbone peptides’ permeability and target binding affinity. Org. Biomol. Chem. 2017, 15, 541544. [Google Scholar] [CrossRef]
- Luther, A.; Moehle, K.; Chevalier, E.; Dale, G.; Obrecht, D. Protein epitope mimetic macrocycles as biopharmaceuticals. Curr. Opin. Chem. Biol. 2017, 38, 45–51. [Google Scholar] [CrossRef]
- Obrecht, D.; Chevalier, E.; Moehle, K.; Robinson, J.A. Beta-hairpin protein epitope mimetic technology in drug discovery. Drug Discov. Today Technol. 2012, 9, e1–e70. [Google Scholar] [CrossRef] [Green Version]
- Cochran, A.G.; Tong, R.T.; Starovasnik, M.A.; Park, E.J.; McDowell, R.S.; Theaker, J.E.; Skelton, N.J. A minimal peptide scaffold for beta-turn display: Optimizing a strand position in disulfide-cyclized beta-hairpins. J. Am. Chem. Soc. 2001, 123, 625–632. [Google Scholar] [CrossRef]
- Cochran, A.G.; Skelton, N.J.; Starovasnik, M.A. Tryptophan zippers: Stable, monomeric beta -hairpins. Proc. Natl. Acad. Sci. USA 2001, 98, 5578–5583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Campbell, R.E. An engineered tryptophan zipper-type peptide as a molecular recognition scaffold. J. Pept. Sci. 2009, 15, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Alvarado, M.; Blanco, F.J.; Serrano, L. De novo design and structural analysis of a model beta-hairpin peptide system. Nat. Struct. Biol. 1996, 3, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Stanger, H.E.; Gellman, S.H. Rules for antiparallel β-sheet design: D-Pro-Gly is superior to L-Asn-Gly for β-hairpin nucleation. J. Am. Chem. Soc. 1998, 120, 4236–4237. [Google Scholar] [CrossRef]
- Masterson, L.R.; Etienne, M.A.; Porcelli, F.; Barany, G.; Hammer, R.P.; Veglia, G. Nonstereogenic alpha-aminoisobutyryl-glycyl dipeptidyl unit nucleates type I’ beta-turn in linear peptides in aqueous solution. Biopolymers 2007, 88, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Raghothama, S.; Sridharan, R.; Balaram, P. Tuning the beta-turn segment in designed peptide beta-hairpins: Construction of a stable type I’ beta-turn nucleus and hairpin-helix transition promoting segments. Biopolymers 2007, 88, 350–361. [Google Scholar] [CrossRef]
- Raghavender, U.S.; Aravinda, S.; Rai, R.; Shamala, N.; Balaram, P. Peptide hairpin nucleation with the obligatory Type I’ beta-turn Aib-DPro segment. Org. Biomol. Chem. 2010, 8, 3133–3135. [Google Scholar] [CrossRef] [PubMed]
- Favre, M.; Moehle, K.; Jiang, L.; Pfeiffer, B.; Robinson, J.A. Structural mimicry of canonical conformations in antibody hypervariable loops using cyclic peptides containing a heterochiral diproline template. J. Am. Chem. Soc. 1999, 121, 2679–2685. [Google Scholar] [CrossRef]
- Descours, A.; Moehle, K.; Renard, A.; Robinson, J.A. A new family of beta-hairpin mimetics based on a trypsin inhibitor from sunflower seeds. Chembiochem 2002, 3, 318–323. [Google Scholar] [CrossRef]
- Dias, R.L.; Fasan, R.; Moehle, K.; Renard, A.; Obrecht, D.; Robinson, J.A. Protein ligand design: From phage display to synthetic protein epitope mimetics in human antibody Fc-binding peptidomimetics. J. Am. Chem. Soc. 2006, 128, 2726–2732. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Ishiguro, K.; Takahashi, T.; Mihara, H. A novel beta-loop scaffold of phage-displayed peptides for highly specific affinities. Mol. Biosyst. 2011, 7, 2558–2562. [Google Scholar] [CrossRef] [PubMed]
- Pastor, M.T.; Lopez de la Paz, M.; Lacroix, E.; Serrano, L.; Perez-Paya, E. Combinatorial approaches: A new tool to search for highly structured beta-hairpin peptides. Proc. Natl. Acad. Sci. USA 2002, 99, 614–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, G.; Mulligan, V.K.; Bahl, C.D.; Gilmore, J.M.; Harvey, P.J.; Cheneval, O.; Buchko, G.W.; Pulavarti, S.V.; Kaas, Q.; Eletsky, A.; et al. Accurate de novo design of hyperstable constrained peptides. Nature 2016, 538, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef]
- Bozovičar, K.; Bratkovič, T. Evolving a peptide: Library platforms and diversification strategies. Int. J. Mol. Sci. 2020, 21, 215. [Google Scholar] [CrossRef] [Green Version]
- Rentero Rebollo, I.; Heinis, C. Phage selection of bicyclic peptides. Methods 2013, 60, 46–54. [Google Scholar] [CrossRef]
- Chen, S.; Rentero Rebollo, I.; Buth, S.A.; Morales-Sanfrutos, J.; Touati, J.; Leiman, P.G.; Heinis, C. Bicyclic peptide ligands pulled out of cysteine-rich peptide libraries. J. Am. Chem. Soc. 2013, 135, 6562–6569. [Google Scholar] [CrossRef]
- Chen, S.; Heinis, C. Phage selection of bicyclic peptides based on two disulfide bridges. Methods Mol. Biol. 2015, 1248, 119–137. [Google Scholar]
- Guillen Schlippe, Y.V.; Hartman, M.C.T.; Josephson, K.; Szostak, J.W. In vitro selection of highly modified cyclic peptides that act as tight binding inhibitors. J. Am. Chem. Soc. 2012, 134, 10469–10477. [Google Scholar] [CrossRef] [PubMed]
- Cervettini, D.; Tang, S.; Fried, S.D.; Willis, J.C.W.; Funke, L.F.H.; Colwell, L.J.; Chin, J.W. Rapid discovery and evolution of orthogonal aminoacyl-tRNA synthetase-tRNA pairs. Nat. Biotechnol. 2020, 38, 989–999. [Google Scholar] [CrossRef]
- Iwasaki, K.; Goto, Y.; Katoh, T.; Suga, H. Selective thioether macrocyclization of peptides having the N-terminal 2-chloroacetyl group and competing two or three cysteine residues in translation. Org. Biomol. Chem. 2012, 10, 5783–5786. [Google Scholar] [CrossRef] [PubMed]
- Okuma, R.; Kuwahara, T.; Yoshikane, T.; Watanabe, M.; Dranchak, P.; Inglese, J.; Shuto, S.; Goto, Y.; Suga, H. A macrocyclic peptide library with a structurally constrained cyclopropane-containing building block leads to thiol-independent inhibitors of phosphoglycerate mutase. Chem. Asian J. 2020, 15, 2631–2636. [Google Scholar] [CrossRef]
- Gonzalez-Navarro, H.; Mora, P.; Pastor, M.; Serrano, L.; Mingarro, I.; Perez-Paya, E. Identification of peptides that neutralize bacterial endotoxins using beta-hairpin conformationally restricted libraries. Mol. Divers 2000, 5, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, Z.; Steinmann, J.; Van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic antibiotics target outer-membrane biogenesis in P. aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, P.; Puijk, W.C.; Meloen, R.H. Functional reconstruction and synthetic mimicry of a conformational epitope using CLIPS technology. J. Mol. Recognit. 2007, 20, 283–299. [Google Scholar] [CrossRef]
- Iqbal, E.S.; Richardson, S.L.; Abrigo, N.A.; Dods, K.K.; Osorio Franco, H.E.; Gerrish, H.S.; Kotapati, H.K.; Morgan, I.M.; Masterson, D.S.; Hartman, M.C.T. A new strategy for the in vitro selection of stapled peptide inhibitors by mRNA display. Chem. Commun. (Camb) 2019, 55, 8959–8962. [Google Scholar] [CrossRef] [Green Version]
- Anananuchatkul, T.; Tsutsumi, H.; Miki, T.; Mihara, H. hDM2 protein-binding peptides screened from stapled alpha-helical peptide phage display libraries with different types of staple linkers. Bioorg. Med. Chem. Lett. 2020, 30, 127605. [Google Scholar] [CrossRef] [PubMed]
- Anananuchatkul, T.; Chang, I.V.; Miki, T.; Tsutsumi, H.; Mihara, H. Construction of a stapled alpha-helix peptide library displayed on phage for the screening of galectin-3-binding peptide ligands. ACS Omega 2020, 5, 5666–5674. [Google Scholar] [CrossRef]
- Timmerman, P.; Barderas, R.; Desmet, J.; Altschuh, D.; Shochat, S.; Hollestelle, M.J.; Höppener, J.W.M.; Monasterio, A.; Ignacio Casal, J.; Meloen, R.H. A combinatorial approach for the design of complementarity-determining region-derived peptidomimetics with in vitro anti-tumoral activity. J. Biol. Chem. 2009, 284, 34126–34134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, G.; Brown, A.; Mudd, G.; Huxley, P.; Van Rietschoten, K.; Pavan, S.; Chen, L.; Watcham, S.; Lahdenranta, J.; Keen, N. MMAE delivery using the bicycle toxin conjugate BT5528. Mol. Cancer Ther. 2020, 19, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Gowland, C.; Berry, P.; Errington, J.; Jeffrey, P.; Bennett, G.; Godfrey, L.; Pittman, M.; Niewiarowski, A.; Symeonides, N.S.; Veal, G.J. Development of a LC-MS/MS method for the quantification of toxic payload DM1 cleaved from BT1718 in a phase I study. Bioanalysis 2021, 13, 101–113. [Google Scholar] [CrossRef]
- Bicycle Therapeutics Programs. Available online: https://www.bicycletherapeutics.com/programs (accessed on 29 January 2021).
- Bernhagen, D.; Jungbluth, V.; Gisbert Quilis, N.; Dostalek, J.; White, P.B.; Jalink, K.; Timmerman, P. High-affinity alpha5beta1-integrin-selective bicyclic RGD peptides identified via screening of designed random libraries. ACS Comb. Sci. 2019, 21, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Bernhagen, D.; Jungbluth, V.; Quilis, N.G.; Dostalek, J.; White, P.B.; Jalink, K.; Timmerman, P. Bicyclic RGD peptides with exquisite selectivity for the integrin alphavbeta3 receptor using a “random design” approach. ACS Comb. Sci. 2019, 21, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamada, K.; Nakajima, S.; Ogawa, N.; Inada, M.; Shibasaki, H.; Sato, A.; Takasawa, R.; Yoshimori, A.; Suzuki, Y.; Watanabe, N.; et al. Papaverine identified as an inhibitor of high mobility group box 1/receptor for advanced glycation end-products interaction suppresses high mobility group box 1-mediated inflammatory responses. Biochem. Biophys. Res. Commun. 2019, 511, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Sclavons, C.; Burtea, C.; Boutry, S.; Laurent, S.; Vander Elst, L.; Muller, R.N. Phage display screening for tumor necrosis factor-alpha-binding peptides: Detection of inflammation in a mouse model of hepatitis. Int. J. Pept. 2013, 2013, 348409. [Google Scholar] [CrossRef] [Green Version]
- Bezer, S.; Matsumoto, M.; Lodewyk, M.W.; Lee, S.J.; Tantillo, D.J.; Gagne, M.R.; Waters, M.L. Identification and optimization of short helical peptides with novel reactive functionality as catalysts for acyl transfer by reactive tagging. Org. Biomol. Chem. 2014, 12, 1488–1494. [Google Scholar] [CrossRef]
- Matsumoto, M.; Lee, S.J.; Gagne, M.R.; Waters, M.L. Cross-strand histidine-aromatic interactions enhance acyl-transfer rates in beta-hairpin peptide catalysts. Org. Biomol. Chem. 2014, 12, 8711–8718. [Google Scholar] [CrossRef]
- Matsumoto, M.; Lee, S.J.; Waters, M.L.; Gagne, M.R. A catalyst selection protocol that identifies biomimetic motifs from beta-hairpin libraries. J. Am. Chem. Soc. 2014, 136, 15817–15820. [Google Scholar] [CrossRef]
- Cary, D.R.; Ohuchi, M.; Reid, P.C.; Masuya, K. Constrained peptides in drug discovery and development. J. Synth. Organ. Chem. Jpn. 2017, 75, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Shenderovich, M.D.; Liao, S.; Qian, X.; Hruby, V.J. A three-dimensional model of the delta-opioid pharmacophore: Comparative molecular modeling of peptide and nonpeptide ligands. Biopolymers 2000, 53, 565–580. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Kouznetsova, V.L.; Biswas, N.; Mahata, S.K.; O’Connor, D.T. Development of a pharmacophore model for the catecholamine release-inhibitory peptide catestatin: Virtual screening and functional testing identify novel small molecule therapeutics of hypertension. Bioorg. Med. Chem. 2013, 21, 5855–5869. [Google Scholar] [CrossRef] [PubMed]











| Chemistry | Residues Involved | Compatible Arrangement | Does Stereochemistry of the Staple Handles Need to be Considered? | Comment |
|---|---|---|---|---|
| alkene ring-closing metathesis | (homo)serine O-allyl ethers [59] | [i, i + 4] | no | all hydrocarbon staple |
| α,α-disubstituted residues with olefinic side chains (R or S configuration, 5 or 8 atoms long) [60] | [i, i + 4] and [i, i + 7] | yes (S5/S5 for [i, i + 4], S8/R5 or S5/R8 for [i, i + 7]) | ||
| lactamisation | lysine and glutamate, or ornithine and aspartate [61] | only compatible with [i, i + 4] arrangement | no | requires extra orthogonal protective groups for amino and carboxy groups for on resin lactamisation |
| cycloadditions | azide and alkyne group containing residues with 4 + 2 or 4 + 3 methylene units long side chains [62] | [i, i + 4] | no | well-established click reaction (Cu(I)-catalyzed azide-alkyne cycloaddition) |
| tetrazole and alkene group containing residues [63] | UV-induced cycloaddition between tetrazoles and alkenes to yield fluorescent pyrazoline tethers | |||
| disulfide bridges | thiol group containing residues [64] | [i, i + 7] | yes (combination of D and L-residues) | chronologically the oldest technique, requires acetamidomethyl protecting groups for thiols, staple unstable (prone to reduction) |
| thioether bridges | cysteine and an alpha-bromo amide group containing residue [65] | [i, i + 3] and [i, i + 4] | no | staple stable, higher helicity achieved with [i, i + 3] arrangement |
| two (homo)cysteines + dichloroacetone crosslinker [66] | [i, i + 4] | no | bis-alkylating crosslinker amenable to further derivation via oxime ligation (e.g., fluorophore or biotin coupling) | |
| cysteines + perfluoroaromatic crosslinker (e.g., hexafluorobenzene) [48] | [i, i + 4] | no | regioselective reaction (para-disubstituted staple) proceeding under mild conditions in high yield even for unprotected peptides |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozovičar, K.; Bratkovič, T. Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties. Int. J. Mol. Sci. 2021, 22, 1611. https://doi.org/10.3390/ijms22041611
Bozovičar K, Bratkovič T. Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties. International Journal of Molecular Sciences. 2021; 22(4):1611. https://doi.org/10.3390/ijms22041611
Chicago/Turabian StyleBozovičar, Krištof, and Tomaž Bratkovič. 2021. "Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties" International Journal of Molecular Sciences 22, no. 4: 1611. https://doi.org/10.3390/ijms22041611
APA StyleBozovičar, K., & Bratkovič, T. (2021). Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties. International Journal of Molecular Sciences, 22(4), 1611. https://doi.org/10.3390/ijms22041611