
Pharmacology of Free Fatty Acid Receptors and Their Allosteric Modulators


Abstract
:1. Introduction
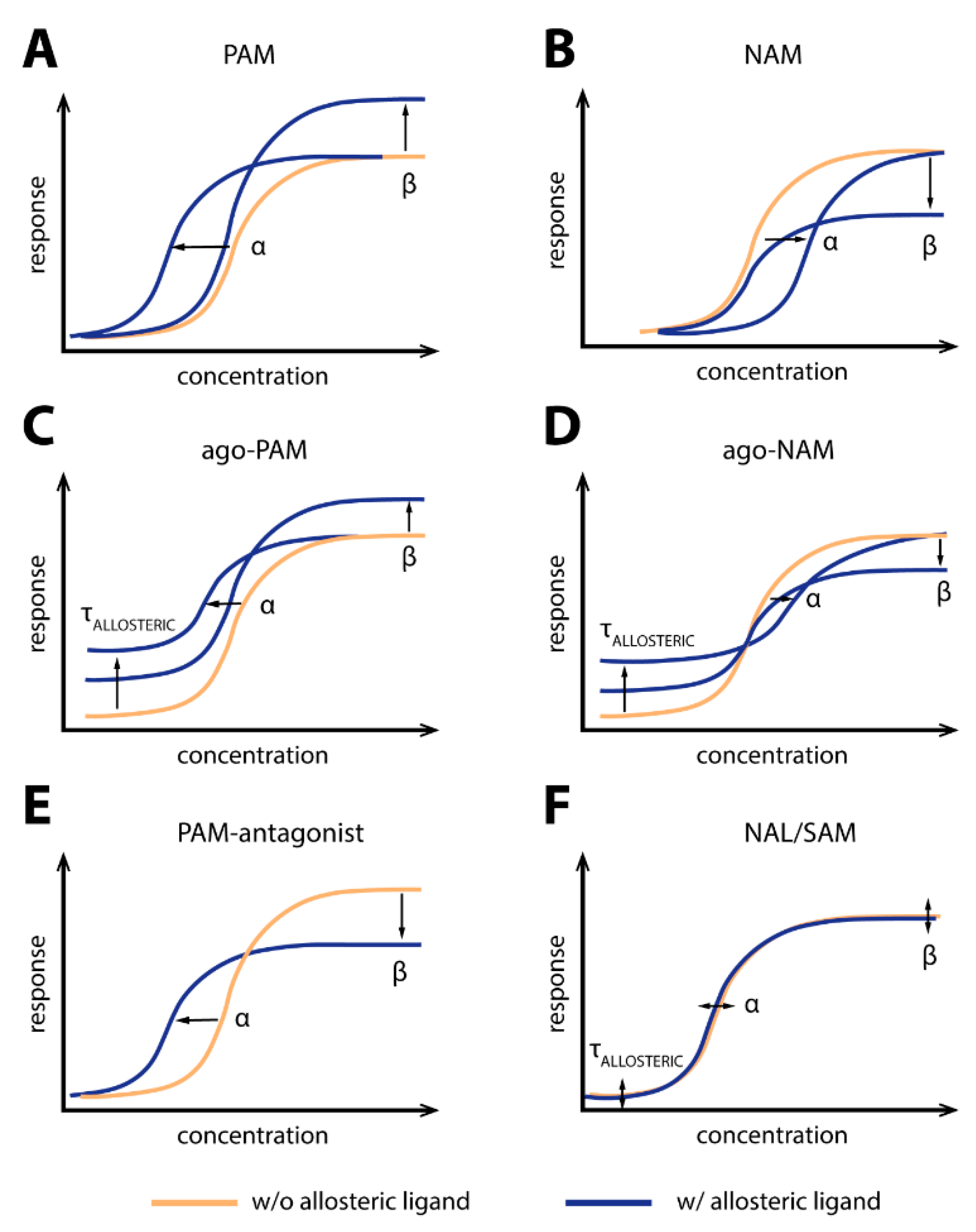
1.1. Allosterism at G Protein-Coupled Receptors
1.2. G protein-Coupled Receptors Activated by Free Fatty Acids (FFARs)
2. Free Fatty Acid Receptor 1 (FFAR1)
2.1. Introduction
2.2. Pancreatic β-Cell Function
2.3. Enteroendocrine Function
2.4. Nervous System
2.5. Bone Function
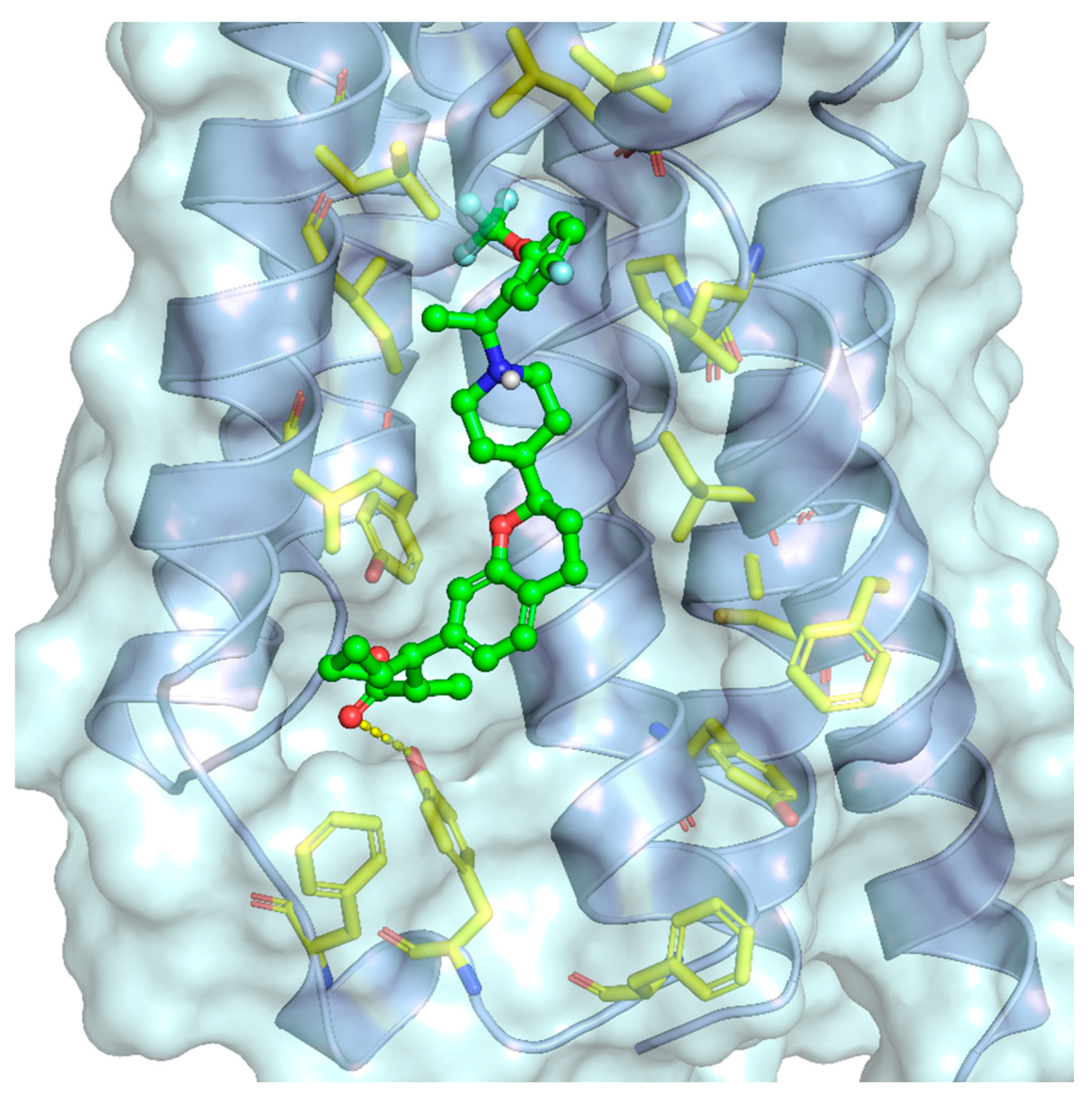
2.6. Molecular Receptor Pharmacology and Drug Discovery Efforts
3. Free Fatty Acid Receptors 2 and 3 (FFAR2 and FFAR3)
3.1. Introduction and Molecular Receptor Pharmacology
3.2. Enteroendocrine Function
3.3. Pancreatic β-Cells
3.4. Metabolic Functions
3.5. Inflammation and Immune Function
3.6. Nervous System
4. Free Fatty Acid Receptor 4 (FFAR4)
5. GPR84
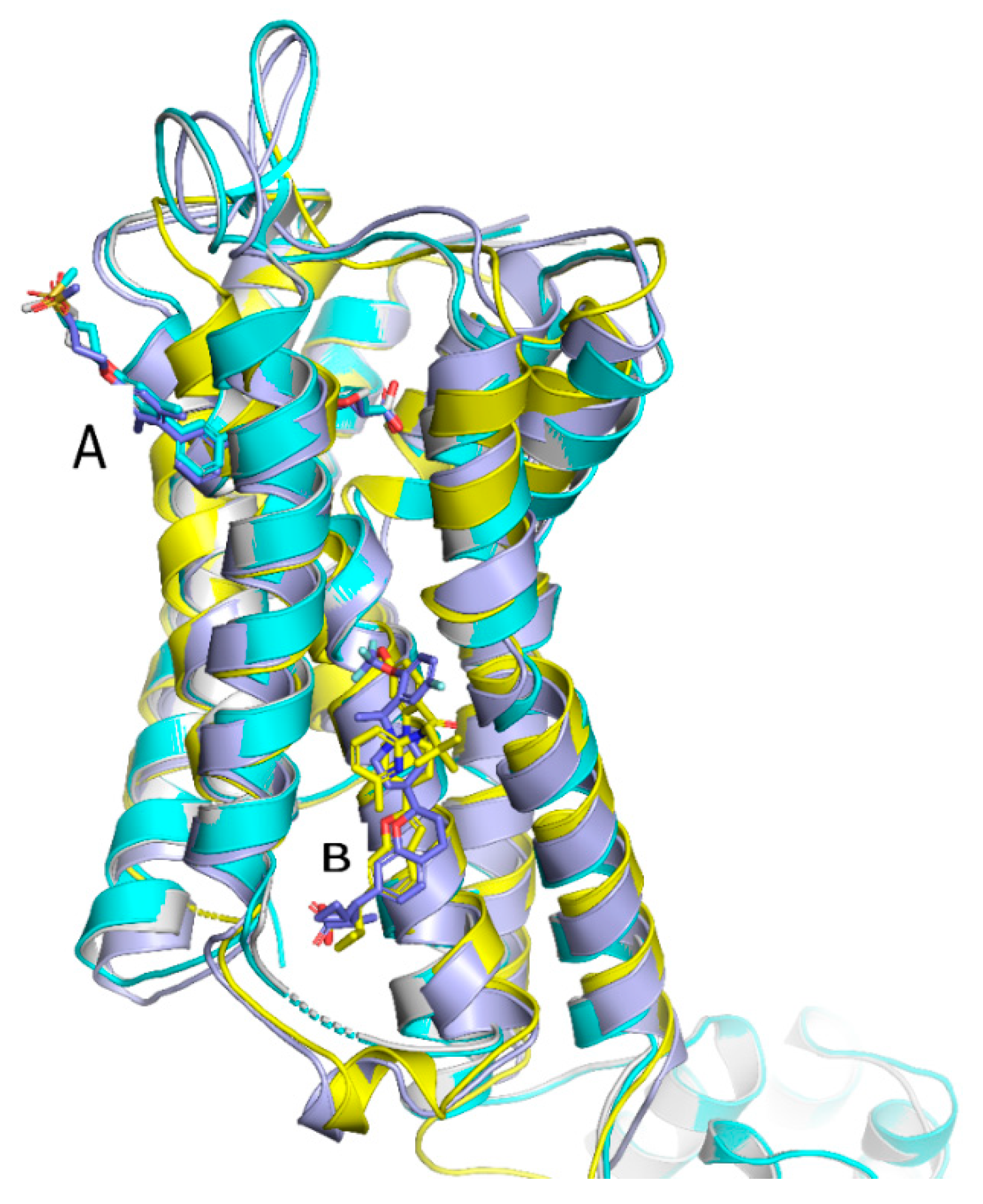
6. Structural Considerations
7. Opportunities and Challenges of Allosteric GPCR Ligands
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna Deepak, R.; Abdullah, A.; Talwar, P.; Fan, H.; Ravanan, P. Identification of FDA-approved drugs as novel allosteric inhibitors of human Exec. caspases. Proteins Struct. Funct. Bioinform. 2018, 86, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T.; Strachan, R.T. PAM-antagonists: A better way to block pathological receptor signaling? Trends Pharmacol. Sci. 2018, 39, 748–765. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A.; Kenakin, T. G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 2002, 54, 323–374. [Google Scholar] [CrossRef]
- Jakubík, J.; Randáková, A.; Chetverikov, N.; El-Fakahany, E.E.; Doležal, V. The operational model of allosteric modulation of pharmacological agonism. Sci. Rep. 2020, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; Casadó, V.; Devi, L.A.; Filizola, M.; Jockers, R.; Lohse, M.J.; Milligan, G.; Pin, J.-P.; Guitart, X. G protein–coupled receptor oligomerization revisited: Functional and pharmacological perspectives. Pharmacol. Rev. 2014, 66, 413–434. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Moreira, I.S.; Urizar, E.; Weinstein, H.; Javitch, J.A. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat. Chem. Biol. 2009, 5, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Park, P.S.-H.; Sum, C.S.; Pawagi, A.B.; Wells, J.W. Cooperativity and oligomeric status of cardiac muscarinic cholinergic receptors. Biochemistry 2002, 41, 5588–5604. [Google Scholar] [CrossRef] [PubMed]
- Ciruela, F.; Casadó, V.; Rodrigues, R.J.; Luján, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1–A2A receptor heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef] [Green Version]
- Ferre, S.; Von Euler, G.; Johansson, B.; Fredholm, B.B.; Fuxe, K. Stimulation of high-affinity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proc. Natl. Acad. Sci. USA 1991, 88, 7238–7241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcellino, D.; Ferré, S.; Casadó, V.; Cortés, A.; Le Foll, B.; Mazzola, C.; Drago, F.; Saur, O.; Stark, H.; Soriano, A. Identification of dopamine D1–D3 receptor heteromers: Indications for a role of synergistic D1–D3 receptor interactions in the striatum. J. Biol. Chem. 2008, 283, 26016–26025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Maeso, J.; Ang, R.L.; Yuen, T.; Chan, P.; Weisstaub, N.V.; López-Giménez, J.F.; Zhou, M.; Okawa, Y.; Callado, L.F.; Milligan, G. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 2008, 452, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleno, R.; Devost, D.; Pétrin, D.; Zhang, A.; Bourque, K.; Shinjo, Y.; Aoki, J.; Inoue, A.; Hébert, T.E. Conformational biosensors reveal allosteric interactions between heterodimeric AT1 angiotensin and prostaglandin F2α receptors. J. Biol. Chem. 2017, 292, 12139–12152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, Z.; Xiong, D.; Wu, M.; Ding, J.L. FFAR2-FFAR3 receptor heteromerization modulates short-chain fatty acid sensing. FASEB J. 2018, 32, 289–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawzdargo, M.; George, S.R.; Nguyen, T.; Xu, S.; Kolakowski Jr, L.F.; O’dowd, B.F. A cluster of four novel human G protein-coupled receptor genes occurring in close proximity to CD22 gene on chromosome 19q13. 1. Biochem. Biophys. Res. Commun. 1997, 239, 543–547. [Google Scholar] [CrossRef]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef] [Green Version]
- Kotarsky, K.; Nilsson, N.E.; Flodgren, E.; Owman, C.; Olde, B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem. Biophys. Res. Commun. 2003, 301, 406–410. [Google Scholar] [CrossRef]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [Green Version]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.-Y.; Lannoy, V.; Decobecq, M.-E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, N.E.; Kotarsky, K.; Owman, C.; Olde, B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem. Biophys. Res. Commun. 2003, 303, 1047–1052. [Google Scholar] [CrossRef]
- Fredriksson, R.; Höglund, P.J.; Gloriam, D.E.; Lagerström, M.C.; Schiöth, H.B. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 2003, 554, 381–388. [Google Scholar] [CrossRef]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Wittenberger, T.; Schaller, H.C.; Hellebrand, S. An expressed sequence tag (EST) data mining strategy succeeding in the discovery of new G-protein coupled receptors. J. Mol. Biol. 2001, 307, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Cooper, P.R.; Potter, S.L.; Mueck, B.; Jarai, G. Cloning and expression analysis of a novel G-protein-coupled receptor selectively expressed on granulocytes. J. Leukoc. Biol. 2001, 69, 1045–1052. [Google Scholar] [PubMed]
- Wang, J.; Wu, X.; Simonavicius, N.; Tian, H.; Ling, L. Medium-chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J. Biol. Chem. 2006, 281, 34457–34464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godinho, R.O.; Duarte, T.; Pacini, E.S.A. New perspectives in signaling mediated by receptors coupled to stimulatory G protein: The emerging significance of cAMP efflux and extracellular cAMP-adenosine pathway. Front. Pharmacol. 2015, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Engelstoft, M.S.; Park, W.-M.; Sakata, I.; Kristensen, L.V.; Husted, A.S.; Osborne-Lawrence, S.; Piper, P.K.; Walker, A.K.; Pedersen, M.H.; Nøhr, M.K. Seven transmembrane G protein-coupled receptor repertoire of gastric ghrelin cells. Mol. Metab. 2013, 2, 376–392. [Google Scholar] [CrossRef] [PubMed]
- Stone, V.M.; Dhayal, S.; Brocklehurst, K.J.; Lenaghan, C.; Winzell, M.S.; Hammar, M.; Xu, X.; Smith, D.M.; Morgan, N.G. GPR120 (FFAR4) is preferentially expressed in pancreatic delta cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia 2014, 57, 1182–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watterson, K.R.; Hansen, S.V.; Hudson, B.D.; Alvarez-Curto, E.; Raihan, S.Z.; Azevedo, C.M.; Martin, G.; Dunlop, J.; Yarwood, S.J.; Ulven, T. Probe-dependent negative allosteric modulators of the long-chain free fatty acid receptor FFA4. Mol. Pharmacol. 2017, 91, 630–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Rüttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Schröder, R.; Janssen, N.; Schmidt, J.; Kebig, A.; Merten, N.; Hennen, S.; Müller, A.; Blättermann, S.; Mohr-Andrä, M.; Zahn, S. Deconvolution of complex G protein–coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat. Biotechnol. 2010, 28, 943–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnon, L.; Leduc, M.; Thibodeau, J.-F.; Zhang, M.-Z.; Grouix, B.; Sarra-Bournet, F.; Gagnon, W.; Hince, K.; Tremblay, M.; Geerts, L. A newly discovered antifibrotic pathway regulated by two fatty acid receptors: GPR40 and GPR84. Am. J. Pathol. 2018, 188, 1132–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillaiyar, T.; Köse, M.; Namasivayam, V.; Sylvester, K.; Borges, G.; Thimm, D.; von Kügelgen, I.; Müller, C.E. 6-(Ar) Alkylamino-substituted uracil derivatives: Lipid mimetics with potent activity at the orphan G Protein-Coupled Receptor 84 (GPR84). ACS Omega 2018, 3, 3365–3383. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free fatty acid receptors in health and disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.-Y.; Jun, H.-S. Fatty acid-induced lipotoxicity in pancreatic beta-cells during development of type 2 diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Steneberg, P.; Rubins, N.; Bartoov-Shifman, R.; Walker, M.D.; Edlund, H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005, 1, 245–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristinsson, H.; Smith, D.M.; Bergsten, P.; Sargsyan, E. FFAR1 is involved in both the acute and chronic effects of palmitate on insulin secretion. Endocrinology 2013, 154, 4078–4088. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Wang, T.; Zhou, Y.; Liu, H.; Jiang, H.; Zhu, W.; Wang, H. DC260126: A small-molecule antagonist of GPR40 that protects against pancreatic β-cells dysfunction in db/db mice. PLoS ONE 2013, 8, e66744. [Google Scholar] [CrossRef] [Green Version]
- Latour, M.G.; Alquier, T.; Oseid, E.; Tremblay, C.; Jetton, T.L.; Luo, J.; Lin, D.C.-H.; Poitout, V. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes 2007, 56, 1087–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebede, M.; Alquier, T.; Latour, M.G.; Semache, M.; Tremblay, C.; Poitout, V. The fatty acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat feeding. Diabetes 2008, 57, 2432–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, H.; Hoos, L.M.; Liu, L.; Tetzloff, G.; Hu, W.; Abbondanzo, S.J.; Vassileva, G.; Gustafson, E.L.; Hedrick, J.A.; Davis, H.R. Lack of FFAR1/GPR40 does not protect mice from high-fat diet–induced metabolic disease. Diabetes 2008, 57, 2999–3006. [Google Scholar] [CrossRef] [Green Version]
- Vilas-Boas, E.A.; Karabacz, N.; Marsiglio-Librais, G.N.; Valle, M.M.R.; Nalbach, L.; Ampofo, E.; Morgan, B.; Carpinelli, A.R.; Roma, L.P. Chronic activation of GPR40 does not negatively impact upon BRIN-BD11 pancreatic β-cell physiology and function. Pharmacol. Rep. 2020, 72, 1725–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, R.; Kaiser, G.; Gerst, F.; Christiansen, E.; Due-Hansen, M.E.; Grundmann, M.; Machicao, F.; Peter, A.; Kostenis, E.; Ulven, T. Reevaluation of fatty acid receptor 1 as a drug target for the stimulation of insulin secretion in humans. Diabetes 2013, 62, 2106–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panse, M.; Gerst, F.; Kaiser, G.; Teutsch, C.-A.; Dölker, R.; Wagner, R.; Häring, H.-U.; Ullrich, S. Activation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) by free fatty acid receptor 1 (FFAR1/GPR40) protects from palmitate-induced beta cell death, but plays no role in insulin secretion. Cell. Physiol. Biochem. 2015, 35, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.H.; Kleinfeld, A.M. Unbound free fatty acid profiles in human plasma and the unexpected absence of unbound palmitoleate. J. Lipid Res. 2017, 58, 578–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tunaru, S.; Bonnavion, R.; Brandenburger, I.; Preussner, J.; Thomas, D.; Scholich, K.; Offermanns, S. 20-HETE promotes glucose-stimulated insulin secretion in an autocrine manner through FFAR1. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Edfalk, S.; Steneberg, P.; Edlund, H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 2008, 57, 2280–2287. [Google Scholar] [CrossRef] [Green Version]
- Liou, A.P.; Lu, X.; Sei, Y.; Zhao, X.; Pechhold, S.; Carrero, R.J.; Raybould, H.E.; Wank, S. The G-protein− coupled receptor GPR40 directly mediates long-chain fatty acid− induced secretion of cholecystokinin. Gastroenterology 2011, 140, 903–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, L.W.; Kuhre, R.E.; Janus, C.; Svendsen, B.; Holst, J.J. Vascular, but not luminal, activation of FFAR 1 (GPR 40) stimulates GLP-1 secretion from isolated perfused rat small intestine. Physiol. Rep. 2015, 3, e12551. [Google Scholar] [CrossRef] [Green Version]
- Psichas, A.; Larraufie, P.F.; Goldspink, D.A.; Gribble, F.M.; Reimann, F. Chylomicrons stimulate incretin secretion in mouse and human cells. Diabetologia 2017, 60, 2475–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauge, M.; Vestmar, M.A.; Husted, A.S.; Ekberg, J.P.; Wright, M.J.; Di Salvo, J.; Weinglass, A.B.; Engelstoft, M.S.; Madsen, A.N.; Lückmann, M. GPR40 (FFAR1)–combined Gs and Gq signaling in vitro is associated with robust incretin secretagogue action ex vivo and in vivo. Mol. Metab. 2015, 4, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, F.; Nishinaka, T.; Yamashita, T.; Nakamoto, K.; Kurihara, T.; Hirasawa, A.; Kasuya, F.; Miyata, A.; Tokuyama, S. GPR40/FFAR1 deficient mice increase noradrenaline levels in the brain and exhibit abnormal behavior. J. Pharmacol. Sci. 2016, 132, 249–254. [Google Scholar] [CrossRef]
- Nakamoto, K.; Aizawa, F.; Miyagi, K.; Yamashita, T.; Mankura, M.; Koyama, Y.; Kasuya, F.; Hirasawa, A.; Kurihara, T.; Miyata, A. Dysfunctional GPR40/FFAR1 signaling exacerbates pain behavior in mice. PLoS ONE 2017, 12, e0180610. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.-F.; Wu, H.-Y.; Tang, X.-Q.; Ali, U.; Liu, H.; Wang, Y.-X. Activation of GPR40 produces mechanical antiallodynia via the spinal glial interleukin-10/β-endorphin pathway. J. Neuroinflammation 2019, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Mieczkowska, A.; Baslé, M.F.; Chappard, D.; Mabilleau, G. Thiazolidinediones induce osteocyte apoptosis by a G protein-coupled receptor 40-dependent mechanism. J. Biol. Chem. 2012, 287, 23517–23526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wauquier, F.; Philippe, C.; Léotoing, L.; Mercier, S.; Davicco, M.-J.; Lebecque, P.; Guicheux, J.; Pilet, P.; Miot-Noirault, E.; Poitout, V. The free fatty acid receptor G protein-coupled receptor 40 (GPR40) protects from bone loss through inhibition of osteoclast differentiation. J. Biol. Chem. 2013, 288, 6542–6551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, A.D.; Bertrand, G.; Vivot, K.; Carpentier, É.; Tremblay, C.; Ghislain, J.; Bouvier, M.; Poitout, V. β-Arrestin recruitment and biased agonism at free fatty acid receptor 1. J. Biol. Chem. 2015, 290, 21131–21140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rives, M.-L.; Rady, B.; Swanson, N.; Zhao, S.; Qi, J.; Arnoult, E.; Bakaj, I.; Mancini, A.; Breton, B.; Lee, S.P. GPR40-mediated Gα12 activation by allosteric full agonists highly efficacious at potentiating glucose-stimulated insulin secretion in human islets. Mol. Pharmacol. 2018, 93, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Xu, X.; Huang, W.; Qian, H. Free fatty acid receptor 1 (FFAR1) as an emerging therapeutic target for type 2 diabetes mellitus: Recent progress and prevailing challenges. Med. Res. Rev. 2018, 38, 381–425. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, Z.; Zhang, L. Current status of GPR40/FFAR1 modulators in medicinal chemistry (2016–2019): A patent review. Expert Opin. Ther. Pat. 2020, 30, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.-H.; Guo, Q.; Luo, J.; Zhang, J.; Nguyen, K.; Chen, M.; Tran, T.; Dransfield, P.J.; Brown, S.P.; Houze, J. Identification and pharmacological characterization of multiple allosteric binding sites on the free fatty acid 1 receptor. Mol. Pharmacol. 2012, 82, 843–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Swaminath, G.; Cao, Q.; Yang, L.; Guo, Q.; Salomonis, H.; Lu, J.; Houze, J.B.; Dransfield, P.J.; Wang, Y. Activation of FFA1 mediates GLP-1 secretion in mice. Evidence for allosterism at FFA1. Mol. Cell. Endocrinol. 2013, 369, 119–129. [Google Scholar] [CrossRef]
- Ueno, H.; Ito, R.; Abe, S.-i.; Ogino, H.; Maruyama, M.; Miyashita, H.; Miyamoto, Y.; Moritoh, Y.; Tsujihata, Y.; Takeuchi, K. GPR40 full agonism exerts feeding suppression and weight loss through afferent vagal nerve. PLoS ONE 2019, 14, e0222653. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.V.; Marcinak, J.F.; Raanan, M.G.; Cao, C. Combining the G-protein-coupled receptor 40 agonist fasiglifam with sitagliptin improves glycaemic control in patients with type 2 diabetes with or without metformin: A randomized, 12-week trial. Diabetes Obes. Metab. 2017, 19, 1127–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Yoshida, S.; Minoura, H.; Negoro, K.; Shimaya, A.; Shimokawa, T.; Shibasaki, M. Novel GPR40 agonist AS2575959 exhibits glucose metabolism improvement and synergistic effect with sitagliptin on insulin and incretin secretion. Life Sci. 2014, 94, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Gorski, J.N.; Pachanski, M.J.; Mane, J.; Plummer, C.W.; Souza, S.; Thomas-Fowlkes, B.S.; Ogawa, A.M.; Weinglass, A.B.; Di Salvo, J.; Cheewatrakoolpong, B. GPR40 reduces food intake and body weight through GLP-1. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E37–E47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachanski, M.J.; Kirkland, M.E.; Kosinski, D.T.; Mane, J.; Cheewatrakoolpong, B.; Xue, J.; Szeto, D.; Forrest, G.; Miller, C.; Bunzel, M. GPR40 partial agonists and AgoPAMs: Differentiating effects on glucose and hormonal secretions in the rodent. PLoS ONE 2017, 12, e0186033. [Google Scholar] [CrossRef] [Green Version]
- Yashiro, H.; Tsujihata, Y.; Takeuchi, K.; Hazama, M.; Johnson, P.R.; Rorsman, P. The effects of TAK-875, a selective G protein-coupled receptor 40/free fatty acid 1 agonist, on insulin and glucagon secretion in isolated rat and human islets. J. Pharmacol. Exp. Ther. 2012, 340, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabuki, C.; Komatsu, H.; Tsujihata, Y.; Maeda, R.; Ito, R.; Matsuda-Nagasumi, K.; Sakuma, K.; Miyawaki, K.; Kikuchi, N.; Takeuchi, K. A novel antidiabetic drug, fasiglifam/TAK-875, acts as an ago-allosteric modulator of FFAR1. PLoS ONE 2013, 8, e76280. [Google Scholar] [CrossRef]
- Kaku, K.; Enya, K.; Nakaya, R.; Ohira, T.; Matsuno, R. Efficacy and safety of fasiglifam (TAK-875), a G protein-coupled receptor 40 agonist, in J apanese patients with type 2 diabetes inadequately controlled by diet and exercise: A randomized, double-blind, placebo-controlled, phase III trial. Diabetes Obes. Metab. 2015, 17, 675–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhong, K.; Guo, Z.; Zhong, D.; Chen, X. Fasiglifam (TAK-875) inhibits hepatobiliary transporters: A possible factor contributing to fasiglifam-induced liver injury. Drug Metab. Dispos. 2015, 43, 1751–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, A.; Poitout, V. GPR40 agonists for the treatment of type 2 diabetes: Life after ‘TAKing’a hit. Diabetes Obes. Metab. 2015, 17, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, C.; Yang, J.; Zhou, J.; Ye, Z.; Feng, D.; Yue, N.; Tong, J.; Huang, W.; Qian, H. Design, synthesis and biological evaluation of novel FFA1/GPR40 agonists: New breakthrough in an old scaffold. Eur. J. Med. Chem. 2019, 179, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Miyamoto, Y.; Hirata, Y.; Watanabe, K.; Hitomi, Y.; Yoshitomi, Y.; Aida, J.; Noguchi, N.; Takakura, N.; Takami, K. Design and Identification of a GPR40 Full Agonist (SCO-267) Possessing a 2-Carbamoylphenyl Piperidine Moiety. J. Med. Chem. 2020, 63, 10352–10379. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Meegalla, S.K.; Lanter, J.C.; Winters, M.P.; Zhao, S.; Littrell, J.; Qi, J.; Rady, B.; Lee, P.S.; Liu, J. Discovery of a GPR40 Superagonist: The Impact of Aryl Propionic Acid α-Fluorination. ACS Med. Chem. Lett. 2018, 10, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qiu, Q.; Geng, X.; Yang, J.; Huang, W.; Qian, H. Free fatty acid receptor agonists for the treatment of type 2 diabetes: Drugs in preclinical to phase II clinical development. Expert Opin. Investig. Drugs 2016, 25, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Hirayama, M.; Hiroi, S.; Kaku, K. GPR40-induced insulin secretion by the novel agonist TAK-875: First clinical findings in patients with type 2 diabetes. Diabetes Obes. Metab. 2012, 14, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.-H.; Zhang, J.; Zhuang, R.; Li, F.; Nguyen, K.; Chen, M.; Tran, T.; Lopez, E.; Lu, J.Y.L.; Li, X.N. AMG 837: A novel GPR40/FFA1 agonist that enhances insulin secretion and lowers glucose levels in rodents. PLoS ONE 2011, 6, e27270. [Google Scholar] [CrossRef]
- Hyde, A.M.; Liu, Z.; Kosjek, B.; Tan, L.; Klapars, A.; Ashley, E.R.; Zhong, Y.-L.; Alvizo, O.; Agard, N.J.; Liu, G. Synthesis of the GPR40 partial agonist MK-8666 through a kinetically controlled dynamic enzymatic ketone reduction. Org. Lett. 2016, 18, 5888–5891. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Byrne, N.; Wang, J.; Bricogne, G.; Brown, F.K.; Chobanian, H.R.; Colletti, S.L.; Di Salvo, J.; Thomas-Fowlkes, B.; Guo, Y. Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nat. Struct. Mol. Biol. 2017, 24, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Vaddady, P.; Railkar, R.; Musser, B.; Cote, J.; Ederveen, A.; Krefetz, D.; DeNoia, E.; Free, A.; Morrow, L. Leveraging a Clinical Phase Ib Proof-of-Concept Study for the GPR40 Agonist MK-8666 in Patients with Type 2 Diabetes for Model-Informed Phase II Dose Selection. Clin. Transl. Sci. 2017, 10, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.P.; Dransfield, P.J.; Vimolratana, M.; Jiao, X.; Zhu, L.; Pattaropong, V.; Sun, Y.; Liu, J.; Luo, J.; Zhang, J. Discovery of AM-1638: A potent and orally bioavailable GPR40/FFA1 full agonist. ACS Med. Chem. Lett. 2012, 3, 726–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.D.; Chau, B.; Rodgers, L.; Lu, F.; Wilbur, K.L.; Otto, K.A.; Chen, Y.; Song, M.; Riley, J.P.; Yang, H.-C. Structural basis for GPR40 allosteric agonism and incretin stimulation. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milligan, G.; Shimpukade, B.; Ulven, T.; Hudson, B.D. Complex pharmacology of free fatty acid receptors. Chem. Rev. 2017, 117, 67–110. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Smith, N.J.; Christiansen, E.; Tikhonova, I.G.; Grundmann, M.; Hudson, B.D.; Ward, R.J.; Drewke, C.; Milligan, G.; Kostenis, E. Selective orthosteric free fatty acid receptor 2 (FFA2) agonists identification of the structural and chemical requirements for selective activation of FFA2 versus FFA3. J. Biol. Chem. 2011, 286, 10628–10640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, B.D.; Due-Hansen, M.E.; Christiansen, E.; Hansen, A.M.; Mackenzie, A.E.; Murdoch, H.; Pandey, S.K.; Ward, R.J.; Marquez, R.; Tikhonova, I.G. Defining the molecular basis for the first potent and selective orthosteric agonists of the FFA2 free fatty acid receptor. J. Biol. Chem. 2013, 288, 17296–17312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.; Schwandner, R.; Swaminath, G.; Weiszmann, J.; Cardozo, M.; Greenberg, J.; Jaeckel, P.; Ge, H.; Wang, Y.; Jiao, X. Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol. Pharmacol. 2008, 74, 1599–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jiao, X.; Kayser, F.; Liu, J.; Wang, Z.; Wanska, M.; Greenberg, J.; Weiszmann, J.; Ge, H.; Tian, H. The first synthetic agonists of FFA2: Discovery and SAR of phenylacetamides as allosteric modulators. Bioorganic Med. Chem. Lett. 2010, 20, 493–498. [Google Scholar] [CrossRef]
- Swaminath, G.; Jaeckel, P.; Guo, Q.; Cardozo, M.; Weiszmann, J.; Lindberg, R.; Wang, Y.; Schwandner, R.; Li, Y. Mutational analysis of G-protein coupled receptor–FFA2. Biochem. Biophys. Res. Commun. 2011, 405, 122–127. [Google Scholar] [CrossRef]
- Smith, N.J.; Ward, R.J.; Stoddart, L.A.; Hudson, B.D.; Kostenis, E.; Ulven, T.; Morris, J.C.; Tränkle, C.; Tikhonova, I.G.; Adams, D.R. Extracellular loop 2 of the free fatty acid receptor 2 mediates allosterism of a phenylacetamide ago-allosteric modulator. Mol. Pharmacol. 2011, 80, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundmann, M.; Tikhonova, I.G.; Hudson, B.D.; Smith, N.J.; Mohr, K.; Ulven, T.; Milligan, G.; Kenakin, T.; Kostenis, E. A molecular mechanism for sequential activation of a G protein-coupled receptor. Cell Chem. Biol. 2016, 23, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundmann, M.; Kostenis, E. Temporal bias: Time-encoded dynamic GPCR signaling. Trends Pharmacol. Sci. 2017, 38, 1110–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantis, C.; Ooms, F.; Bernard, J. Novel Amino Acid Derivatives and Their Use as GPR43 Receptor Modulators. International Patent Application WO/2011/092284, 28 January 2011. [Google Scholar]
- Park, B.-O.; Kim, S.H.; Kong, G.Y.; Kim, D.H.; Kwon, M.S.; Lee, S.U.; Kim, M.-O.; Cho, S.; Lee, S.; Lee, H.-J. Selective novel inverse agonists for human GPR43 augment GLP-1 secretion. Eur. J. Pharmacol. 2016, 771, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Saniere, L.; Raymond, M.; Pizzonero, M. Azetidine Derivatives Useful for the Treatment of Metabolic and Inflammatory Diseases. International Patent Application WO 2012/098033 A1, 10 January 2012. [Google Scholar]
- Namour, F.; Galien, R.; Van Kaem, T.; Van der Aa, A.; Vanhoutte, F.; Beetens, J.; Van’t Klooster, G. Safety, pharmacokinetics and pharmacodynamics of GLPG0974, a potent and selective FFA2 antagonist, in healthy male subjects. Br. J. Clin. Pharmacol. 2016, 82, 139–148. [Google Scholar] [CrossRef]
- Nøhr, M.K.; Pedersen, M.H.; Gille, A.; Egerod, K.L.; Engelstoft, M.S.; Husted, A.S.; Sichlau, R.M.; Grunddal, K.V.; Seier Poulsen, S.; Han, S. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology 2013, 154, 3552–3564. [Google Scholar] [CrossRef] [PubMed]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein–coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caengprasath, N.; Gonzalez-Abuin, N.; Shchepinova, M.; Ma, Y.; Inoue, A.; Tate, E.W.; Frost, G.; Hanyaloglu, A.C. Internalization-dependent free fatty acid receptor 2 signaling is essential for propionate-induced anorectic gut hormone release. Iscience 2020, 23, 101449. [Google Scholar] [CrossRef]
- Bolognini, D.; Barki, N.; Butcher, A.J.; Hudson, B.D.; Sergeev, E.; Molloy, C.; Moss, C.E.; Bradley, S.J.; Le Gouill, C.; Bouvier, M. Chemogenetics defines receptor-mediated functions of short chain free fatty acids. Nat. Chem. Biol. 2019, 15, 489–498. [Google Scholar] [CrossRef]
- Pfeil, E.M.; Brands, J.; Merten, N.; Vögtle, T.; Vescovo, M.; Rick, U.; Albrecht, I.-M.; Heycke, N.; Kawakami, K.; Ono, Y. Heterotrimeric G Protein Subunit Gαq Is a Master Switch for Gβγ-Mediated Calcium Mobilization by Gi-Coupled GPCRs. Mol. Cell 2020, 80, 940–954. [Google Scholar] [CrossRef]
- Liu, J.-L.; Segovia, I.; Yuan, X.-L.; Gao, Z.-h. Controversial Roles of Gut Microbiota-Derived Short-Chain Fatty Acids (SCFAs) on Pancreatic β-Cell Growth and Insulin Secretion. Int. J. Mol. Sci. 2020, 21, 910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Ahmed, K.; Gille, A.; Lu, S.; Gröne, H.-J.; Tunaru, S.; Offermanns, S. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat. Med. 2015, 21, 173–177. [Google Scholar] [CrossRef] [PubMed]
- McNelis, J.C.; Lee, Y.S.; Mayoral, R.; van der Kant, R.; Johnson, A.M.; Wollam, J.; Olefsky, J.M. GPR43 potentiates β-cell function in obesity. Diabetes 2015, 64, 3203–3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priyadarshini, M.; Layden, B.T. FFAR3 modulates insulin secretion and global gene expression in mouse islets. Islets 2015, 7, e1045182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priyadarshini, M.; Villa, S.R.; Fuller, M.; Wicksteed, B.; Mackay, C.R.; Alquier, T.; Poitout, V.; Mancebo, H.; Mirmira, R.G.; Gilchrist, A. An acetate-specific GPCR, FFAR2, regulates insulin secretion. Mol. Endocrinol. 2015, 29, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Veprik, A.; Laufer, D.; Weiss, S.; Rubins, N.; Walker, M.D. GPR41 modulates insulin secretion and gene expression in pancreatic β-cells and modifies metabolic homeostasis in fed and fasting states. FASEB J. 2016, 30, 3860–3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, H.; Li, X.; Weiszmann, J.; Wang, P.; Baribault, H.; Chen, J.-L.; Tian, H.; Li, Y. Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology 2008, 149, 4519–4526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolognini, D.; Dedeo, D.; Milligan, G. Metabolic and inflammatory functions of short chain fatty acid receptors. Curr. Opin. Endocr. Metab. Res. 2020, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, J.; Ohue-Kitano, R.; Mukouyama, H.; Nishida, A.; Watanabe, K.; Igarashi, M.; Irie, J.; Tsujimoto, G.; Satoh-Asahara, N.; Itoh, H. Ketone body receptor GPR43 regulates lipid metabolism under ketogenic conditions. Proc. Natl. Acad. Sci. USA 2019, 116, 23813–23821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, H.; Kamikado, K.; Aoki, R.; Suganuma, N.; Nishijima, T.; Nakatani, A.; Kimura, I. Bifidobacterium animalis subsp. lactis GCL2505 modulates host energy metabolism via the short-chain fatty acid receptor GPR43. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, L.; Viardot, A.; Tsakmaki, A.; Stolarczyk, E.; Howard, J.K.; Cani, P.D.; Everard, A.; Sleeth, M.L.; Psichas, A.; Anastasovskaj, J. Fermentable carbohydrate stimulates FFAR2-dependent colonic PYY cell expansion to increase satiety. Mol. Metab. 2017, 6, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjursell, M.; Admyre, T.; Göransson, M.; Marley, A.E.; Smith, D.M.; Oscarsson, J.; Bohlooly-Y, M. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E211–E220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Chassaing, B.; Singh, V.; Pellizzon, M.; Ricci, M.; Fythe, M.D.; Kumar, M.V.; Gewirtz, A.T. Fiber-mediated nourishment of gut microbiota protects against diet-induced obesity by restoring IL-22-mediated colonic health. Cell Host Microbe 2018, 23, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, E.; Lavoie, S.; Fonseca-Pereira, D.; Bae, S.; Michaud, M.; Hoveyda, H.R.; Fraser, G.L.; Comeau, C.A.G.; Glickman, J.N.; Fuller, M.H. Metabolite-sensing receptor Ffar2 regulates colonic group 3 innate lymphoid cells and gut immunity. Immunity 2019, 51, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Sun, M.; Chen, F.; Cao, A.T.; Liu, H.; Zhao, Y.; Huang, X.; Xiao, Y.; Yao, S.; Zhao, Q. Microbiota metabolite short-chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol. 2017, 10, 946–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Xiao, Y.; Huang, X.; Chen, F.; Sun, M.; Bilotta, A.J.; Xu, L.; Lu, Y.; Yao, S.; Zhao, Q. Microbiota metabolite short-chain fatty acids facilitate mucosal adjuvant activity of cholera toxin through GPR43. J. Immunol. 2019, 203, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Singer, J.; Kwan, T.K.; Loh, Y.W.; Wang, C.; Tan, J.; Li, Y.J.; Lai, S.W.C.; Macia, L.; Alexander, S.I. Gut Microbial Metabolites Induce Donor-Specific Tolerance of Kidney Allografts through Induction of T Regulatory Cells by Short-Chain Fatty Acids. J. Am. Soc. Nephrol. 2020, 31, 1445–1461. [Google Scholar] [CrossRef]
- Li, Y.J.; Chen, X.; Kwan, T.K.; Loh, Y.W.; Singer, J.; Liu, Y.; Ma, J.; Tan, J.; Macia, L.; Mackay, C.R. Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid–Mediated Activation of G Protein–Coupled Receptors GPR43 and GPR109A. J. Am. Soc. Nephrol. 2020, 31, 1267–1281. [Google Scholar] [CrossRef]
- Huang, W.; Man, Y.; Gao, C.; Zhou, L.; Gu, J.; Xu, H.; Wan, Q.; Long, Y.; Chai, L.; Xu, Y. Short-Chain Fatty Acids Ameliorate Diabetic Nephropathy via GPR43-Mediated Inhibition of Oxidative Stress and NF-κB Signaling. Oxidative Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Mikami, D.; Kimura, H.; Kamiyama, K.; Morikawa, Y.; Yokoi, S.; Kasuno, K.; Takahashi, N.; Taniguchi, T.; Iwano, M. Short-chain fatty acids, GPR41 and GPR43 ligands, inhibit TNF-α-induced MCP-1 expression by modulating p38 and JNK signaling pathways in human renal cortical epithelial cells. Biochem. Biophys. Res. Commun. 2017, 486, 499–505. [Google Scholar] [CrossRef]
- Kaye, D.M.; Shihata, W.A.; Jama, H.A.; Tsyganov, K.; Ziemann, M.; Kiriazis, H.; Horlock, D.; Vijay, A.; Giam, B.; Vinh, A. Deficiency of Prebiotic Fiber and Insufficient Signaling Through Gut Metabolite-Sensing Receptors Leads to Cardiovascular Disease. Circulation 2020, 141, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Fachi, J.L.; Sécca, C.; Rodrigues, P.B.; de Mato, F.C.P.; Di Luccia, B.; de Souza Felipe, J.; Pral, L.P.; Rungue, M.; de Melo Rocha, V.; Sato, F.T. Acetate coordinates neutrophil and ILC3 responses against C. difficile through FFAR2. J. Exp. Med. 2020, 217, e20190489. [Google Scholar] [CrossRef] [PubMed]
- Galvão, I.; Tavares, L.P.; Corrêa, R.O.; Fachi, J.L.; Rocha, V.M.; Rungue, M.; Garcia, C.C.; Cassali, G.; Ferreira, C.M.; Martins, F.S. The metabolic sensor GPR43 receptor plays a role in the control of Klebsiella pneumoniae infection in the lung. Front. Immunol. 2018, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Sencio, V.; Barthelemy, A.; Tavares, L.P.; Machado, M.G.; Soulard, D.; Cuinat, C.; Queiroz-Junior, C.M.; Noordine, M.-L.; Salomé-Desnoulez, S.; Deryuter, L. Gut dysbiosis during influenza contributes to pulmonary pneumococcal superinfection through altered short-chain fatty acid production. Cell Rep. 2020, 30, 2934–2947. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Jiang, L.; Wang, J.; Zhang, J.; Kong, F.; Li, Q.; Yan, Y.; Huang, S.; Zhao, Y.; Liang, L. The G protein-coupled receptor FFAR2 promotes internalization during influenza A virus entry. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Vinolo, M.A.; Ferguson, G.J.; Kulkarni, S.; Damoulakis, G.; Anderson, K.; Bohlooly-Y, M.; Stephens, L.; Hawkins, P.T.; Curi, R. SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS ONE 2011, 6, e21205. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Sina, C.; Gavrilova, O.; Förster, M.; Till, A.; Derer, S.; Hildebrand, F.; Raabe, B.; Chalaris, A.; Scheller, J.; Rehmann, A. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J. Immunol. 2009, 183, 7514–7522. [Google Scholar] [CrossRef]
- Frei, R.; Nordlohne, J.; Hüser, U.; Hild, S.; Schmidt, J.; Eitner, F.; Grundmann, M. Allosteric targeting of the FFA2 receptor (GPR43) restores responsiveness of desensitized human neutrophils. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lind, S.; Holdfeldt, A.; Mårtensson, J.; Sundqvist, M.; Björkman, L.; Forsman, H.; Dahlgren, C. Functional selective ATP receptor signaling controlled by the free fatty acid receptor 2 through a novel allosteric modulation mechanism. FASEB J. 2019, 33, 6887–6903. [Google Scholar] [CrossRef] [PubMed]
- Mårtensson, J.; Holdfeldt, A.; Sundqvist, M.; Gabl, M.; Kenakin, T.P.; Björkman, L.; Forsman, H.; Dahlgren, C. Neutrophil priming that turns natural FFA2R agonists into potent activators of the superoxide generating NADPH-oxidase. J. Leukoc. Biol. 2018, 104, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.T.; Macia, L.; Galvão, I.; Martins, F.S.; Canesso, M.C.C.; Amaral, F.A.; Garcia, C.C.; Maslowski, K.M.; De Leon, E.; Shim, D. A role for gut microbiota and the metabolite-sensing receptor GPR43 in a murine model of gout. Arthritis Rheumatol. 2015, 67, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Kamp, M.E.; Shim, R.; Nicholls, A.J.; Oliveira, A.C.; Mason, L.J.; Binge, L.; Mackay, C.R.; Wong, C.H. G protein-coupled receptor 43 modulates neutrophil recruitment during acute inflammation. PLoS ONE 2016, 11, e0163750. [Google Scholar] [CrossRef]
- Nakajima, A.; Nakatani, A.; Hasegawa, S.; Irie, J.; Ozawa, K.; Tsujimoto, G.; Suganami, T.; Itoh, H.; Kimura, I. The short chain fatty acid receptor GPR43 regulates inflammatory signals in adipose tissue M2-type macrophages. PLoS ONE 2017, 12, e0179696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in obesity and inflammation–protective or causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkman, L.; Mårtensson, J.; Winther, M.; Gabl, M.; Holdfeldt, A.; Uhrbom, M.; Bylund, J.; Hansen, A.H.; Pandey, S.K.; Ulven, T. The neutrophil response induced by an agonist for free fatty acid receptor 2 (GPR43) Is primed by tumor necrosis factor alpha and by receptor uncoupling from the cytoskeleton but attenuated by tissue recruitment. Mol. Cell. Biol. 2016, 36, 2583–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, S.; Holdfeldt, A.; Mårtensson, J.; Sundqvist, M.; Kenakin, T.P.; Björkman, L.; Forsman, H.; Dahlgren, C. Interdependent allosteric free fatty acid receptor 2 modulators synergistically induce functional selective activation and desensitization in neutrophils. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2020, 1867, 118689. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaji, I.; Akiba, Y.; Furuyama, T.; Adelson, D.W.; Iwamoto, K.; Watanabe, M.; Kuwahara, A.; Kaunitz, J.D. Free fatty acid receptor 3 activation suppresses neurogenic motility in rat proximal colon. Neurogastroenterol. Motil. 2018, 30, e13157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, I.; Miyamoto, J.; Ohue-Kitano, R.; Watanabe, K.; Yamada, T.; Onuki, M.; Aoki, R.; Isobe, Y.; Kashihara, D.; Inoue, D. Maternal gut microbiota in pregnancy influences offspring metabolic phenotype in mice. Science 2020, 367. [Google Scholar] [CrossRef]
- Bolognini, D.; Moss, C.E.; Nilsson, K.; Petersson, A.U.; Donnelly, I.; Sergeev, E.; König, G.M.; Kostenis, E.; Kurowska-Stolarska, M.; Miller, A. A novel allosteric activator of free fatty acid 2 receptor displays unique Gi-functional bias. J. Biol. Chem. 2016, 291, 18915–18931. [Google Scholar] [CrossRef] [Green Version]
- Leonard, J.N.; Chu, Z.L.; Bruce, M.A.; Boatman, P.D. GPR41 and Modulators Thereof for the Treatment of Insulin-Related Disorders. U.S. Patent 2008/0312277 A1, 18 December 2008. [Google Scholar]
- Hudson, B.D.; Christiansen, E.; Murdoch, H.; Jenkins, L.; Hansen, A.H.; Madsen, O.; Ulven, T.; Milligan, G. Complex pharmacology of novel allosteric free fatty acid 3 receptor ligands. Mol. Pharmacol. 2014, 86, 200–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulven, T.; Christiansen, E. Dietary fatty acids and their potential for controlling metabolic diseases through activation of FFA4/GPR120. Annu. Rev. Nutr. 2015, 35, 239–263. [Google Scholar] [CrossRef]
- Robinson, L.E.; Mazurak, V.C. N-3 polyunsaturated fatty acids: Relationship to inflammation in healthy adults and adults exhibiting features of metabolic syndrome. Lipids 2013, 48, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Huang, T.; Zheng, J.; Wu, K.; Li, D. Effect of marine-derived n-3 polyunsaturated fatty acids on C-reactive protein, interleukin 6 and tumor necrosis factor α: A meta-analysis. PLoS ONE 2014, 9, e88103. [Google Scholar] [CrossRef] [Green Version]
- Watson, S.-J.; Brown, A.J.; Holliday, N.D. Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol. Pharmacol. 2012, 81, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Curto, E.; Inoue, A.; Jenkins, L.; Raihan, S.Z.; Prihandoko, R.; Tobin, A.B.; Milligan, G. Targeted elimination of G proteins and arrestins defines their specific contributions to both intensity and duration of G protein-coupled receptor signaling. J. Biol. Chem. 2016, 291, 27147–27159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, S.J.; Larsen, L.K.; Hansen, G.; Chelur, S.; Larsen, P.J.; Vrang, N. Expression of the fatty acid receptor GPR120 in the gut of diet-induced-obese rats and its role in GLP-1 secretion. PLoS ONE 2014, 9, e88227. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-inflammatory and Insulin-Sensitizing Effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.; Zhang, Q.; Murgolo, N.; Hosted, T.; Duffy, R. Cloning, expression, and pharmacological characterization of the GPR120 free fatty acid receptor from cynomolgus monkey: Comparison with human GPR120 splice variants. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2009, 154, 419–426. [Google Scholar] [CrossRef]
- Lee, K.-P.; Park, S.-J.; Kang, S.; Koh, J.-M.; Sato, K.; Chung, H.-Y.; Okajima, F.; Im, D.-S. ω-3 polyunsaturated fatty acids accelerate airway repair by activating FFA4 in club cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L835–L844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, C.; Hong, Y.-H.; Iga, T.; Hishikawa, D.; Suzuki, Y.; Song, S.-H.; Choi, K.-C.; Adachi, T.; Hirasawa, A.; Tsujimoto, G. The regulation of adipogenesis through GPR120. Biochem. Biophys. Res. Commun. 2007, 354, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Huang, Q.; Jie, Q.; Lu, W.-G.; Wang, L.; Li, X.-J.; Sun, Z.; Hu, Y.-Q.; Chen, L.; Liu, B.-H. GPR120: A bi-potential mediator to modulate the osteogenic and adipogenic differentiation of BMMSCs. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houthuijzen, J.M.; Oosterom, I.; Hudson, B.D.; Hirasawa, A.; Daenen, L.G.; McLean, C.M.; Hansen, S.V.; van Jaarsveld, M.T.; Peeper, D.S.; Sadatmand, S.J. Fatty acid 16: 4 (n-3) stimulates a GPR120-induced signaling cascade in splenic macrophages to promote chemotherapy resistance. FASEB J. 2017, 31, 2195–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, B.D.; Shimpukade, B.; Mackenzie, A.E.; Butcher, A.J.; Pediani, J.D.; Christiansen, E.; Heathcote, H.; Tobin, A.B.; Ulven, T.; Milligan, G. The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol. Pharmacol. 2013, 84, 710–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, D.Y.; Walenta, E.; Akiyama, T.E.; Lagakos, W.S.; Lackey, D.; Pessentheiner, A.R.; Sasik, R.; Hah, N.; Chi, T.J.; Cox, J.M.; et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat. Med. 2014, 20, 942–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briscoe, C.P.; Peat, A.J.; McKeown, S.C.; Corbett, D.F.; Goetz, A.S.; Littleton, T.R.; McCoy, D.C.; Kenakin, T.P.; Andrews, J.L.; Ammala, C. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: Identification of agonist and antagonist small molecules. Br. J. Pharmacol. 2006, 148, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimpukade, B.; Hudson, B.D.; Hovgaard, C.K.; Milligan, G.; Ulven, T. Discovery of a potent and selective GPR120 agonist. J. Med. Chem. 2012, 55, 4511–4515. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.D.; Shimpukade, B.; Milligan, G.; Ulven, T. The molecular basis of ligand interaction at free fatty acid receptor 4 (FFA4/GPR120). J. Biol. Chem. 2014, 289, 20345–20358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeger, A.L.; Cipollina, C.; Cole, M.P.; Woodcock, S.R.; Bonacci, G.; Rudolph, T.K.; Rudolph, V.; Freeman, B.A.; Schopfer, F.J. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat. Chem. Biol. 2010, 6, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ruan, X.Z.; Powis, S.H.; Fernando, R.; Mon, W.Y.; Wheeler, D.C.; Moorhead, J.F.; Varghese, Z. EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: Evidence for a PPAR-γ–dependent mechanism. Kidney Int. 2005, 67, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelliah, M.; Chu, H.D.; Cox, J.M.; Debenham, J.S.; Eagen, K.; Lan, P.; London, C.; Plotkin, M.A.; Shah, U.; Sinz, C.J. Substituted Spiropiperidinyl Compounds Useful as GPR120 Agonists. U.S. Patent 9,708,270 B2, 18 July 2017. [Google Scholar]
- Azevedo, C.M.; Watterson, K.R.; Wargent, E.T.; Hansen, S.V.; Hudson, B.D.; Kępczyńska, M.A.; Dunlop, J.; Shimpukade, B.; Christiansen, E.; Milligan, G. Non-acidic free fatty acid receptor 4 agonists with antidiabetic activity. J. Med. Chem. 2016, 59, 8868–8878. [Google Scholar] [CrossRef] [PubMed]
- Sparks, S.M.; Chen, G.; Collins, J.L.; Danger, D.; Dock, S.T.; Jayawickreme, C.; Jenkinson, S.; Laudeman, C.; Leesnitzer, M.A.; Liang, X. Identification of diarylsulfonamides as agonists of the free fatty acid receptor 4 (FFA4/GPR120). Bioorganic Med. Chem. Lett. 2014, 24, 3100–3103. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Olefsky, J.M.; Osborn, O. Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol. Sci. 2011, 32, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, A.; Hirasawa, A.; Poulain-Godefroy, O.; Bonnefond, A.; Hara, T.; Yengo, L.; Kimura, I.; Leloire, A.; Liu, N.; Iida, K. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 2012, 483, 350–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halder, S.; Kumar, S.; Sharma, R. The therapeutic potential of GPR120: A patent review. Expert Opin. Ther. Pat. 2013, 23, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Macielag, M.J. GPR120 agonists for the treatment of diabetes: A patent review (2014 present). Expert Opin. Ther. Pat. 2020, 30, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cai, C.; Winters, M.; Wells, M.; Wall, M.; Lanter, J.; Sui, Z.; Ma, J.; Novack, A.; Nashashibi, I. Design, synthesis and SAR of a novel series of heterocyclic phenylpropanoic acids as GPR120 agonists. Bioorganic Med. Chem. Lett. 2017, 27, 3272–3278. [Google Scholar] [CrossRef] [PubMed]
- Winters, M.P.; Sui, Z.; Wall, M.; Wang, Y.; Gunnet, J.; Leonard, J.; Hua, H.; Yan, W.; Suckow, A.; Bell, A. Discovery of N-arylpyrroles as agonists of GPR120 for the treatment of type II diabetes. Bioorganic Med. Chem. Lett. 2018, 28, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Igari, S.-I.; Hirasawa, A.; Hata, M.; Ishiguro, M.; Fujieda, H.; Itoh, Y.; Hirano, T.; Nakagawa, H.; Ogura, M. Identification of G protein-coupled receptor 120-selective agonists derived from PPARγ agonists. J. Med. Chem. 2008, 51, 7640–7644. [Google Scholar] [CrossRef]
- Lombardo, M.; Bender, K.; London, C.; Plotkin, M.A.; Kirkland, M.; Mane, J.; Pachanski, M.; Geissler, W.; Cummings, J.; Habulihaz, B. Discovery of benzofuran propanoic acid GPR120 agonists: From uHTS hit to mechanism-based pharmacodynamic effects. Bioorganic Med. Chem. Lett. 2016, 26, 5724–5728. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.L.; Velazquez, F.; Jayne, C.; Shah, U.; Miao, S.; Ashley, E.R.; Madeira, M.; Akiyama, T.E.; Di Salvo, J.; Suzuki, T. Discovery of chromane propionic acid analogues as selective agonists of GPR120 with in vivo activity in rodents. ACS Med. Chem. Lett. 2017, 8, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikhonova, I.G. Application of gpcr structures for modelling of free fatty acid receptors. In Free Fatty Acid Receptors; Springer: Berlin/Heidelberg, Germany, 2016; pp. 57–77. [Google Scholar]
- Al Mahmud, Z.; Jenkins, L.; Ulven, T.; Labéguère, F.; Gosmini, R.; De Vos, S.; Hudson, B.D.; Tikhonova, I.G.; Milligan, G. Three classes of ligands each bind to distinct sites on the orphan G protein-coupled receptor GPR84. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, S.; Yamamoto, A.; Okada, T.; Matsumura, E.; Nose, E.; Kogure, K.; Kojima, S.; Haga, T. Identification of surrogate ligands for orphan G protein-coupled receptors. Life Sci. 2003, 74, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Du Toit, E.; Browne, L.; Irving-Rodgers, H.; Massa, H.M.; Fozzard, N.; Jennings, M.P.; Peak, I.R. Effect of GPR84 deletion on obesity and diabetes development in mice fed long chain or medium chain fatty acid rich diets. Eur. J. Nutr. 2018, 57, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Gaidarov, I.; Anthony, T.; Gatlin, J.; Chen, X.; Mills, D.; Solomon, M.; Han, S.; Semple, G.; Unett, D.J. Embelin and its derivatives unravel the signaling, proinflammatory and antiatherogenic properties of GPR84 receptor. Pharmacol. Res. 2018, 131, 185–198. [Google Scholar] [CrossRef]
- Recio, C.; Lucy, D.; Purvis, G.S.; Iveson, P.; Zeboudj, L.; Iqbal, A.J.; Lin, D.; O’Callaghan, C.; Davison, L.; Griesbach, E. Activation of the immune-metabolic receptor GPR84 enhances inflammation and phagocytosis in macrophages. Front. Immunol. 2018, 9, 1419. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Takaishi, S.; Nagasaki, M.; Onozawa, Y.; Iino, I.; Maeda, H.; Komai, T.; Oda, T. Medium-chain fatty acid-sensing receptor, GPR84, is a proinflammatory receptor. J. Biol. Chem. 2013, 288, 10684–10691. [Google Scholar] [CrossRef] [Green Version]
- Köse, M.; Pillaiyar, T.; Namasivayam, V.; De Filippo, E.; Sylvester, K.; Ulven, T.; von Kügelgen, I.; Müller, C.E. An agonist radioligand for the proinflammatory lipid-activated G protein-coupled receptor GPR84 providing structural insights. J. Med. Chem. 2019, 63, 2391–2410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, H.; Li, J.; Xie, X. Discovery and Characterization of a Novel Small-Molecule Agonist for Medium-Chain Free Fatty Acid Receptor G Protein–Coupled Receptor 84. J. Pharmacol. Exp. Ther. 2016, 357, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, G.A.; Arneson, D.W.; Putnam, W.C.; Smith, H.J.; Gray, J.C.; Sullivan, D.K.; Mayo, M.S.; Crowell, J.A.; Hurwitz, A. Single-dose and multiple-dose administration of indole-3-carbinol to women: Pharmacokinetics based on 3, 3′-diindolylmethane. Cancer Epidemiol. Prev. Biomark. 2006, 15, 2477–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labéguère, F.G.; Newsome, G.J.R.; Alvey, L.J.; Sanière, L.R.M.; Fletcher, S.R. Novel Dihydropyrimidinoisoquinolinones and Pharmaceutical Compositions Thereof for the Treatment of Inflammatory Disorders. U.S. Patent 2019/0002458 A1, 3 January 2019. [Google Scholar]
- Nagasaki, H.; Kondo, T.; Fuchigami, M.; Hashimoto, H.; Sugimura, Y.; Ozaki, N.; Arima, H.; Ota, A.; Oiso, Y.; Hamada, Y. Inflammatory changes in adipose tissue enhance expression of GPR84, a medium-chain fatty acid receptor: TNFα enhances GPR84 expression in adipocytes. FEBS Lett. 2012, 586, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Saniere, L.; Marsais, F.; Jagerschmidt, C.; Meurisse, S.; Cuzic, S.; Shoji, K.; Clement-Lacroix, P.; Van Osselaer, N.; De Vos, S. Characterization of GLPG1205 in Mouse Fibrosis Models: A Potent and Selective Antagonist of GPR84 for Treatment of Idiopathic Pulmonary Fibrosis. In A19. Less Idiopathic: Structural and Functional Abnormalities in IPF; American Thoracic Society: New York, NY, USA, 2019; p. A1046. [Google Scholar]
- Montgomery, M.K.; Osborne, B.; Brandon, A.E.; O’Reilly, L.; Fiveash, C.E.; Brown, S.H.; Wilkins, B.P.; Samsudeen, A.; Yu, J.; Devanapalli, B. Regulation of mitochondrial metabolism in murine skeletal muscle by the medium-chain fatty acid receptor Gpr84. FASEB J. 2019, 33, 12264–12276. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, C.; Pagé, J.; Bédard, A.; Tremblay, P.; Vallières, L. G protein-coupled receptor 84, a microglia-associated protein expressed in neuroinflammatory conditions. Glia 2007, 55, 790–800. [Google Scholar] [CrossRef]
- Puengel, T.; De Vos, S.; Hundertmark, J.; Kohlhepp, M.; Guldiken, N.; Pujuguet, P.; Auberval, M.; Marsais, F.; Shoji, K.F.; Saniere, L. The medium-chain fatty acid receptor GPR84 mediates myeloid cell infiltration promoting steatohepatitis and fibrosis. J. Clin. Med. 2020, 9, 1140. [Google Scholar] [CrossRef] [PubMed]
- Labéguère, F.; Dupont, S.; Alvey, L.; Soulas, F.; Newsome, G.; Tirera, A.; Quenehen, V.; Mai, T.T.T.; Deprez, P.; Blanqué, R. Discovery of 9-cyclopropylethynyl-2-((S)-1-[1, 4] dioxan-2-ylmethoxy)-6, 7-dihydropyrimido [6, 1-a] isoquinolin-4-one (GLPG1205), a unique GPR84 negative allosteric modulator undergoing evaluation in a phase II clinical trial. J. Med. Chem. 2020, 63, 13526–13545. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.; Manganas, H.; Ryerson, C.J.; Shapera, S.; Cantin, A.M.; Hernandez, P.; Turcotte, E.E.; Parker, J.M.; Moran, J.E.; Albert, G.R. PMaternal gut microbiota in pregnancy influences offspring metabolic phesis. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chung, S.; Li, Z.; Overstreet, J.M.; Gagnon, L.; Grouix, B.; Leduc, M.; Laurin, P.; Zhang, M.-Z.; Harris, R.C. Fatty acid receptor modulator PBI-4050 inhibits kidney fibrosis and improves glycemic control. JCI Insight 2018, 3, e120365. [Google Scholar] [CrossRef] [Green Version]
- Thibodeau, J.-F.; Simard, J.-C.; Holterman, C.E.; Blais, A.; Cloutier, M.-P.; Medeiros, T.; Leduc, M.; Grouix, B.; Leblond, F.A.; Burger, D. PBI-4050 via GPR40 activation improves adenine-induced kidney injury in mice. Clin. Sci. 2019, 133, 1587–1602. [Google Scholar] [CrossRef] [PubMed]
- Sum, C.S.; Tikhonova, I.G.; Neumann, S.; Engel, S.; Raaka, B.M.; Costanzi, S.; Gershengorn, M.C. Identification of residues important for agonist recognition and activation in GPR40. J. Biol. Chem. 2007, 282, 29248–29255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sum, C.S.; Tikhonova, I.G.; Costanzi, S.; Gershengorn, M.C. Two arginine-glutamate ionic locks near the extracellular surface of FFAR1 gate receptor activation. J. Biol. Chem. 2009, 284, 3529–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
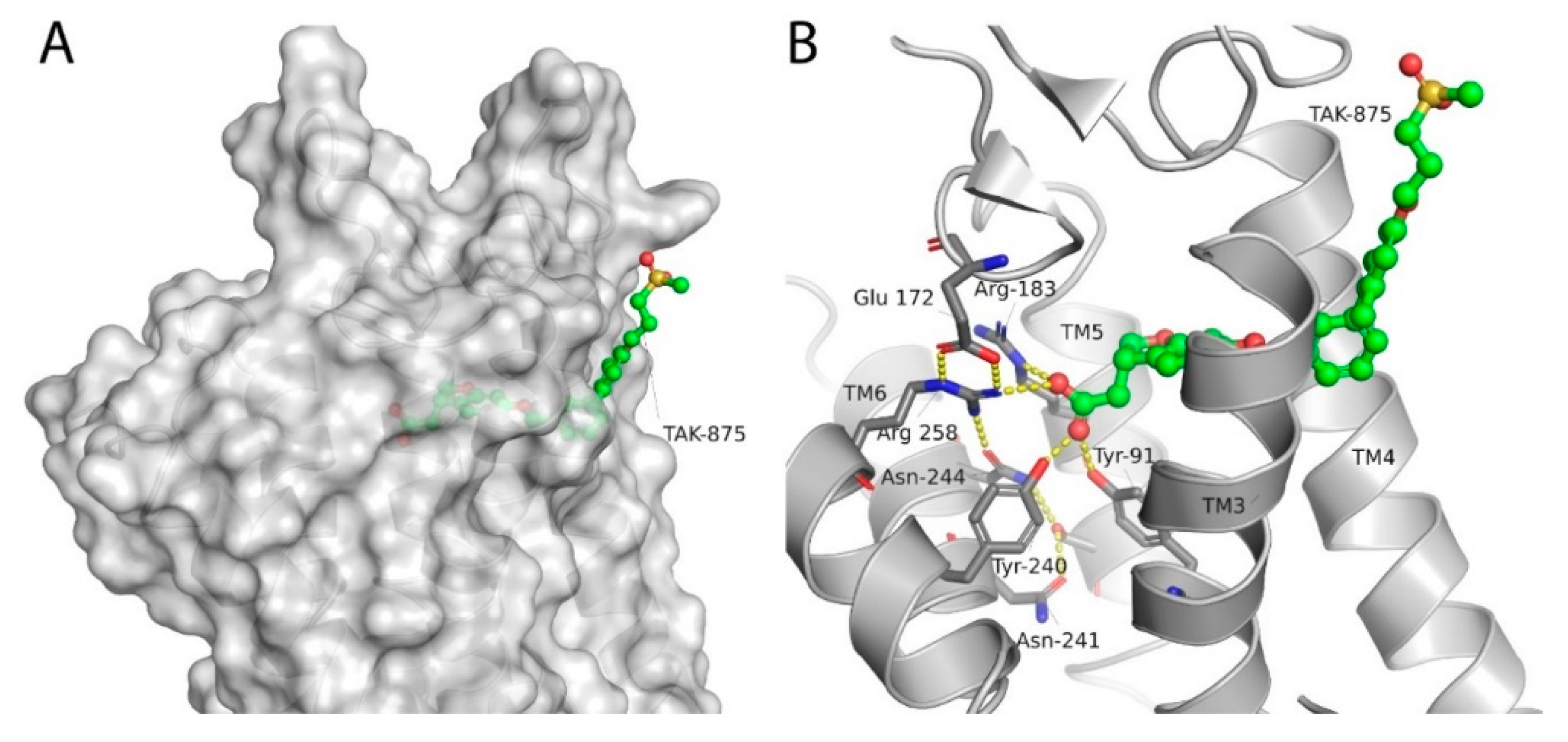
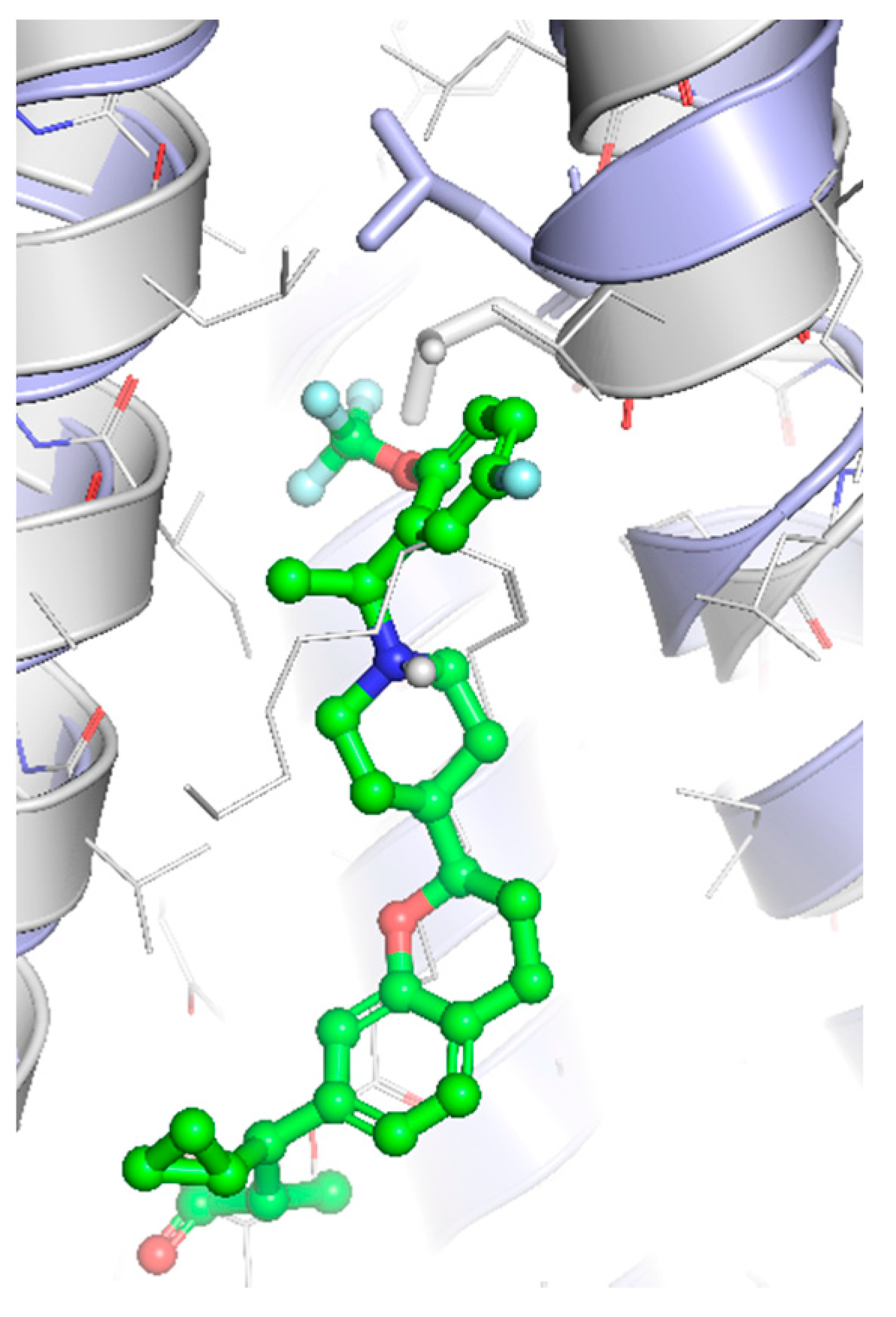
- Srivastava, A.; Yano, J.; Hirozane, Y.; Kefala, G.; Gruswitz, F.; Snell, G.; Lane, W.; Ivetac, A.; Aertgeerts, K.; Nguyen, J. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 2014, 513, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Elsevier: Amsterdam, The Netherlands, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenakin, T. Biased receptor signaling in drug discovery. Pharmacol. Rev. 2019, 71, 267–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobingier, B.T.; von Zastrow, M. When trafficking and signaling mix: How subcellular location shapes G protein-coupled receptor activation of heterotrimeric G proteins. Traffic 2019, 20, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebelman, M.P.; Crudden, C.; Pegtel, D.M.; Smit, M.J. The Convergence of Extracellular Vesicle and GPCR Biology. Trends Pharmacol. Sci. 2020, 41, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Quarles, L.D.; Sherrard, D.J.; Adler, S.; Rosansky, S.J.; McCary, L.C.; Liu, W.; Turner, S.A.; Bushinsky, D.A. The calcimimetic AMG 073 as a potential treatment for secondary hyperparathyroidism of end-stage renal disease. J. Am. Soc. Nephrol. 2003, 14, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springthorpe, B.; Bailey, A.; Barton, P.; Birkinshaw, T.N.; Bonnert, R.V.; Brown, R.C.; Chapman, D.; Dixon, J.; Guile, S.D.; Humphries, R.G. From ATP to AZD6140: The discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorganic Med. Chem. Lett. 2007, 17, 6013–6018. [Google Scholar] [CrossRef]
- Surin, A.; Pshenichkin, S.; Grajkowska, E.; Surina, E.; Wroblewski, J.T. Cyclothiazide selectively inhibits mGluR1 receptors interacting with a common allosteric site for non-competitive antagonists. Neuropharmacology 2007, 52, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J. Structure of the CCR5 chemokine receptor–HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Wold, E.A.; Chen, J.; Cunningham, K.A.; Zhou, J. Allosteric modulation of class A GPCRs: Targets, agents, and emerging concepts. J. Med. Chem. 2018, 62, 88–127. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Schubert, M.; Stichel, J.; Weaver, D.; Beck-Sickinger, A.G.; Meiler, J. Discovery of small-molecule modulators of the human Y4 receptor. PLoS ONE 2016, 11, e0157146. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Opportunities and challenges in the discovery of allosteric modulators of GPCRs. In Computational Methods for GPCR Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2018; pp. 297–319. [Google Scholar]
- Bennett, K.A.; Doré, A.S.; Christopher, J.A.; Weiss, D.R.; Marshall, F.H. Structures of mGluRs shed light on the challenges of drug development of allosteric modulators. Curr. Opin. Pharmacol. 2015, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.E.; Schiedel, A.C.; Baqi, Y. Allosteric modulators of rhodopsin-like G protein-coupled receptors: Opportunities in drug development. Pharmacol. Ther. 2012, 135, 292–315. [Google Scholar] [CrossRef] [PubMed]
- Pizzonero, M.; Dupont, S.; Babel, M.; Beaumont, S.; Bienvenu, N.; Blanque, R.; Cherel, L.; Christophe, T.; Crescenzi, B.; De Lemos, E. Discovery and optimization of an azetidine chemical series as a free fatty acid receptor 2 (FFA2) antagonist: From hit to clinic. J. Med. Chem. 2014, 57, 10044–10057. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, E.; Hansen, A.H.; Pandey, S.K.; MacKenzie, A.E.; Hudson, B.D.; Ulven, T.; Milligan, G. Non-equivalence of key positively charged residues of the free fatty acid 2 receptor in the recognition and function of agonist versus antagonist ligands. J. Biol. Chem. 2016, 291, 303–317. [Google Scholar] [CrossRef] [Green Version]
- Vermeire, S.; Kojecky, V.; Knoflícek, V.; Reinisch, W.; Van Kaem, T.; Namour, F.; Beetens, J.; Vanhoutte, F. GLPG0974, an FFA2 antagonist, in ulcerative colitis: Efficacy and safety in a multicenter proof-of-concept study. J. Crohn’s Colitis 2015, 9 (Suppl. 1), S39. [Google Scholar]






| Receptor | Major Coupling Proteins | Major Expression Sites | Main Function |
|---|---|---|---|
| FFAR1 | Gαq/11 Gαi/o Gαs Gα12/13 β-arrestin | Pancreas (β-cells) Intestine (L, K, I cells) Bone Central nervous system Immune cells (Monocytes) | Insulin secretion Gut hormone secretion Bone remodeling Pain perception Macrophage M2 differentiation |
| FFAR2 | Gαq/11 Gαi/o Gα12/13 β-arrestin | PMNs (Neutrophils, Eosinophils) Lymphocytes Monocytes Pancreas (β-cells) Intestine (L cells, IECs) White adipose tissue | Immune cell activation Treg expansion Cytokine secretion Insulin release Gut hormone secretion, immune-modulatory Reduction in lipolysis, lipid accumulation, and insulin resistance |
| FFAR3 | Gαi/o β-arrestin | Peripheral nervous system Pancreas (β-cell) Intestine (L, K cells) Immune tissue (DCs, thymus) | Increase in heart rate, energy expenditure, reduction of gut motility Inhibition of insulin secretion Gut hormone release Decrease Th2 response, increase Treg differentiation |
| FFAR4 | Gαq/11 Gαi/o β-arrestin | Adipose tissue Macrophages Lung Intestine (K, I cells) Bone | Differentiation, browning Anti-inflammatory Epithelial repair Gut hormone release Bone formation |
| GPR84 | Gαi/o β-arrestin | Immune cells (Neutrophils, Eosinophils, macrophages) Lung, Liver, Muscle, and Adipose tissues | Proinflammatory cell responses |
| Name | Structure | References |
|---|---|---|
| Partial allosteric agonists | ||
| TAK-875/fasiglifam |  | Takeda Pharmaceuticals [70,71,78] |
| AM 837 |  | Amgen [62,79] |
| MK8666 |  | Merck [68,80,81,82] |
| Full allosteric agonists | ||
| AM 1638 |  | Amgen [62,83] |
| AP8 |  | Merck [81] |
| Compound 1 |  | Eli Lilly [84] |
| Name | Structure | References |
|---|---|---|
| FFA2 allosteric agonists | ||
| AMG 7703/4-CMTB |  | Amgen [88,91,92,105,107] |
| Compound 58 |  | Amgen [89,141] |
| AZ1729 |  | AstraZeneca [145] |
| FFA3 allosteric agonist | ||
| AR420626 |  | Arena Pharmaceuticals [146,147] |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grundmann, M.; Bender, E.; Schamberger, J.; Eitner, F. Pharmacology of Free Fatty Acid Receptors and Their Allosteric Modulators. Int. J. Mol. Sci. 2021, 22, 1763. https://doi.org/10.3390/ijms22041763
Grundmann M, Bender E, Schamberger J, Eitner F. Pharmacology of Free Fatty Acid Receptors and Their Allosteric Modulators. International Journal of Molecular Sciences. 2021; 22(4):1763. https://doi.org/10.3390/ijms22041763
Chicago/Turabian StyleGrundmann, Manuel, Eckhard Bender, Jens Schamberger, and Frank Eitner. 2021. "Pharmacology of Free Fatty Acid Receptors and Their Allosteric Modulators" International Journal of Molecular Sciences 22, no. 4: 1763. https://doi.org/10.3390/ijms22041763
APA StyleGrundmann, M., Bender, E., Schamberger, J., & Eitner, F. (2021). Pharmacology of Free Fatty Acid Receptors and Their Allosteric Modulators. International Journal of Molecular Sciences, 22(4), 1763. https://doi.org/10.3390/ijms22041763