
The MHC Class-I Transactivator NLRC5: Implications to Cancer Immunology and Potential Applications to Cancer Immunotherapy


Abstract
:1. Introduction
2. Cancer Immunogenicity and Tumor Antigenic Peptides
3. Processing and Presentation of Cancer Antigenic Peptides by MHC-I
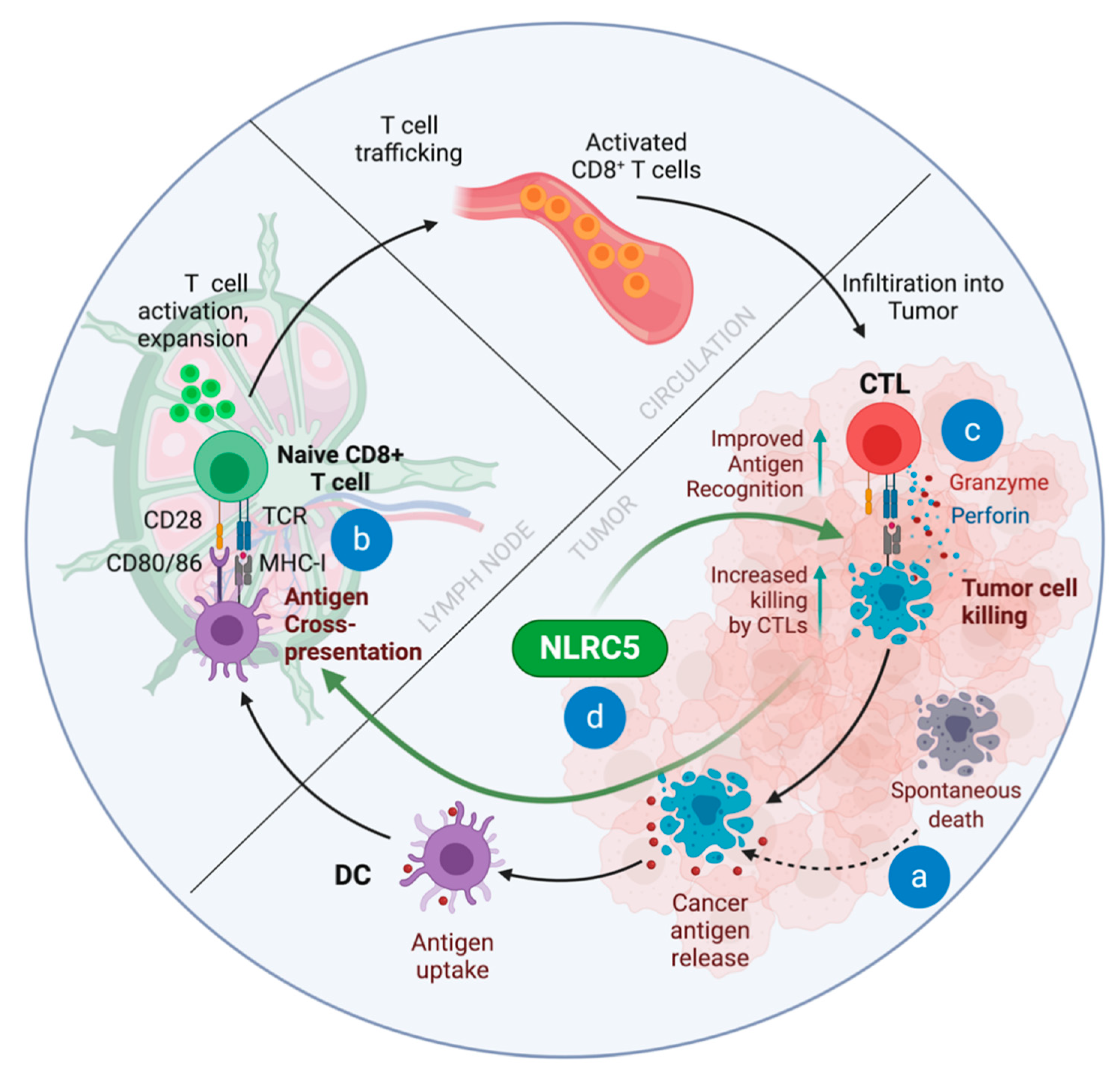
4. The Cancer-Immunity Cycle and ‘Immune Invisibility’ of Cancers
5. Defective MHC-I Expression in Cancers
6. Loss of NLRC5 Expression Frequently Underlies Reduced MHC-I Expression in Cancers
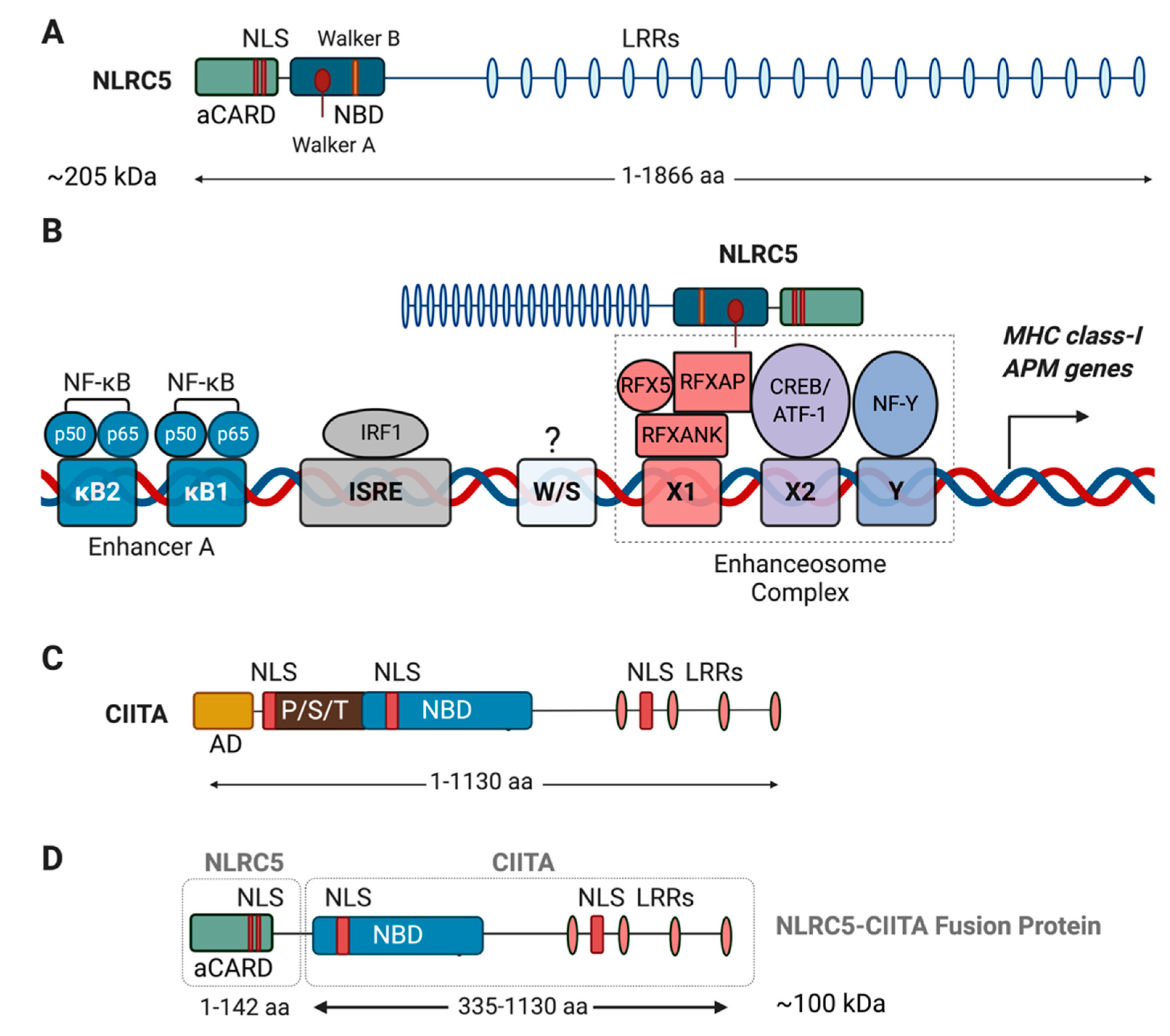
7. Structure and Transcriptional Coactivator Function of NLRC5
8. Role of NLRC5 in MHC-I Expression, CD8+ T Cell Development and Functions
9. Induction of Butyrophilins by NLRC5 and γδ T Cell Activation
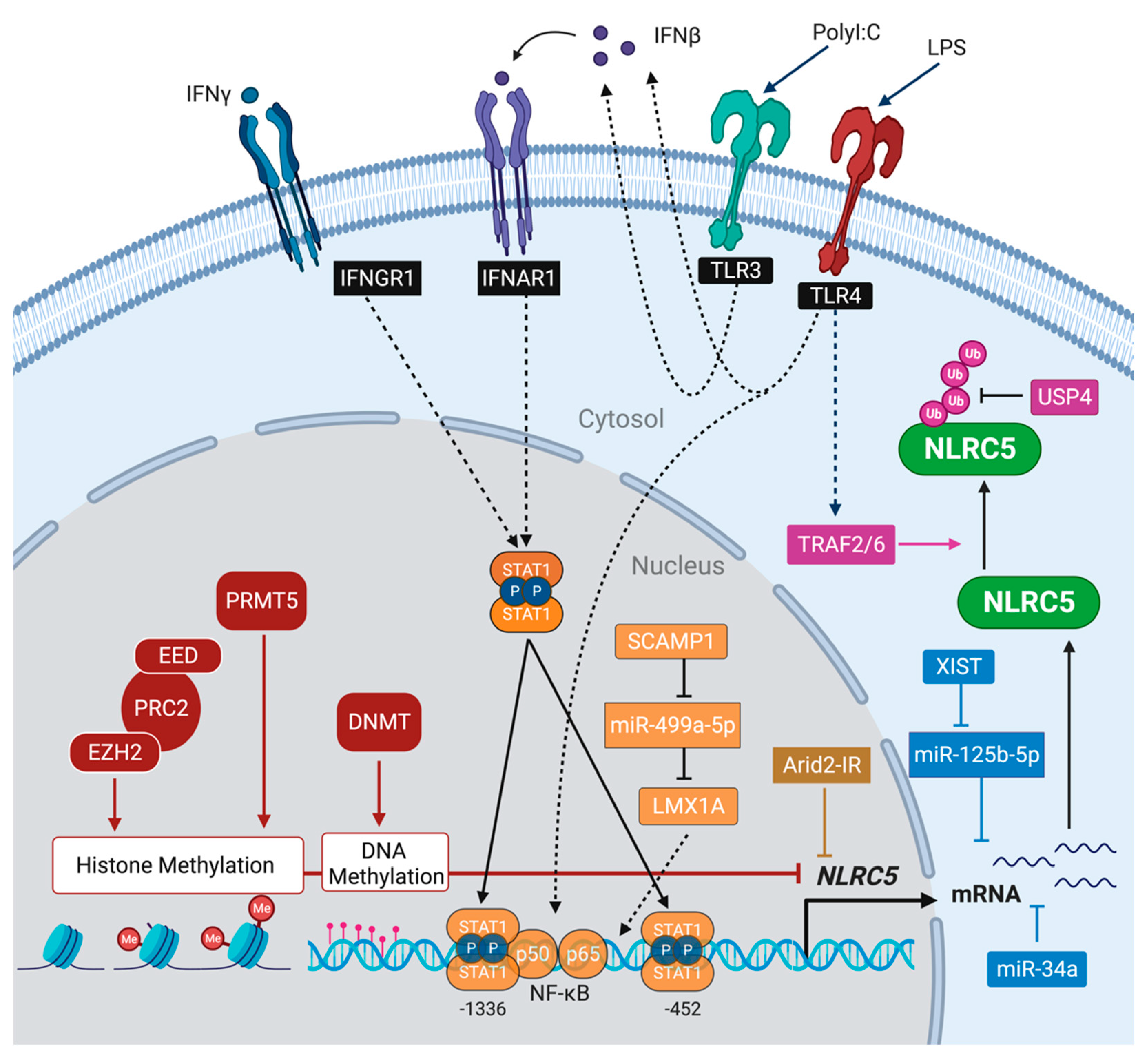
10. Regulation of NLRC5 Expression
11. Impact of NLRC5 on Antitumor Immunity
12. Tumor Promoting Potential of NLRC5
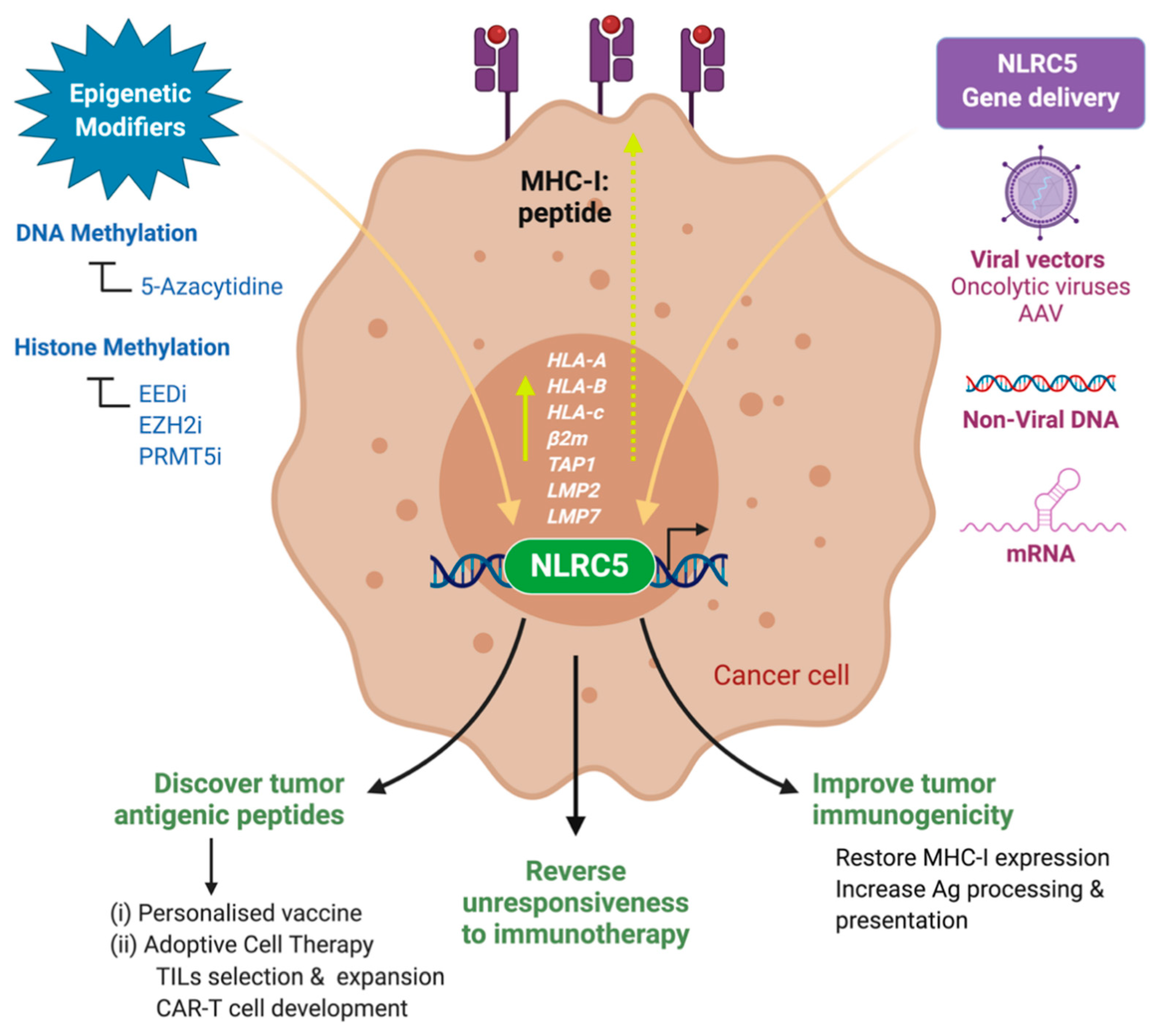
13. Restoring MHC-I Expression in Cancers
14. NLRC5-Independent MHC-I Expression
15. Role of NLRC5 in Cancer Immune Surveillance
16. Exploiting NLRC5 for Cancer Immunotherapy
16.1. Restoring Cancer Immunogenicity
16.2. Identification of Immunogenic Peptides
16.3. Biomarker to Predict Responsiveness to Immune Checkpoint Therapy
17. Conclusions
18. Outstanding Questions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couzin-Frankel, J. Cancer Immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [Green Version]
- Ledford, H.; Else, H.; Warren, M. Cancer immunologists scoop medicine Nobel prize. Nature 2018, 562, 20–21. [Google Scholar] [CrossRef]
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin. Orthop. Relat. Res. 1991, 3–11. [Google Scholar] [PubMed]
- Lenis, A.T.; Lec, P.M.; Chamie, K.; Mshs, M.D. Bladder Cancer: A Review. JAMA 2020, 324, 1980–1991. [Google Scholar] [CrossRef]
- Pettenati, C.; Ingersoll, M.A. Mechanisms of BCG immunotherapy and its outlook for bladder cancer. Nat. Rev. Urol. 2018, 15, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L. On immunosurveillance in human cancer. Yale J. Biol. Med. 1982, 55, 329–333. [Google Scholar] [PubMed]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Fishman, M.; Dutcher, J.P.; Clark, J.I.; Alva, A.; Miletello, G.P.; Curti, B.; Agarwal, N.; Hauke, R.; Mahoney, K.M.; Moon, H.; et al. Overall survival by clinical risk category for high dose interleukin-2 (HD IL-2) treated patients with metastatic renal cell cancer (mRCC): Data from the PROCLAIM(SM) registry. J. Immunother. Cancer 2019, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J. Adoptive cellular therapies: The current landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D. Does the immune system see tumors as foreign or self? Annu. Rev. Immunol. 2003, 21, 807–839. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Ochsenbein, A.F.; Sierro, S.; Odermatt, B.; Pericin, M.; Karrer, U.; Hermans, J.; Hemmi, S.; Hengartner, H.; Zinkernagel, R.M. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature 2001, 411, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D. T cells and tumours. Nature 2001, 411, 1010–1012. [Google Scholar] [CrossRef]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef]
- Fearon, D.T. Immune-Suppressing Cellular Elements of the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2017, 1, 241–255. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Mechanisms of Tumor Cell–Intrinsic Immune Evasion. Annu. Rev. Cancer Biol. 2018, 2, 213–228. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicklin, D.J.; Marincola, F.M.; Ferrone, S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol. Med. Today 1999, 5, 178–186. [Google Scholar] [CrossRef]
- Garrido, F.; Algarra, I.; Garcia-Lora, A.M. The escape of cancer from T lymphocytes: Immunoselection of MHC class I loss variants harboring structural-irreversible “hard” lesions. Cancer Immunol. Immunother. CII 2010, 59, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Lampen, M.H.; van Hall, T. Strategies to counteract MHC-I defects in tumors. Curr. Opin. Immunol. 2011, 23, 293–298. [Google Scholar] [CrossRef]
- Meissner, T.B.; Liu, Y.J.; Lee, K.H.; Li, A.; Biswas, A.; van Eggermond, M.C.; van den Elsen, P.J.; Kobayashi, K.S. NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J. Immunol. 2012, 188, 4951–4958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.S.; van den Elsen, P.J. NLRC5: A key regulator of MHC class I-dependent immune responses. Nat. Rev. Immunol. 2012, 12, 813–820. [Google Scholar] [CrossRef]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, G.M.; Bobbala, D.; Serrano, D.; Mayhue, M.; Champagne, A.; Saucier, C.; Steimle, V.; Kufer, T.A.; Menendez, A.; Ramanathan, S.; et al. NLRC5 elicits antitumor immunity by enhancing processing and presentation of tumor antigens to CD8(+) T lymphocytes. Oncoimmunology 2016, 5, e1151593. [Google Scholar] [CrossRef] [Green Version]
- Gross, L. Intradermal Immunization of C3H Mice against a Sarcoma That Originated in an Animal of the Same Line. Cancer Res. 1943, 3, 326–333. [Google Scholar]
- Delorme, E.J.; Alexander, P. Treatment of Primary Fibrosarcoma in the Rat with Immune Lymphocytes. Lancet 1964, 2, 117–120. [Google Scholar] [CrossRef]
- Cheever, M.A.; Greenberg, P.D.; Fefer, A. Tumor neutralization, immunotherapy, and chemoimmunotherapy of a Friend leukemia with cells secondarily sensitized in vitro: II. Comparison of cells cultured with and without tumor to noncultured immune cells. J. Immunol. 1978, 121, 2220–2227. [Google Scholar]
- Eberlein, T.J.; Rosenstein, M.; Rosenberg, S.A. Regression of a disseminated syngeneic solid tumor by systemic transfer of lymphoid cells expanded in interleukin 2. J. Exp. Med. 1982, 156, 385–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gansbacher, B.; Bannerji, R.; Daniels, B.; Zier, K.; Cronin, K.; Gilboa, E. Retroviral vector-mediated gamma-interferon gene transfer into tumor cells generates potent and long lasting antitumor immunity. Cancer Res. 1990, 50, 7820–7825. [Google Scholar] [PubMed]
- Townsend, S.E.; Allison, J.P. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science 1993, 259, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [Green Version]
- Hock, H.; Dorsch, M.; Kunzendorf, U.; Qin, Z.; Diamantstein, T.; Blankenstein, T. Mechanisms of rejection induced by tumor cell-targeted gene transfer of interleukin 2, interleukin 4, interleukin 7, tumor necrosis factor, or interferon gamma. Proc. Natl. Acad. Sci. USA 1993, 90, 2774–2778. [Google Scholar] [CrossRef] [Green Version]
- Knuth, A.; Danowski, B.; Oettgen, H.F.; Old, L.J. T-cell-mediated cytotoxicity against autologous malignant melanoma: Analysis with interleukin 2-dependent T-cell cultures. Proc. Natl. Acad. Sci. USA 1984, 81, 3511–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degiovanni, G.; Lahaye, T.; Herin, M.; Hainaut, P.; Boon, T. Antigenic heterogeneity of a human melanoma tumor detected by autologous CTL clones. Eur. J. Immunol. 1988, 18, 671–676. [Google Scholar] [CrossRef]
- Topalian, S.L.; Kasid, A.; Rosenberg, S.A. Immunoselection of a human melanoma resistant to specific lysis by autologous tumor-infiltrating lymphocytes. Possible mechanisms for immunotherapeutic failures. J. Immunol. 1990, 144, 4487–4495. [Google Scholar]
- Tanaka, Y.; Tevethia, M.J.; Kalderon, D.; Smith, A.E.; Tevethia, S.S. Clustering of antigenic sites recognized by cytotoxic T lymphocyte clones in the amino terminal half of SV40 T antigen. Virology 1988, 162, 427–436. [Google Scholar] [CrossRef]
- Klarnet, J.P.; Kern, D.E.; Okuno, K.; Holt, C.; Lilly, F.; Greenberg, P.D. FBL-reactive CD8+ cytotoxic and CD4+ helper T lymphocytes recognize distinct Friend murine leukemia virus-encoded antigens. J. Exp. Med. 1989, 169, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Kast, W.M.; Melief, C.J. Fine peptide specificity of cytotoxic T lymphocytes directed against adenovirus-induced tumours and peptide-MHC binding. Int. J. Cancer Suppl. 1991, 6, 90–94. [Google Scholar] [CrossRef]
- Darrow, T.L.; Slingluff, C.L., Jr.; Seigler, H.F. The role of HLA class I antigens in recognition of melanoma cells by tumor-specific cytotoxic T lymphocytes. Evidence for shared tumor antigens. J. Immunol. 1989, 142, 3329–3335. [Google Scholar] [PubMed]
- Kawakami, Y.; Zakut, R.; Topalian, S.L.; Stotter, H.; Rosenberg, S.A. Shared human melanoma antigens. Recognition by tumor-infiltrating lymphocytes in HLA-A2.1-transfected melanomas. J. Immunol. 1992, 148, 638–643. [Google Scholar] [PubMed]
- Boon, T.; Cerottini, J.C.; Van den Eynde, B.; van der Bruggen, P.; Van Pel, A. Tumor antigens recognized by T lymphocytes. Annu. Rev. Immunol. 1994, 12, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef]
- Van den Eynde, B.; Lethe, B.; Van Pel, A.; De Plaen, E.; Boon, T. The gene coding for a major tumor rejection antigen of tumor P815 is identical to the normal gene of syngeneic DBA/2 mice. J. Exp. Med. 1991, 173, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Traversari, C.; van der Bruggen, P.; Van den Eynde, B.; Hainaut, P.; Lemoine, C.; Ohta, N.; Old, L.; Boon, T. Transfection and expression of a gene coding for a human melanoma antigen recognized by autologous cytolytic T lymphocytes. Immunogenetics 1992, 35, 145–152. [Google Scholar] [CrossRef]
- Jerome, K.R.; Domenech, N.; Finn, O.J. Tumor-specific cytotoxic T cell clones from patients with breast and pancreatic adenocarcinoma recognize EBV-immortalized B cells transfected with polymorphic epithelial mucin complementary DNA. J. Immunol. 1993, 151, 1654–1662. [Google Scholar]
- Van Bleek, G.M.; Nathenson, S.G. Isolation of an endogenously processed immunodominant viral peptide from the class I H-2Kb molecule. Nature 1990, 348, 213–216. [Google Scholar] [CrossRef]
- Falk, K.; Rotzschke, O.; Rammensee, H.G. Cellular peptide composition governed by major histocompatibility complex class I molecules. Nature 1990, 348, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Wallny, H.J.; Deres, K.; Faath, S.; Jung, G.; Van Pel, A.; Boon, T.; Rammensee, H.G. Identification and quantification of a naturally presented peptide as recognized by cytotoxic T lymphocytes specific for an immunogenic tumor variant. Int. Immunol. 1992, 4, 1085–1090. [Google Scholar] [CrossRef]
- Slingluff, C.L., Jr.; Cox, A.L.; Henderson, R.A.; Hunt, D.F.; Engelhard, V.H. Recognition of human melanoma cells by HLA-A2.1-restricted cytotoxic T lymphocytes is mediated by at least six shared peptide epitopes. J. Immunol. 1993, 150, 2955–2963. [Google Scholar]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Chan, T.A. Immunogenic peptide discovery in cancer genomes. Curr. Opin. Genet. Dev. 2015, 30, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W. Plumbing the sources of endogenous MHC class I peptide ligands. Curr. Opin. Immunol. 2007, 19, 79–86. [Google Scholar] [CrossRef]
- Granados, D.P.; Laumont, C.M.; Thibault, P.; Perreault, C. The nature of self for T cells-a systems-level perspective. Curr. Opin. Immunol. 2015, 34, 1–8. [Google Scholar] [CrossRef]
- Laumont, C.M.; Daouda, T.; Laverdure, J.P.; Bonneil, E.; Caron-Lizotte, O.; Hardy, M.P.; Granados, D.P.; Durette, C.; Lemieux, S.; Thibault, P.; et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun. 2016, 7, 10238. [Google Scholar] [CrossRef]
- Shao, W.; Pedrioli, P.G.A.; Wolski, W.; Scurtescu, C.; Schmid, E.; Vizcaino, J.A.; Courcelles, M.; Schuster, H.; Kowalewski, D.; Marino, F.; et al. The SysteMHC Atlas project. Nucleic Acids Res. 2018, 46, D1237–D1247. [Google Scholar] [CrossRef]
- Zhao, Q.; Laverdure, J.P.; Lanoix, J.; Durette, C.; Cote, C.; Bonneil, E.; Laumont, C.M.; Gendron, P.; Vincent, K.; Courcelles, M.; et al. Proteogenomics Uncovers a Vast Repertoire of Shared Tumor-Specific Antigens in Ovarian Cancer. Cancer Immunol. Res. 2020, 8, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, F.; Textoris-Taube, K.; Keller, C.; Golnik, R.; Vigneron, N.; Van den Eynde, B.J.; Schuler-Thurner, B.; Schadendorf, D.; Lorenz, F.K.; Uckert, W.; et al. Proteasomes generate spliced epitopes by two different mechanisms and as efficiently as non-spliced epitopes. Sci. Rep. 2016, 6, 24032. [Google Scholar] [CrossRef] [PubMed]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.; Heck, A.J.; Mishto, M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016, 354, 354–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anton, L.C.; Yewdell, J.W. Translating DRiPs: MHC class I immunosurveillance of pathogens and tumors. J. Leukoc. Biol. 2014, 95, 551–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platteel, A.C.M.; Liepe, J.; van Eden, W.; Mishto, M.; Sijts, A. An Unexpected Major Role for Proteasome-Catalyzed Peptide Splicing in Generation of T Cell Epitopes: Is There Relevance for Vaccine Development? Front. Immunol. 2017, 8, 1441. [Google Scholar] [CrossRef] [Green Version]
- Rolfs, Z.; Solntsev, S.K.; Shortreed, M.R.; Frey, B.L.; Smith, L.M. Global Identification of Post-Translationally Spliced Peptides with Neo-Fusion. J. Proteome Res. 2019, 18, 349–358. [Google Scholar] [CrossRef]
- Germain, R.N. MHC-dependent antigen processing and peptide presentation: Providing ligands for T lymphocyte activation. Cell 1994, 76, 287–299. [Google Scholar] [CrossRef]
- Vyas, J.M.; Van der Veen, A.G.; Ploegh, H.L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 2008, 8, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Linsley, P.S.; Ledbetter, J.A. The role of the CD28 receptor during T cell responses to antigen. Annu. Rev. Immunol. 1993, 11, 191–212. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Walunas, T.L.; Bluestone, J.A. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 1996, 14, 233–258. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.K.; Meidenbauer, N.; Dworacki, G.; Kanaya, H.; Whiteside, T.L. Generation of tumor-specific T-lymphocytes by cross-priming with human dendritic cells ingesting apoptotic tumor cells. Cancer Res. 2000, 60, 3542–3549. [Google Scholar]
- Den Haan, J.M.; Bevan, M.J. Antigen presentation to CD8+ T cells: Cross-priming in infectious diseases. Curr. Opin. Immunol. 2001, 13, 437–441. [Google Scholar] [CrossRef]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2001, 2, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yewdell, J.W.; Reits, E.; Neefjes, J. Making sense of mass destruction: Quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 2003, 3, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Eggensperger, S.; Tampe, R. The transporter associated with antigen processing: A key player in adaptive immunity. Biol. Chem. 2015, 396, 1059–1072. [Google Scholar] [CrossRef]
- Hulpke, S.; Tampe, R. The MHC I loading complex: A multitasking machinery in adaptive immunity. Trends Biochem. Sci. 2013, 38, 412–420. [Google Scholar] [CrossRef]
- Kloetzel, P.M. The proteasome and MHC class I antigen processing. Biochim. Biophys. Acta 2004, 1695, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Abarca-Heidemann, K.; Friederichs, S.; Klamp, T.; Boehm, U.; Guethlein, L.A.; Ortmann, B. Regulation of the expression of mouse TAP-associated glycoprotein (tapasin) by cytokines. Immunol. Lett. 2002, 83, 197–207. [Google Scholar] [CrossRef]
- Seliger, B.; Ruiz-Cabello, F.; Garrido, F. IFN inducibility of major histocompatibility antigens in tumors. Adv. Cancer Res. 2008, 101, 249–276. [Google Scholar] [CrossRef]
- Abdel-Wahab, Z.; Dar, M.M.; Hester, D.; Vervaert, C.; Gangavalli, R.; Barber, J.; Darrow, T.L.; Seigler, H.F. Effect of irradiation on cytokine production, MHC antigen expression, and vaccine potential of interleukin-2 and interferon-gamma gene-modified melanoma cells. Cell. Immunol. 1996, 171, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Wollscheid, U.; Momburg, F.; Blankenstein, T.; Huber, C. Characterization of the major histocompatibility complex class I deficiencies in B16 melanoma cells. Cancer Res. 2001, 61, 1095–1099. [Google Scholar] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Clouthier, D.L.; Ohashi, P.S. Costimulation, a surprising connection for immunotherapy. Science 2017, 355, 1373–1374. [Google Scholar] [CrossRef]
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC class I antigens, immune surveillance, and tumor immune escape. J. Cell. Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef]
- Bukur, J.; Jasinski, S.; Seliger, B. The role of classical and non-classical HLA class I antigens in human tumors. Semin. Cancer Biol. 2012, 22, 350–358. [Google Scholar] [CrossRef]
- Parham, P.; Lomen, C.E.; Lawlor, D.A.; Ways, J.P.; Holmes, N.; Coppin, H.L.; Salter, R.D.; Wan, A.M.; Ennis, P.D. Nature of polymorphism in HLA-A, -B, and -C molecules. Proc. Natl. Acad. Sci. USA 1988, 85, 4005–4009. [Google Scholar] [CrossRef] [Green Version]
- Bjorkman, P.J.; Parham, P. Structure, function, and diversity of class I major histocompatibility complex molecules. Annu. Rev. Biochem. 1990, 59, 253–288. [Google Scholar] [CrossRef]
- Shawar, S.M.; Vyas, J.M.; Rodgers, J.R.; Rich, R.R. Antigen presentation by major histocompatibility complex class I-B molecules. Annu. Rev. Immunol. 1994, 12, 839–880. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, M.P.; Adams, E.; Altman, J.D.; Birnbaum, M.E.; Boggiano, C.; Casorati, G.; Chien, Y.H.; Conley, A.; Eckle, S.B.G.; Fruh, K.; et al. Casting a wider net: Immunosurveillance by nonclassical MHC molecules. PLoS Pathog. 2019, 15, e1007567. [Google Scholar] [CrossRef] [Green Version]
- Koopman, L.A.; Corver, W.E.; van der Slik, A.R.; Giphart, M.J.; Fleuren, G.J. Multiple genetic alterations cause frequent and heterogeneous human histocompatibility leukocyte antigen class I loss in cervical cancer. J. Exp. Med. 2000, 191, 961–976. [Google Scholar] [CrossRef]
- Paschen, A.; Mendez, R.M.; Jimenez, P.; Sucker, A.; Ruiz-Cabello, F.; Song, M.; Garrido, F.; Schadendorf, D. Complete loss of HLA class I antigen expression on melanoma cells: A result of successive mutational events. Int. J. Cancer. 2003, 103, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Maeurer, M.J.; Ferrone, S. TAP off—Tumors on. Immunol. Today 1997, 18, 292–299. [Google Scholar] [CrossRef]
- Benitez, R.; Godelaine, D.; Lopez-Nevot, M.A.; Brasseur, F.; Jimenez, P.; Marchand, M.; Oliva, M.R.; van Baren, N.; Cabrera, T.; Andry, G.; et al. Mutations of the beta2-microglobulin gene result in a lack of HLA class I molecules on melanoma cells of two patients immunized with MAGE peptides. Tissue Antigens 1998, 52, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.L.; Gabrilovich, D.; Tampe, R.; Girgis, K.R.; Nadaf, S.; Carbone, D.P. A functionally defective allele of TAP1 results in loss of MHC class I antigen presentation in a human lung cancer. Nat. Genet. 1996, 13, 210–213. [Google Scholar] [CrossRef]
- Seliger, B.; Ritz, U.; Abele, R.; Bock, M.; Tampe, R.; Sutter, G.; Drexler, I.; Huber, C.; Ferrone, S. Immune escape of melanoma: First evidence of structural alterations in two distinct components of the MHC class I antigen processing pathway. Cancer Res. 2001, 61, 8647–8650. [Google Scholar]
- Sokol, L.; Koelzer, V.H.; Rau, T.T.; Karamitopoulou, E.; Zlobec, I.; Lugli, A. Loss of tapasin correlates with diminished CD8(+) T-cell immunity and prognosis in colorectal cancer. J. Transl. Med. 2015, 13, 279. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, T.; Mendez, R.; Del Campo, A.; Jimenez, P.; Aptsiauri, N.; Garrido, F.; Ruiz-Cabello, F. Distinct mechanisms of loss of IFN-gamma mediated HLA class I inducibility in two melanoma cell lines. BMC Cancer 2007, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Respa, A.; Bukur, J.; Ferrone, S.; Pawelec, G.; Zhao, Y.; Wang, E.; Marincola, F.M.; Seliger, B. Association of IFN-gamma signal transduction defects with impaired HLA class I antigen processing in melanoma cell lines. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2668–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campoli, M.; Chang, C.C.; Ferrone, S. HLA class I antigen loss, tumor immune escape and immune selection. Vaccine 2002, 20 (Suppl. 4), A40–A45. [Google Scholar] [CrossRef]
- Cabrera, T.; Lara, E.; Romero, J.M.; Maleno, I.; Real, L.M.; Ruiz-Cabello, F.; Valero, P.; Camacho, F.M.; Garrido, F. HLA class I expression in metastatic melanoma correlates with tumor development during autologous vaccination. Cancer Immunol. Immunother. CII 2007, 56, 709–717. [Google Scholar] [CrossRef]
- Carretero, R.; Romero, J.M.; Ruiz-Cabello, F.; Maleno, I.; Rodriguez, F.; Camacho, F.M.; Real, L.M.; Garrido, F.; Cabrera, T. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics 2008, 60, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B. Molecular mechanisms of MHC class I abnormalities and APM components in human tumors. Cancer Immunol. Immunother. CII 2008, 57, 1719–1726. [Google Scholar] [CrossRef]
- Garrido, F.; Cabrera, T.; Aptsiauri, N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer. 2010, 127, 249–256. [Google Scholar] [CrossRef]
- Nie, Y.; Yang, G.; Song, Y.; Zhao, X.; So, C.; Liao, J.; Wang, L.D.; Yang, C.S. DNA hypermethylation is a mechanism for loss of expression of the HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis 2001, 22, 1615–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigalotti, L.; Fratta, E.; Coral, S.; Tanzarella, S.; Danielli, R.; Colizzi, F.; Fonsatti, E.; Traversari, C.; Altomonte, M.; Maio, M. Intratumor heterogeneity of cancer/testis antigens expression in human cutaneous melanoma is methylation-regulated and functionally reverted by 5-aza-2’-deoxycytidine. Cancer Res. 2004, 64, 9167–9171. [Google Scholar] [CrossRef] [Green Version]
- Manning, J.; Indrova, M.; Lubyova, B.; Pribylova, H.; Bieblova, J.; Hejnar, J.; Simova, J.; Jandlova, T.; Bubenik, J.; Reinis, M. Induction of MHC class I molecule cell surface expression and epigenetic activation of antigen-processing machinery components in a murine model for human papilloma virus 16-associated tumours. Immunology 2008, 123, 218–227. [Google Scholar] [CrossRef]
- Campoli, M.; Ferrone, S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008, 27, 5869–5885. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.N.; Gregorie, C.J.; Tomasi, T.B. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol. Immunother. CII 2008, 57, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlkova, V.; Stepanek, I.; Hruskova, V.; Senigl, F.; Mayerova, V.; Sramek, M.; Simova, J.; Bieblova, J.; Indrova, M.; Hejhal, T.; et al. Epigenetic regulations in the IFNgamma signalling pathway: IFNgamma-mediated MHC class I upregulation on tumour cells is associated with DNA demethylation of antigen-presenting machinery genes. Oncotarget 2014, 5, 6923–6935. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.W.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Coscoy, L.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. USA 2000, 97, 8051–8056. [Google Scholar] [CrossRef] [Green Version]
- Ishido, S.; Wang, C.; Lee, B.S.; Cohen, G.B.; Jung, J.U. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 2000, 74, 5300–5309. [Google Scholar] [CrossRef]
- Stevenson, P.G.; Efstathiou, S.; Doherty, P.C.; Lehner, P.J. Inhibition of MHC class I-restricted antigen presentation by gamma 2-herpesviruses. Proc. Natl. Acad. Sci. USA 2000, 97, 8455–8460. [Google Scholar] [CrossRef] [Green Version]
- Bartee, E.; Mansouri, M.; Hovey Nerenberg, B.T.; Gouveia, K.; Fruh, K. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol. 2004, 78, 1109–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boname, J.M.; Thomas, M.; Stagg, H.R.; Xu, P.; Peng, J.; Lehner, P.J. Efficient internalization of MHC I requires lysine-11 and lysine-63 mixed linkage polyubiquitin chains. Traffic 2010, 11, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Duncan, L.M.; Nathan, J.A.; Lehner, P.J. Stabilization of an E3 ligase-E2-ubiquitin complex increases cell surface MHC class I expression. J. Immunol. 2010, 184, 6978–6985. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Li, S.; Shu, H.B. The Membrane-Associated MARCH E3 Ligase Family: Emerging Roles in Immune Regulation. Front. Immunol. 2019, 10, 1751. [Google Scholar] [CrossRef] [Green Version]
- De Angelis Rigotti, F.; De Gassart, A.; Pforr, C.; Cano, F.; N’Guessan, P.; Combes, A.; Camossetto, V.; Lehner, P.J.; Pierre, P.; Gatti, E. MARCH9-mediated ubiquitination regulates MHC I export from the TGN. Immunol. Cell Biol. 2017, 95, 753–764. [Google Scholar] [CrossRef] [PubMed]
- El-Jawhari, J.J.; El-Sherbiny, Y.M.; Scott, G.B.; Morgan, R.S.; Prestwich, R.; Bowles, P.A.; Blair, G.E.; Tanaka, T.; Rabbitts, T.H.; Meade, J.L.; et al. Blocking oncogenic RAS enhances tumour cell surface MHC class I expression but does not alter susceptibility to cytotoxic lymphocytes. Mol. Immunol. 2014, 58, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef]
- Meissner, T.B.; Li, A.; Biswas, A.; Lee, K.H.; Liu, Y.J.; Bayir, E.; Iliopoulos, D.; van den Elsen, P.J.; Kobayashi, K.S. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. USA 2010, 107, 13794–13799. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, J.G.; Sorbara, M.T.; Girardin, S.E.; Philpott, D.J. What is new with Nods? Curr. Opin. Immunol. 2011, 23, 29–34. [Google Scholar] [CrossRef]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-like receptors: Versatile cytosolic sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef] [Green Version]
- Steimle, V.; Otten, L.A.; Zufferey, M.; Mach, B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 1993, 75, 135–146. [Google Scholar] [CrossRef]
- Steimle, V.; Siegrist, C.A.; Mottet, A.; Lisowska-Grospierre, B.; Mach, B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science 1994, 265, 106–109. [Google Scholar] [CrossRef]
- Cui, J.; Zhu, L.; Xia, X.; Wang, H.Y.; Legras, X.; Hong, J.; Ji, J.; Shen, P.; Zheng, S.; Chen, Z.J.; et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 2010, 141, 483–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benko, S.; Magalhaes, J.G.; Philpott, D.J.; Girardin, S.E. NLRC5 limits the activation of inflammatory pathways. J. Immunol. 2010, 185, 1681–1691. [Google Scholar] [CrossRef] [Green Version]
- Neerincx, A.; Lautz, K.; Menning, M.; Kremmer, E.; Zigrino, P.; Hosel, M.; Buning, H.; Schwarzenbacher, R.; Kufer, T.A. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J. Biol. Chem. 2010, 285, 26223–26232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, P.; Singh, N.; Kumar, A.; Neerincx, A.; Kremmer, E.; Cao, W.; Davis, W.G.; Katz, J.M.; Gangappa, S.; Lin, R.; et al. NLRC5 interacts with RIG-I to induce a robust antiviral response against influenza virus infection. Eur. J. Immunol. 2015, 45, 758–772. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Pandey, S.; Zou, J.; Kumagai, Y.; Takahashi, K.; Akira, S.; Kawai, T. NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. J. Immunol. 2011, 186, 994–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, G.R.; Truax, A.D.; Davis, B.K.; Zhang, L.; Brickey, W.J.; Ting, J.P. Regulation of class I major histocompatibility complex (MHC) by nucleotide-binding domain, leucine-rich repeat-containing (NLR) proteins. J. Biol. Chem. 2012, 287, 24294–24303. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Qian, Y. Expression regulation and function of NLRC5. Protein Cell 2013, 4, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Cui, J.; Li, Q.; Zou, J.; Wang, H.Y.; Wang, R.F. Enhanced TLR-induced NF-kappaB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res. 2012, 22, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Chonwerawong, M.; Ferrand, J.; Chaudhry, H.M.; Higgins, C.; Tran, L.S.; Lim, S.S.; Walker, M.M.; Bhathal, P.S.; Dev, A.; Moore, G.T.; et al. Innate Immune Molecule NLRC5 Protects Mice from Helicobacter-induced Formation of Gastric Lymphoid Tissue. Gastroenterology 2020, 159, 169–182.e168. [Google Scholar] [CrossRef]
- Benko, S.; Kovacs, E.G.; Hezel, F.; Kufer, T.A. NLRC5 Functions beyond MHC I Regulation-What Do We Know So Far? Front. Immunol. 2017, 8, 150. [Google Scholar] [CrossRef]
- Davis, B.K.; Roberts, R.A.; Huang, M.T.; Willingham, S.B.; Conti, B.J.; Brickey, W.J.; Barker, B.R.; Kwan, M.; Taxman, D.J.; Accavitti-Loper, M.A.; et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J. Immunol. 2011, 186, 1333–1337. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Wang, Y.; Chen, F.; Huang, Y.; Zhu, S.; Leng, Q.; Wang, H.; Shi, Y.; Qian, Y. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res. 2012, 22, 836–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triantafilou, K.; Kar, S.; van Kuppeveld, F.J.; Triantafilou, M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Meissner, T.B.; Kawai, T.; Kobayashi, K.S. Cutting edge: Impaired MHC class I expression in mice deficient for Nlrc5/class I transactivator. J. Immunol. 2012, 189, 516–520. [Google Scholar] [CrossRef] [Green Version]
- Staehli, F.; Ludigs, K.; Heinz, L.X.; Seguin-Estevez, Q.; Ferrero, I.; Braun, M.; Schroder, K.; Rebsamen, M.; Tardivel, A.; Mattmann, C.; et al. NLRC5 deficiency selectively impairs MHC class I- dependent lymphocyte killing by cytotoxic T cells. J. Immunol. 2012, 188, 3820–3828. [Google Scholar] [CrossRef] [Green Version]
- Meissner, T.B.; Li, A.; Kobayashi, K.S. NLRC5: A newly discovered MHC class I transactivator (CITA). Microbes Infect. 2012, 14, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuenzel, S.; Till, A.; Winkler, M.; Hasler, R.; Lipinski, S.; Jung, S.; Grotzinger, J.; Fickenscher, H.; Schreiber, S.; Rosenstiel, P. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J. Immunol. 2010, 184, 1990–2000. [Google Scholar] [CrossRef]
- Ng, A.C.; Eisenberg, J.M.; Heath, R.J.; Huett, A.; Robinson, C.M.; Nau, G.J.; Xavier, R.J. Human leucine-rich repeat proteins: A genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4631–4638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutte, P.G.; Jurt, S.; Grutter, M.G.; Zerbe, O. Unusual structural features revealed by the solution NMR structure of the NLRC5 caspase recruitment domain. Biochemistry 2014, 53, 3106–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neerincx, A.; Rodriguez, G.M.; Steimle, V.; Kufer, T.A. NLRC5 controls basal MHC class I gene expression in an MHC enhanceosome-dependent manner. J. Immunol. 2012, 188, 4940–4950. [Google Scholar] [CrossRef] [PubMed]
- Meissner, T.B.; Li, A.; Liu, Y.J.; Gagnon, E.; Kobayashi, K.S. The nucleotide-binding domain of NLRC5 is critical for nuclear import and transactivation activity. Biochem. Biophys. Res. Commun. 2012, 418, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobin, S.J.; van Zutphen, M.; Westerheide, S.D.; Boss, J.M.; van den Elsen, P.J. The MHC-specific enhanceosome and its role in MHC class I and beta(2)-microglobulin gene transactivation. J. Immunol. 2001, 167, 5175–5184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Elsen, P.J. Expression regulation of major histocompatibility complex class I and class II encoding genes. Front. Immunol. 2011, 2, 48. [Google Scholar] [CrossRef] [Green Version]
- Ludigs, K.; Seguin-Estevez, Q.; Lemeille, S.; Ferrero, I.; Rota, G.; Chelbi, S.; Mattmann, C.; MacDonald, H.R.; Reith, W.; Guarda, G. NLRC5 exclusively transactivates MHC class I and related genes through a distinctive SXY module. PLoS Genet. 2015, 11, e1005088. [Google Scholar] [CrossRef] [Green Version]
- Gobin, S.J.; Peijnenburg, A.; Keijsers, V.; van den Elsen, P.J. Site alpha is crucial for two routes of IFN gamma-induced MHC class I transactivation: The ISRE-mediated route and a novel pathway involving CIITA. Immunity 1997, 6, 601–611. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.K.; Chin, K.C.; Olsen, J.C.; Skinner, C.A.; Dey, A.; Ozato, K.; Ting, J.P. Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity 1997, 6, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Hobart, M.; Ramassar, V.; Goes, N.; Urmson, J.; Halloran, P.F. IFN regulatory factor-1 plays a central role in the regulation of the expression of class I and II MHC genes in vivo. J. Immunol. 1997, 158, 4260–4269. [Google Scholar] [PubMed]
- Gobin, S.J.; Keijsers, V.; van Zutphen, M.; van den Elsen, P.J. The role of enhancer A in the locus-specific transactivation of classical and nonclassical HLA class I genes by nuclear factor kappa B. J. Immunol. 1998, 161, 2276–2283. [Google Scholar]
- Israel, A.; Le Bail, O.; Hatat, D.; Piette, J.; Kieran, M.; Logeat, F.; Wallach, D.; Fellous, M.; Kourilsky, P. TNF stimulates expression of mouse MHC class I genes by inducing an NF kappa B-like enhancer binding activity which displaces constitutive factors. EMBO J. 1989, 8, 3793–3800. [Google Scholar] [CrossRef]
- Gobin, S.J.; van den Elsen, P.J. The regulation of HLA class I expression: Is HLA-G the odd one out? Semin. Cancer Biol. 1999, 9, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.L.; Ting, J.P. Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 2006, 27, 405–412. [Google Scholar] [CrossRef]
- Lupfer, C.R.; Stokes, K.L.; Kuriakose, T.; Kanneganti, T.D. Deficiency of the NOD-Like Receptor NLRC5 Results in Decreased CD8(+) T Cell Function and Impaired Viral Clearance. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Ferrero, R.L.; Girardin, S.E.; Gommerman, J.L.; Philpott, D.J. NLRC5 deficiency has a moderate impact on immunodominant CD8(+) T cell responses during rotavirus infection of adult mice. Immunol. Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Guerder, S.; Hong, S.C.; van Ewijk, W.; Flavell, R.A. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity 1996, 4, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.S.; Malin, M.; Vremec, D.; Chang, C.H.; Boyd, R.; Benoist, C.; Mathis, D. Mice lacking the transcription factor CIITA--a second look. Int. Immunol. 1998, 10, 1957–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhammadi, M.; Mathe, J.; Dumont-Lagace, M.; Kobayashi, K.S.; Gaboury, L.; Brochu, S.; Perreault, C. IFN-lambda Enhances Constitutive Expression of MHC Class I Molecules on Thymic Epithelial Cells. J. Immunol. 2020, 205, 1268–1280. [Google Scholar] [CrossRef]
- Kobayashi, K.S. NLRC5/CITA: A novel regulator of class I major histocompatibility complex genes. J. Immunodefic. Disord. 2012, 1. [Google Scholar] [CrossRef]
- Xu, H.C.; Grusdat, M.; Pandyra, A.A.; Polz, R.; Huang, J.; Sharma, P.; Deenen, R.; Kohrer, K.; Rahbar, R.; Diefenbach, A.; et al. Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 2014, 40, 949–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludigs, K.; Jandus, C.; Utzschneider, D.T.; Staehli, F.; Bessoles, S.; Dang, A.T.; Rota, G.; Castro, W.; Zehn, D.; Vivier, E.; et al. NLRC5 shields T lymphocytes from NK-cell-mediated elimination under inflammatory conditions. Nat. Commun. 2016, 7, 10554. [Google Scholar] [CrossRef] [PubMed]
- Rota, G.; Ludigs, K.; Siegert, S.; Tardivel, A.; Morgado, L.; Reith, W.; De Gassart, A.; Guarda, G. T Cell Priming by Activated Nlrc5-Deficient Dendritic Cells Is Unaffected despite Partially Reduced MHC Class I Levels. J. Immunol. 2016, 196, 2939–2946. [Google Scholar] [CrossRef] [Green Version]
- Dang, A.T.; Strietz, J.; Zenobi, A.; Khameneh, H.J.; Brandl, S.M.; Lozza, L.; Conradt, G.; Kaufmann, S.H.E.; Reith, W.; Kwee, I.; et al. NLRC5 promotes transcription of BTN3A1-3 genes and Vgamma9Vdelta2 T cell-mediated killing. iScience 2021, 24, 101900. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of gammadelta T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Vantourout, P.; Laing, A.; Woodward, M.J.; Zlatareva, I.; Apolonia, L.; Jones, A.W.; Snijders, A.P.; Malim, M.H.; Hayday, A.C. Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing gammadelta T cell biology. Proc. Natl. Acad. Sci. USA 2018, 115, 1039–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnett, H.A.; Viney, J.L. Immune modulation by butyrophilins. Nat. Rev. Immunol. 2014, 14, 559–569. [Google Scholar] [CrossRef]
- Sarter, K.; Leimgruber, E.; Gobet, F.; Agrawal, V.; Dunand-Sauthier, I.; Barras, E.; Mastelic-Gavillet, B.; Kamath, A.; Fontannaz, P.; Guery, L.; et al. Btn2a2, a T cell immunomodulatory molecule coregulated with MHC class II genes. J. Exp. Med. 2016, 213, 177–187. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.C.; Kersbergen, A.; Lam, E.Y.N.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019, 36, 385–401.e388. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, H.; Feng, Y.; Li, Y.; Tamiya, H.; Tocci, S.; Ronai, Z.A. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 arginine methyltransferase: Many roles in development, cancer and beyond. Cell Mol. Life Sci. 2015, 72, 2041–2059. [Google Scholar] [CrossRef]
- Zebertavage, L.K.; Alice, A.; Crittenden, M.R.; Gough, M.J. Transcriptional Upregulation of NLRC5 by Radiation Drives STING- and Interferon-Independent MHC-I Expression on Cancer Cells and T Cell Cytotoxicity. Sci. Rep. 2020, 10, 7376. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.C.; Liu, L.F. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE 2012, 7, e32542. [Google Scholar] [CrossRef]
- Chung, S.A.; Nititham, J.; Elboudwarej, E.; Quach, H.L.; Taylor, K.E.; Barcellos, L.F.; Criswell, L.A. Genome-Wide Assessment of Differential DNA Methylation Associated with Autoantibody Production in Systemic Lupus Erythematosus. PLoS ONE 2015, 10, e0129813. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.M.; Liu, X.P.; Chang, G.B.; Xu, L.; Bi, Y.L.; Wang, H.Z.; Zhang, Y.; Zhu, P.F.; Wu, Y.; Chen, G.H. Characterization of the NLRC5 promoter in chicken: SNPs, regulatory elements and CpG islands. Anim. Genet. 2016, 47, 579–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agudo, J.; Park, E.S.; Rose, S.A.; Alibo, E.; Sweeney, R.; Dhainaut, M.; Kobayashi, K.S.; Sachidanandam, R.; Baccarini, A.; Merad, M.; et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 2018, 48, 271–285.e275. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Alvarez, B.; Rodriguez, R.M.; Calvanese, V.; Blanco-Gelaz, M.A.; Suhr, S.T.; Ortega, F.; Otero, J.; Cibelli, J.B.; Moore, H.; Fraga, M.F.; et al. Epigenetic mechanisms regulate MHC and antigen processing molecules in human embryonic and induced pluripotent stem cells. PLoS ONE 2010, 5, e10192. [Google Scholar] [CrossRef] [Green Version]
- Schatton, T.; Schutte, U.; Frank, N.Y.; Zhan, Q.; Hoerning, A.; Robles, S.C.; Zhou, J.; Hodi, F.S.; Spagnoli, G.C.; Murphy, G.F.; et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010, 70, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Maccalli, C.; Volonte, A.; Cimminiello, C.; Parmiani, G. Immunology of cancer stem cells in solid tumours. A review. Eur. J. Cancer 2014, 50, 649–655. [Google Scholar] [CrossRef]
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultan, M.; Coyle, K.M.; Vidovic, D.; Thomas, M.L.; Gujar, S.; Marcato, P. Hide-and-seek: The interplay between cancer stem cells and the immune system. Carcinogenesis 2017, 38, 107–118. [Google Scholar] [CrossRef]
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018, 18, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Yu, C.; Yu, J.; Li, Z.; Lan, H.Y.; Zhou, Q. Arid2-IR promotes NF-kappaB-mediated renal inflammation by targeting NLRC5 transcription. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Zhou, Q.; Huang, X.R.; Yu, J.; Yu, X.; Lan, H.Y. Long Noncoding RNA Arid2-IR Is a Novel Therapeutic Target for Renal Inflammation. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Zong, Z.; Song, Y.; Xue, Y.; Ruan, X.; Liu, X.; Yang, C.; Zheng, J.; Cao, S.; Li, Z.; Liu, Y. Knockdown of LncRNA SCAMP1 suppressed malignant biological behaviours of glioma cells via modulating miR-499a-5p/LMX1A/NLRC5 pathway. J. Cell Mol. Med. 2019, 23, 5048–5062. [Google Scholar] [CrossRef] [Green Version]
- Muhlethaler-Mottet, A.; Otten, L.A.; Steimle, V.; Mach, B. Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J. 1997, 16, 2851–2860. [Google Scholar] [CrossRef]
- LeibundGut-Landmann, S.; Waldburger, J.M.; Krawczyk, M.; Otten, L.A.; Suter, T.; Fontana, A.; Acha-Orbea, H.; Reith, W. Mini-review: Specificity and expression of CIITA, the master regulator of MHC class II genes. Eur. J. Immunol. 2004, 34, 1513–1525. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, P.; Thangaraj, A.; Bendi, V.S.; Buch, S. HIV-1 Tat-mediated microglial inflammation involves a novel miRNA-34a-NLRC5-NFkappaB signaling axis. Brain Behav. Immun. 2019, 80, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, L.; Shen, Z.; Li, Y.; Chen, B.; Wei, W.; Chen, X.; Wang, Q.; Tong, F.; Lou, H.; et al. miR-34a and its novel target, NLRC5, are associated with HPV16 persistence. Infect. Genet. Evol. 2016, 44, 293–299. [Google Scholar] [CrossRef]
- Zong, Y.; Zhang, Y.; Hou, D.; Xu, J.; Cui, F.; Qin, Y.; Sun, X. The lncRNA XIST promotes the progression of breast cancer by sponging miR-125b-5p to modulate NLRC5. Am. J. Transl. Res. 2020, 12, 3501–3511. [Google Scholar]
- Rodino, K.G.; Adcox, H.E.; Martin, R.K.; Patel, V.; Conrad, D.H.; Carlyon, J.A. The Obligate Intracellular Bacterium Orientia tsutsugamushi Targets NLRC5 To Modulate the Major Histocompatibility Complex Class I Pathway. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Cai, C.; Sun, T.; Wang, Q.; Xie, W.; Wang, R.; Cui, J. Reversible ubiquitination shapes NLRC5 function and modulates NF-kappaB activation switch. J. Cell Biol. 2015, 211, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y. A feedforward loop of NLRC5 (de)ubiquitination keeps IKK-NF-kappaB in check. J. Cell Biol. 2015, 211, 941–943. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Dersh, D.; Phelan, J.D.; Gumina, M.E.; Wang, B.; Arbuckle, J.H.; Holly, J.; Kishton, R.J.; Markowitz, T.E.; Seedhom, M.O.; Fridlyand, N.; et al. Genome-wide Screens Identify Lineage- and Tumor-Specific Genes Modulating MHC-I- and MHC-II-Restricted Immunosurveillance of Human Lymphomas. Immunity 2020. [Google Scholar] [CrossRef]
- Bhavsar, A.P.; Brown, N.F.; Stoepel, J.; Wiermer, M.; Martin, D.D.; Hsu, K.J.; Imami, K.; Ross, C.J.; Hayden, M.R.; Foster, L.J.; et al. The Salmonella type III effector SspH2 specifically exploits the NLR co-chaperone activity of SGT1 to subvert immunity. PLoS Pathog. 2013, 9, e1003518. [Google Scholar] [CrossRef] [Green Version]
- Mayor, A.; Martinon, F.; De Smedt, T.; Petrilli, V.; Tschopp, J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007, 8, 497–503. [Google Scholar] [CrossRef]
- Kitagawa, K.; Skowyra, D.; Elledge, S.J.; Harper, J.W.; Hieter, P. SGT1 encodes an essential component of the yeast kinetochore assembly pathway and a novel subunit of the SCF ubiquitin ligase complex. Mol. Cell 1999, 4, 21–33. [Google Scholar] [CrossRef]
- Willhoft, O.; Kerr, R.; Patel, D.; Zhang, W.; Al-Jassar, C.; Daviter, T.; Millson, S.H.; Thalassinos, K.; Vaughan, C.K. The crystal structure of the Sgt1-Skp1 complex: The link between Hsp90 and both SCF E3 ubiquitin ligases and kinetochores. Sci. Rep. 2017, 7, 41626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overwijk, W.W.; Restifo, N.P. B16 as a mouse model for human melanoma. Curr. Protoc. Immunol. 2001, 20. [Google Scholar] [CrossRef]
- Kalbasi, A.; Tariveranmoshabad, M.; Hakimi, K.; Kremer, S.; Campbell, K.M.; Funes, J.M.; Vega-Crespo, A.; Parisi, G.; Champekar, A.; Nguyen, C.; et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in melanoma. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.Y.; He, Y.H.; Chen, C.; Xu, T.; Li, L.; Ni, M.M.; Meng, X.M.; Huang, C.; Li, J. NLRC5 regulates cell proliferation, migration and invasion in hepatocellular carcinoma by targeting the Wnt/beta-catenin signaling pathway. Cancer Lett. 2016, 376, 10–21. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ding, H.; He, Y.; Li, X.; Cheng, Y.; Xu, Q.; Yang, Y.; Liao, G.; Meng, X.; Huang, C.; et al. NLRC5 mediates cell proliferation, migration, and invasion by regulating the Wnt/beta-catenin signalling pathway in clear cell renal cell carcinoma. Cancer Lett. 2019, 444, 9–19. [Google Scholar] [CrossRef]
- He, Y.H.; Li, M.F.; Zhang, X.Y.; Meng, X.M.; Huang, C.; Li, J. NLRC5 promotes cell proliferation via regulating the AKT/VEGF-A signaling pathway in hepatocellular carcinoma. Toxicology 2016, 359–360, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Dong, Z.; Shi, Y.; Sun, S.; Wei, B.; Zhan, L. NLRC5 promotes cell migration and invasion by activating the PI3K/AKT signaling pathway in endometrial cancer. J. Int. Med. Res. 2020, 48, 300060520925352. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, M.; Cao, L.; Cong, L.; Gao, Y.; Lu, J.; Feng, J.; Shen, B.; Liu, D. miR-4319 Suppresses the Growth of Esophageal Squamous Cell Carcinoma Via Targeting NLRC5. Curr. Mol. Pharm. 2020, 13, 144–149. [Google Scholar] [CrossRef]
- Ozcan, M.; Janikovits, J.; von Knebel Doeberitz, M.; Kloor, M. Complex pattern of immune evasion in MSI colorectal cancer. Oncoimmunology 2018, 7, e1445453. [Google Scholar] [CrossRef] [Green Version]
- Alimonti, J.; Zhang, Q.J.; Gabathuler, R.; Reid, G.; Chen, S.S.; Jefferies, W.A. TAP expression provides a general method for improving the recognition of malignant cells in vivo. Nat. Biotechnol. 2000, 18, 515–520. [Google Scholar] [CrossRef]
- Lou, Y.; Basha, G.; Seipp, R.P.; Cai, B.; Chen, S.S.; Moise, A.R.; Jeffries, A.P.; Gopaul, R.S.; Vitalis, T.Z.; Jefferies, W.A. Combining the antigen processing components TAP and Tapasin elicits enhanced tumor-free survival. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1494–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.L.; Zhang, D.; Knight, D.; Odaka, Y.; Glass, J.; Mathis, J.M.; Zhang, Q.J. Priming of immune responses against transporter associated with antigen processing (TAP)-deficient tumours: Tumour direct priming. Immunology 2009, 128, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, A.B.; Aptsiauri, N.; Mendez, R.; Zinchenko, S.; Vales, A.; Paschen, A.; Ward, S.; Ruiz-Cabello, F.; Gonzalez-Aseguinolaza, G.; Garrido, F. Efficient recovery of HLA class I expression in human tumor cells after beta2-microglobulin gene transfer using adenoviral vector: Implications for cancer immunotherapy. Scand. J. Immunol. 2009, 70, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, R.; Martin, A.; Bommarito, D.; Wang, K.; Hansen, S.H.; Freeman, G.J.; Ritz, J. Interferon-gamma-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015, 4, e1008824. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e1512. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- McGray, A.J.; Hallett, R.; Bernard, D.; Swift, S.L.; Zhu, Z.; Teoderascu, F.; Vanseggelen, H.; Hassell, J.A.; Hurwitz, A.A.; Wan, Y.; et al. Immunotherapy-induced CD8+ T cells instigate immune suppression in the tumor. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, M.R. The Interferon-Gamma Paradox in Cancer. J. Interferon Cytokine Res. 2019, 39, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Kang, K.; Giannopoulou, E.; Fang, C.; Ivashkiv, L.B. IFN-gamma Induces Histone 3 Lysine 27 Trimethylation in a Small Subset of Promoters to Stably Silence Gene Expression in Human Macrophages. Cell Rep. 2016, 16, 3121–3129. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Antonczyk, A.; Krist, B.; Sajek, M.; Michalska, A.; Piaszyk-Borychowska, A.; Plens-Galaska, M.; Wesoly, J.; Bluyssen, H.A.R. Direct Inhibition of IRF-Dependent Transcriptional Regulatory Mechanisms Associated with Disease. Front. Immunol. 2019, 10, 1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musella, M.; Manic, G.; De Maria, R.; Vitale, I.; Sistigu, A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology 2017, 6, e1314424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gessani, S.; Conti, L.; Del Corno, M.; Belardelli, F. Type I interferons as regulators of human antigen presenting cell functions. Toxins 2014, 6, 1696–1723. [Google Scholar] [CrossRef]
- Budhwani, M.; Mazzieri, R.; Dolcetti, R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front. Oncol. 2018, 8, 322. [Google Scholar] [CrossRef] [Green Version]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remedios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Wang, X.; Schoenhals, J.E.; Li, A.; Valdecanas, D.R.; Ye, H.; Zang, F.; Tang, C.; Tang, M.; Liu, C.G.; Liu, X.; et al. Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy. Cancer Res. 2017, 77, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Xi, S.; Dyer, K.F.; Kimak, M.; Zhang, Q.; Gooding, W.E.; Chaillet, J.R.; Chai, R.L.; Ferrell, R.E.; Zamboni, B.; Hunt, J.; et al. Decreased STAT1 expression by promoter methylation in squamous cell carcinogenesis. J. Natl. Cancer Inst. 2006, 98, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komyod, W.; Bohm, M.; Metze, D.; Heinrich, P.C.; Behrmann, I. Constitutive suppressor of cytokine signaling 3 expression confers a growth advantage to a human melanoma cell line. Mol. Cancer Res. 2007, 5, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Quintanilla, J.; Seah, I.; Chua, M.; Shah, K. Oncolytic viruses: Overcoming translational challenges. J. Clin. Investig. 2019, 129, 1407–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widschwendter, M.; Jones, A.; Evans, I.; Reisel, D.; Dillner, J.; Sundstrom, K.; Steyerberg, E.W.; Vergouwe, Y.; Wegwarth, O.; Rebitschek, F.G.; et al. Epigenome-based cancer risk prediction: Rationale, opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 292–309. [Google Scholar] [CrossRef]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef]
- Kortenhorst, M.S.; Wissing, M.D.; Rodriguez, R.; Kachhap, S.K.; Jans, J.J.; Van der Groep, P.; Verheul, H.M.; Gupta, A.; Aiyetan, P.O.; van der Wall, E.; et al. Analysis of the genomic response of human prostate cancer cells to histone deacetylase inhibitors. Epigenetics 2013, 8, 907–920. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Kungulovski, G.; Jeltsch, A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2016, 32, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [Green Version]
- Romero, I.; Martinez, M.; Garrido, C.; Collado, A.; Algarra, I.; Garrido, F.; Garcia-Lora, A.M. The tumour suppressor Fhit positively regulates MHC class I expression on cancer cells. J. Pathol. 2012, 227, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Pulido, M.; Chamorro, V.; Romero, I.; Algarra, I.A.S.M.; Collado, A.; Garrido, F.; Garcia-Lora, A.M. Restoration of MHC-I on Tumor Cells by Fhit Transfection Promotes Immune Rejection and Acts as an Individualized Immunotherapeutic Vaccine. Cancers 2020, 12, 1563. [Google Scholar] [CrossRef]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Immunol. 2003, 4, 815. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Bevan, M.J. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature 2011, 471, 629–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyo, C.W.; Williams, L.M.; Moore, Y.; Hyodo, H.; Li, S.S.; Zhao, L.P.; Sageshima, N.; Ishitani, A.; Geraghty, D.E. HLA-E, HLA-F, and HLA-G polymorphism: Genomic sequence defines haplotype structure and variation spanning the nonclassical class I genes. Immunogenetics 2006, 58, 241–251. [Google Scholar] [CrossRef]
- Goodall, K.J.; Nguyen, A.; Sullivan, L.C.; Andrews, D.M. The expanding role of murine class Ib MHC in the development and activation of Natural Killer cells. Mol. Immunol. 2019, 115, 31–38. [Google Scholar] [CrossRef]
- Moser, J.M.; Gibbs, J.; Jensen, P.E.; Lukacher, A.E. CD94-NKG2A receptors regulate antiviral CD8(+) T cell responses. Nat. Immunol. 2002, 3, 189–195. [Google Scholar] [CrossRef]
- Vance, R.E.; Jamieson, A.M.; Cado, D.; Raulet, D.H. Implications of CD94 deficiency and monoallelic NKG2A expression for natural killer cell development and repertoire formation. Proc. Natl. Acad. Sci. USA 2002, 99, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd Hamid, M.; Wang, R.Z.; Yao, X.; Fan, P.; Li, X.; Chang, X.M.; Feng, Y.; Jones, S.; Maldonado-Perez, D.; Waugh, C.; et al. Enriched HLA-E and CD94/NKG2A Interaction Limits Antitumor CD8(+) Tumor-Infiltrating T Lymphocyte Responses. Cancer Immunol. Res. 2019, 7, 1293–1306. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 2019, 129, 2094–2106. [Google Scholar] [CrossRef] [Green Version]
- Ottenhoff, T.H.M.; Joosten, S.A. Mobilizing unconventional T cells. Science 2019, 366, 302–303. [Google Scholar] [CrossRef]
- Wiendl, H.; Mitsdoerffer, M.; Hofmeister, V.; Wischhusen, J.; Bornemann, A.; Meyermann, R.; Weiss, E.H.; Melms, A.; Weller, M. A functional role of HLA-G expression in human gliomas: An alternative strategy of immune escape. J. Immunol. 2002, 168, 4772–4780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouas-Freiss, N.; Moreau, P.; Ferrone, S.; Carosella, E.D. HLA-G proteins in cancer: Do they provide tumor cells with an escape mechanism? Cancer Res. 2005, 65, 10139–10144. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, P.; Masternak, K.; Krawczyk, M.; Reith, W.; Dausset, J.; Carosella, E.D.; Moreau, P. In vivo, RFX5 binds differently to the human leucocyte antigen-E, -F, and -G gene promoters and participates in HLA class I protein expression in a cell type-dependent manner. Immunology 2004, 111, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquelot, N.; Yamazaki, T.; Roberti, M.P.; Duong, C.P.M.; Andrews, M.C.; Verlingue, L.; Ferrere, G.; Becharef, S.; Vetizou, M.; Daillere, R.; et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019, 29, 846–861. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Du Four, S.; Van der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix mRNA Results in T-cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol. Res. 2016, 4, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Van der Jeught, K.; Van Lint, S.; Thielemans, K.; Breckpot, K. Intratumoral delivery of mRNA: Overcoming obstacles for effective immunotherapy. Oncoimmunology 2015, 4, e1005504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Jeught, K.; Joe, P.T.; Bialkowski, L.; Heirman, C.; Daszkiewicz, L.; Liechtenstein, T.; Escors, D.; Thielemans, K.; Breckpot, K. Intratumoral administration of mRNA encoding a fusokine consisting of IFN-beta and the ectodomain of the TGF-beta receptor II potentiates antitumor immunity. Oncotarget 2014, 5, 10100–10113. [Google Scholar] [CrossRef] [Green Version]
- Neerincx, A.; Jakobshagen, K.; Utermohlen, O.; Buning, H.; Steimle, V.; Kufer, T.A. The N-terminal domain of NLRC5 confers transcriptional activity for MHC class I and II gene expression. J. Immunol. 2014, 193, 3090–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data from Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Pitt, J.M.; Vetizou, M.; Daillere, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Yuan, J.; Hegde, P.S.; Clynes, R.; Foukas, P.G.; Harari, A.; Kleen, T.O.; Kvistborg, P.; Maccalli, C.; Maecker, H.T.; Page, D.B.; et al. Novel technologies and emerging biomarkers for personalized cancer immunotherapy. J. Immunother. Cancer 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, S.; Mehnert, J. Biomarkers for Response to Immune Checkpoint Blockade. Annu. Rev. Cancer Biol. 2020, 4, 331–351. [Google Scholar] [CrossRef] [Green Version]
- Lizee, G.; Overwijk, W.W.; Radvanyi, L.; Gao, J.; Sharma, P.; Hwu, P. Harnessing the power of the immune system to target cancer. Annu. Rev. Med. 2013, 64, 71–90. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Kershaw, M.H.; Darcy, P.K. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology 2013, 2, e26286. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Howat, W.J.; Lewis, A.; Jones, P.; Kampf, C.; Ponten, F.; van der Loos, C.M.; Gray, N.; Womack, C.; Warford, A. Antibody validation of immunohistochemistry for biomarker discovery: Recommendations of a consortium of academic and pharmaceutical based histopathology researchers. Methods 2014, 70, 34–38. [Google Scholar] [CrossRef]
- O’Hurley, G.; Sjostedt, E.; Rahman, A.; Li, B.; Kampf, C.; Ponten, F.; Gallagher, W.M.; Lindskog, C. Garbage in, garbage out: A critical evaluation of strategies used for validation of immunohistochemical biomarkers. Mol. Oncol. 2014, 8, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, F.; Liu, Y.; Chen, H.J.; Wen, F.; Zou, B.; Li, D.; Qin, Q.; Liu, X.; Shen, Y.; et al. NLRC5 expression in tumors and its role as a negative prognostic indicator in stage III non-small-cell lung cancer patients. Oncol. Lett. 2015, 10, 1533–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, M.; Zheng, X. High Expression of NLRC5 Is Associated with Prognosis of Gastric Cancer. Open Med. (Wars) 2018, 13, 443–449. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Model Systems | Study Description | Molecular Effects | Ref. |
|---|---|---|---|---|
| NLRC5 as a tumor suppressor | ||||
| Melanoma | Murine B16.F10 cell line stably expressing NLRC5 | NLRC5 limits tumor growth and metastasis in C57BL/6 mice by activating antitumor CD8+ T lymphocytes; | NLRC5 upregulates MHC-I, β2M, PSMB9, PSMB8, TAP1 gene expression; NLRC5 promotes presentation of peptide from gp100 (Pmel-1) tumor antigen | [208] |
| Melanoma | PRMT5 knockdown in B16.F10 and Yummer1.7 cell lines; B16.F10 stably expressing NLRC5 | Induction of endogenous NLRC5 by PRMT5 knockdown, or stable NLRC5 expression inhibited tumor growth in C57BL/6 mice | PRMT5 reduces NLRC5 expression by promoting methylation of Arg residues on histones | [176] |
| Melanoma | Jak1−/−B16 cell line expressing NLRC5 | NLRC5 rendered Jak1−/− B16 cells susceptible to killing by adoptively transferred Pmel-1 TCR transgenic CD8+ T cells in vivo | NLRC5 upregulates MHC-I | [210] |
| Pancreatic adeno-carcinoma (PDAC) | Murine Panc02 cell line expressing a model antigenic peptide SIYRYYGL fused to GFP (Panc02SIY100) | Gamma irradiation induces NLRC5 expression renders Panc02 cells susceptible to anti-PD-L1 in vivo; Stable NLRC5 expression in Panc02SIY100 promotes activation of 2C TCR transgenic CD8+ T lymphocytes | Gamma irradiation induces MHC-I independently of IFN-I signaling | [178] |
| NLRC5 as a tumor promoter | ||||
| Hepatocellular carcinoma (HCC) | Human HCC specimens; HCC cell lines: HepG2, SMMC-7721, BEL-7402; stable expression or knockdown of NLRC5 | HCC specimens and cell lines display elevated NLRC5 expression; NLRC5 promotes cell proliferation, migration and invasion; NLRC5 knockdown has opposite effect & reduces HepG2 tumor growth in nude mice | NLRC5 expression promotes Wnt/β-catenin signaling and c-Myc, CyclinD1, MMP3 expression; β-catenin inhibitor iCRT3 attenuated the effects of NLRC5 overexpression | [211] |
| Hepatocellular carcinoma | HepG2 cell line | NLRC5 overexpression in HepG2 cells promotes cell growth via upregulating VEGF-A | NLRC5 promotes VEGF-A expression via AKT activation | [213] |
| Clear cell renal cell carcinoma (ccRCC) | Human ccRCC specimens Human ccRCC cell lines Caki-1, 786-O and 769-P | ccRCC specimens and cell lines display elevated NLRC5 expression; NLRC5 promotes cell proliferation, migration and invasion; NLRC5 knockdown causes opposite effects and reduces 786-O tumor growth in nude mice | NLRC5 promotes β-catenin, c-Myc, CyclinD1, MMP2, MMP9 expression | [212] |
| Giloma | Human glioma tissues, cell lines U87, U251 | High grade glioma tissues and cell lines display elevated NLRC5 expression due to high levels of lncRNA SCAMP1; NLRC5 knockdown restrains cell proliferation, migration & invasion and increases apoptosis | SCAM1-1 sponges off miR-499a-5p, which targets LMX1A; LM1A binds NLRC5 promoter and is proposed to augment NLRC5 expression to activate the Wnt/β-catenin pathway | [192] |
| Esophageal squamous cell carcinoma (ESCC) | ESCC cell lines | NLRC5 overexpression in ESCC cell lines promotes cell proliferation, colony formation and cell cycle progression | miR-4319 targets NLRC5; low miR-4319 in ESCC upregulates NLRC5 expression | [215] |
| Breast cancer | Breast cancer tissues, cell lines MCF-7, MDA-MB-231 | MCF-7 and MDA-MB-231 show increased proliferation, migration and invasion due to elevated expression of XIST lncRNA, which upregulates NLRC5 | XIST sponges off miR-125b-5p, which targets NLRC5 | [197] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shukla, A.; Cloutier, M.; Appiya Santharam, M.; Ramanathan, S.; Ilangumaran, S. The MHC Class-I Transactivator NLRC5: Implications to Cancer Immunology and Potential Applications to Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 1964. https://doi.org/10.3390/ijms22041964
Shukla A, Cloutier M, Appiya Santharam M, Ramanathan S, Ilangumaran S. The MHC Class-I Transactivator NLRC5: Implications to Cancer Immunology and Potential Applications to Cancer Immunotherapy. International Journal of Molecular Sciences. 2021; 22(4):1964. https://doi.org/10.3390/ijms22041964
Chicago/Turabian StyleShukla, Akhil, Maryse Cloutier, Madanraj Appiya Santharam, Sheela Ramanathan, and Subburaj Ilangumaran. 2021. "The MHC Class-I Transactivator NLRC5: Implications to Cancer Immunology and Potential Applications to Cancer Immunotherapy" International Journal of Molecular Sciences 22, no. 4: 1964. https://doi.org/10.3390/ijms22041964
APA StyleShukla, A., Cloutier, M., Appiya Santharam, M., Ramanathan, S., & Ilangumaran, S. (2021). The MHC Class-I Transactivator NLRC5: Implications to Cancer Immunology and Potential Applications to Cancer Immunotherapy. International Journal of Molecular Sciences, 22(4), 1964. https://doi.org/10.3390/ijms22041964