
Runx3 Induces a Cell Shape Change and Suppresses Migration and Metastasis of Melanoma Cells by Altering a Transcriptional Profile

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Stable Cell Lines by Lentiviral Infection
2.2. Cell Culture and Treatment, Cell Proliferation Assay, and Cell Cycle Assay
2.3. Wound-Healing Assay and Transwell Migration/Invasion Assay
2.4. Subcutaneous Tumor Formation and Pulmonary Metastasis Assay
2.5. Microarray
2.6. RNA Preparation and RT-qPCR
2.7. Western Blotting
2.8. Histology
2.9. Actin Cytoskeleton Staining
2.10. The Cancer Genome Atlas (TCGA) Data Mining and Gene Expression Correlation Analysis
2.11. Statistical Analysis
3. Results
3.1. Runx3 Re-Expression Induces a Cell Shape Change and Suppresses the Migration and Metastasis of B16-F10 Melanoma Cells
3.2. Runx3 Re-Expression Alters a Specific Transcriptional Profile in B16-F10 Melanoma Cells
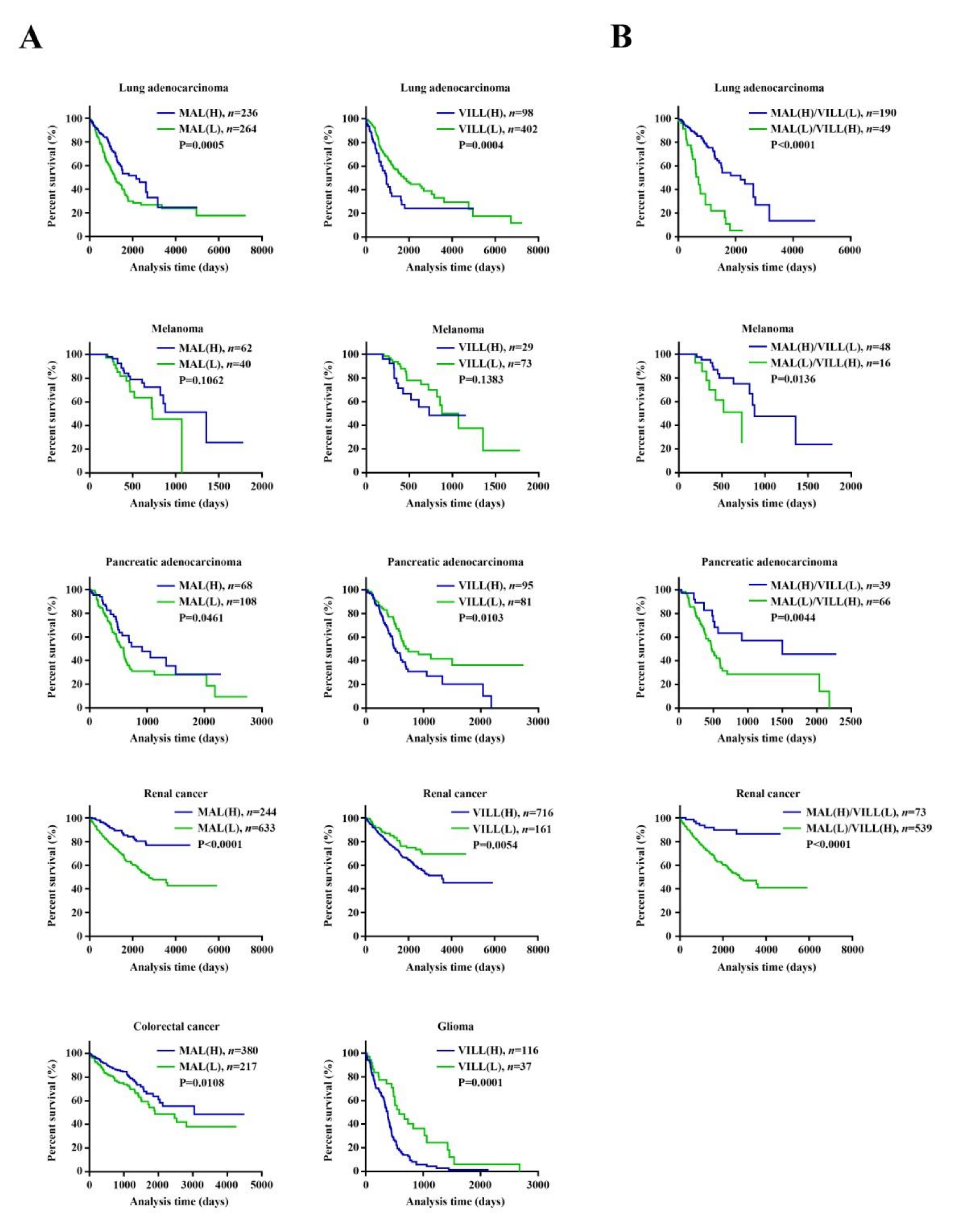
3.3. The Runx3-Altered Transcriptional Profile May Have Prognostic Value for Various Cancers, as Evidenced by MAL and/or VILL
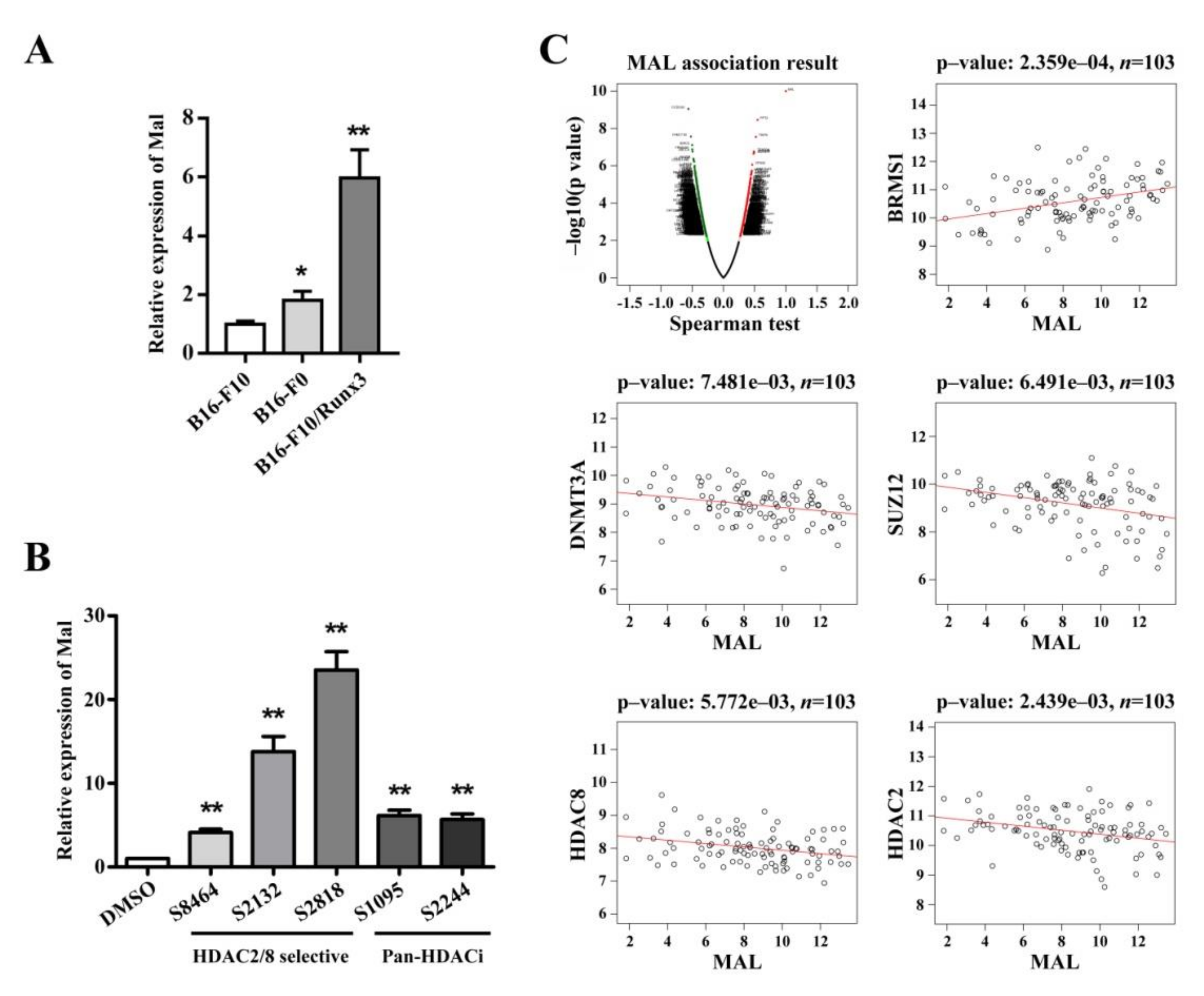
3.4. Mal Gene Expression Is Correlated with Metastatic Potential among Cellular Models and Is Subject to Epigenetic Regulation
3.5. Mal Is Necessary for Suppressing the Migratory and Invasive Traits of Metastasis in Melanoma Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DEG | differentially expressed gene |
| ECM | extracellular matrix |
| EMT | epithelial–mesenchymal transition |
| FPKM | fragments per kilobase of exon per million reads |
| GO | Gene Ontology |
| HDAC | histone deacetylase |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| Mal | myelin and lymphocyte protein |
| PBS | phosphate-buffered saline |
| qRT-PCR | quantitative reverse transcription–polymerase chain reaction |
| Runx3 | runt-related transcription factor-3 |
| TCGA | The Cancer Genome Atlas |
| Vill | vilin-like |
References
- Stoletov, K.; Beatty, P.H.; Lewis, J.D. Novel therapeutic targets for cancer metastasis. Expert Rev. Anticancer 2020, 20, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, I.J.; Kripke, M.L. The challenge of targeting metastasis. Cancer Metastasis Rev. 2015, 34, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Suhail, Y.; Cain, M.P.; Vanaja, K.; Kurywchak, P.A.; Levchenko, A.; Kalluri, R. Systems Biology of Cancer Metastasis. Cell Syst. 2019, 9, 109–127. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Steeg, P.S. Metastasis suppressors: Functional pathways. Lab. Investig. 2018, 98, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.L.; Balasas, T.; Callaghan, J.; Coombes, R.C.; Evans, J.; Hall, J.A.; Kinrade, S.; Jones, D.; Jones, P.S.; Jones, R.; et al. A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 2019, 16, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer. 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac activation and inactivation control plasticity of tumor cell movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren, K.; Pincus, Z.; Allen, G.M.; Barnhart, E.L.; Marriott, G.; Mogilner, A.; Theriot, J.A. Mechanism of shape determination in motile cells. Nature 2008, 453, 475–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnhart, E.L.; Lee, K.C.; Keren, K.; Mogilner, A.; Theriot, J.A. An adhesion-dependent switch between mechanisms that determine motile cell shape. PLoS Biol. 2011, 9, e1001059. [Google Scholar] [CrossRef]
- Yin, Z.; Sadok, A.; Sailem, H.; McCarthy, A.; Xia, X.; Li, F.; Garcia, M.A.; Evans, L.; Barr, A.R.; Perrimon, N.; et al. A screen for morphological complexity identifies regulators of switch-like transitions between discrete cell shapes. Nat. Cell Biol. 2013, 15, 860–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Lakshmanan, M.; Swa, H.L.F.; Chen, J.; Zhang, X.; Ong, Y.S.; Loo, L.S.; Akincilar, S.C.; Gunaratne, J.; Tergaonkar, V.; et al. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat. Commun. 2015, 6, 6184. [Google Scholar] [CrossRef]
- Roszkowska, K.A.; Gizinski, S.; Sady, M.; Gajewski, Z.; Olszewski, M.B. Gain-of-Function Mutations in p53 in Cancer Invasiveness and Metastasis. Int. J. Mol. Sci. 2020, 21, 1334. [Google Scholar]
- Mogilner, A.; Keren, K. The shape of motile cells. Curr. Biol. 2009, 19, R762–R771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardel, M.L.; Schneider, I.C.; Aratyn-Schaus, Y.; Waterman, C.M. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 2010, 26, 315–333. [Google Scholar]
- Chang, C.W.; Kumar, S. Vinculin tension distributions of individual stress fibers within cell-matrix adhesions. J. Cell Sci. 2013, 126, 3021–3030. [Google Scholar] [CrossRef] [Green Version]
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 2017, 17, 131–140. [Google Scholar] [PubMed] [Green Version]
- Wangpu, X.; Lu, J.; Xi, R.; Yue, F.; Sahni, S.; Park, K.C.; Menezes, S.; Huang, M.L.H.; Zheng, M.; Kovacevic, Z.; et al. Targeting the Metastasis Suppressor, N-Myc Downstream Regulated Gene-1, with Novel Di-2-Pyridylketone Thiosemicarbazones: Suppression of Tumor Cell Migration and Cell-Collagen Adhesion by Inhibiting Focal Adhesion Kinase/Paxillin Signaling. Mol. Pharm. 2016, 89, 521–540. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, R.O.; Destree, A.T. Relationships between fibronectin (LETS protein) and actin. Cell 1978, 15, 875–886. [Google Scholar] [CrossRef]
- Lawler, J.; Weinstein, R.; Hynes, R.O. Cell attachment to thrombospondin: The role of ARG-GLY-ASP, calcium, and integrin receptors. J. Cell Biol. 1988, 107, 2351–2361. [Google Scholar] [CrossRef]
- Roberts, D.D. Regulation of tumor growth and metastasis by thrombospondin-1. FASEB J. 1996, 10, 1183–1191. [Google Scholar] [CrossRef]
- Clark, E.A.; Golub, T.R.; Lander, E.S.; Hynes, R.O. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000, 406, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.C.; Bornstein, P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003, 22, 63–71. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Iozzo, R.V. Thrombospondins in physiology and disease: New tricks for old dogs. Matrix Biol. 2012, 31, 152–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naba, A.; Clauser, K.R.; Lamar, J.M.; Carr, S.A.; Hynes, R.O. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife 2014, 3, e01308. [Google Scholar] [CrossRef] [Green Version]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front Oncol. 2015, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, M.; Barker, T.H.; Gibbons, D.L.; Kurie, J.M. The fibrotic tumor stroma. J. Clin. Investig. 2018, 128, 16–25. [Google Scholar] [CrossRef]
- Paolillo, M.; Schinelli, S. Extracellular Matrix Alterations in Metastatic Processes. Int. J. Mol. Sci. 2019, 20, 4947. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.P.; Chang, H.L.; Bamodu, O.A.; Yadav, V.K.; Huang, T.Y.; Wu, A.T.H.; Yeh, C.T.; Tsai, S.H.; Lee, W.H. Collagen 1A1 (COL1A1) Is a Reliable Biomarker and Putative Therapeutic Target for Hepatocellular Carcinogenesis and Metastasis. Cancers 2019, 11, 786. [Google Scholar] [CrossRef] [Green Version]
- Reticker-Flynn, N.E.; Malta, D.F.B.; Winslow, M.M.; Lamar, J.M.; Xu, M.J.; Underhill, G.H.; Hynes, R.O.; Jacks, T.E.; Bhatia, S.N. A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nat. Commun. 2012, 3, 1122. [Google Scholar] [CrossRef] [Green Version]
- Mevel, R.; Draper, J.E.; Lie-A-Ling, M.; Kouskoff, V.; Lacaud, G. RUNX transcription factors: Orchestrators of development. Development 2019, 146, dev148296. [Google Scholar] [CrossRef] [Green Version]
- Lau, Q.C.; Raja, E.; Salto-Tellez, M.; Liu, Q.; Ito, K.; Inoue, M.; Putti, T.C.; Loh, M.; Ko, T.K.; Huang, C.; et al. RUNX3 is frequently inactivated by dual mechanisms of protein mislocalization and promoter hypermethylation in breast cancer. Cancer Res. 2006, 66, 6512–6520. [Google Scholar] [CrossRef] [Green Version]
- Kudo, Y.; Tsunematsu, T.; Takata, T. Oncogenic role of RUNX3 in head and neck cancer. J. Cell Biochem. 2011, 112, 387–393. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, J.W.; Jang, J.W.; Chi, X.Z.; Kim, J.H.; Li, Y.H.; Kim, M.K.; Kim, D.M.; Choi, B.S.; Kim, E.G.; et al. Runx3 inactivation is a crucial early event in the development of lung adenocarcinoma. Cancer Cell 2013, 24, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Bae, S.C.; Chuang, L.S.H. The RUNX family: Developmental regulators in cancer. Nat. Rev. Cancer 2015, 15, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Kitago, M.; Martinez, S.R.; Nakamura, T.; Sim, M.S.; Hoon, D.S. Regulation of RUNX3 tumor suppressor gene expression in cutaneous melanoma. Clin. Cancer Res. 2009, 15, 2988–2994. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.F. Tumor suppressor function of RUNX3 in breast cancer. J. Cell Biochem. 2012, 113, 1470–1477. [Google Scholar] [CrossRef] [Green Version]
- Manandhar, S.; Lee, Y.M. Emerging role of RUNX3 in the regulation of tumor microenvironment. BMB Rep. 2018, 51, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Crosley, P.; Azad, A.K.; Gupta, N.; Gokul, N.; Xu, Z.; Weinfeld, M.; Postovit, L.M.; Pangas, S.A.; Hitt, M.M.; et al. RUNX3 Promotes the Tumorigenic Phenotype in KGN, a Human Granulosa Cell Tumor-Derived Cell Line. Int. J. Mol. Sci. 2019, 20, 3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Date, Y.; Ito, K. Oncogenic RUNX3: A Link between p53 Deficiency and MYC Dysregulation. Mol. Cells 2020, 43, 176–181. [Google Scholar] [PubMed]
- Chang, T.L.; Ito, K.; Ko, T.K.; Liu, Q.; Salto-Tellez, M.; Yeoh, K.G.; Fukamachi, H.; Ito, Y. Claudin-1 has tumor suppressive activity and is a direct target of RUNX3 in gastric epithelial cells. Gastroenterology 2010, 138, 255–265.e3. [Google Scholar] [CrossRef] [Green Version]
- Voon, D.C.; Wang, H.; Koo, J.K.W.; Nguyen, T.A.P.; Hor, Y.T.; Chu, Y.S.; Ito, K.; Fukamachi, H.; Chan, S.L.; Thiery, J.P.; et al. Runx3 protects gastric epithelial cells against epithelial-mesenchymal transition-induced cellular plasticity and tumorigenicity. Stem Cells 2012, 30, 2088–2099. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Shiraha, H.; Nakanishi, Y.; Nishina, S.; Matsubara, M.; Horiguchi, S.; Takaoka, N.; Iwamuro, M.; Kataoka, J.; Kuwaki, K.; et al. Runt-related transcription factor 3 reverses epithelial-mesenchymal transition in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 2537–2546. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, M.; Tan, T.Z.; Syed Sulaiman, N.B.; Lamar, J.M.; Bansal, P.; Cui, J.; Qiao, Y.; Ito, Y. RUNX1 and RUNX3 protect against YAP-mediated EMT, stem-ness and shorter survival outcomes in breast cancer. Oncotarget 2018, 9, 14175–14192. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, L.; Zeng, X.; Fujita, T.; Liu, W. Runx3 inhibits melanoma cell migration through regulation of cell shape change. Cell Biol. Int. 2017, 41, 1048–1055. [Google Scholar] [CrossRef]
- Chen, F.; Liu, X.; Bai, J.; Pei, D.; Zheng, J. The emerging role of RUNX3 in cancer metastasis (Review). Oncol Rep. 2016, 35, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Lotem, J.; Levanon, D.; Negreanu, V.; Bauer, O.; Hantisteanu, S.; Dicken, J.; Groner, Y. Runx3 in Immunity, Inflammation and Cancer. Adv. Exp. Med. Biol. 2017, 962, 369–393. [Google Scholar]
- Kim, B.R.; Na, Y.J.; Kim, J.L.; Jeong, Y.A.; Park, S.H.; Jo, M.J.; Jeong, S.; Kang, S.; Oh, S.C.; Lee, D.H. RUNX3 suppresses metastasis and stemness by inhibiting Hedgehog signaling in colorectal cancer. Cell Death Differ. 2020, 27, 676–694. [Google Scholar] [CrossRef]
- Cao, W.; Zhang, Z.Y.; Xu, Q.; Sun, Q.; Yan, M.; Zhang, J.; Zhang, P.; Han, Z.G.; Chen, W.T. Epigenetic silencing of MAL, a putative tumor suppressor gene, can contribute to human epithelium cell carcinoma. Mol. Cancer 2010, 9, 296. [Google Scholar] [CrossRef] [Green Version]
- Dubash, A.D.; Koetsier, J.L.; Amargo, E.V.; Najor, N.A.; Harmon, R.M.; Green, K.J. The GEF Bcr activates RhoA/MAL signaling to promote keratinocyte differentiation via desmoglein-1. J. Cell Biol. 2013, 202, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Kalmar, A.; Peterfia, B.; Hollosi, P.; Galamb, O.; Spisak, S.; Wichmann, B.; Bodor, A.; Toth, K.; Patai, A.V.; Valcz, G.; et al. DNA hypermethylation and decreased mRNA expression of MAL, PRIMA1, PTGDR and SFRP1 in colorectal adenoma and cancer. BMC Cancer 2015, 15, 736. [Google Scholar] [CrossRef] [Green Version]
- Beder, L.B.; Gunduz, M.; Hotomi, M.; Fujihara, K.; Shimada, J.; Tamura, S.; Gunduz, E.; Fukushima, K.; Yaykasli, K.; Grenman, R.; et al. T-lymphocyte maturation-associated protein gene as a candidate metastasis suppressor for head and neck squamous cell carcinomas. Cancer Sci. 2009, 100, 873–880. [Google Scholar] [CrossRef]
- Fidler, I.J. Selection of successive tumour lines for metastasis. Nat. New Biol. 1973, 242, 148–149. [Google Scholar] [CrossRef]
- Nakamura, K.; Yoshikawa, N.; Yamaguchi, Y.; Kagota, S.; Shinozuka, K.; Kunitomo, M. Characterization of mouse melanoma cell lines by their mortal malignancy using an experimental metastatic model. Life Sci. 2002, 70, 791–798. [Google Scholar] [CrossRef]
- Chen, H.; Shan, J.; Chen, D.; Wang, R.; Qi, W.; Wang, H.; Ke, Y.; Liu, W.; Zeng, X. CtIP promotes G2/M arrest in etoposide-treated HCT116 cells in a p53-independent manner. J. Cell Physiol. 2019, 234, 11871–11881. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [Green Version]
- Nichols, S.E., Jr.; Reams, W.M., Jr. The occurrence and morphogenesis of melanocytes in the connective tissues of the PET/MCV mouse strain. J. Embryol. Exp. Morphol. 1960, 8, 24–32. [Google Scholar]
- Cramer, S.F. The origin of epidermal melanocytes. Implications for the histogenesis of nevi and melanomas. Arch. Pathol. Lab. Med. 1991, 115, 115–119. [Google Scholar]
- Gong, F.; Guo, Y.; Niu, Y.; Jin, J.; Zhang, X.; Shi, X.; Zhang, L.; Li, R.; Chen, L.; Ma, R.Z. Epigenetic silencing of TET2 and TET3 induces an EMT-like process in melanoma. Oncotarget 2017, 8, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Deepak, V.; Wang, L.; Ba, X.; Komori, T.; Zeng, X.; Liu, W. Thrombospondin-1 is a putative target gene of Runx2 and Runx3. Int. J. Mol. Sci. 2013, 14, 14321–14332. [Google Scholar] [CrossRef] [Green Version]
- Murphy-Ullrich, J.E.; Hook, M. Thrombospondin modulates focal adhesions in endothelial cells. J. Cell Biol. 1989, 109, 1309–1319. [Google Scholar] [CrossRef]
- Goodenow, R.S.; Vogel, J.M.; Linsk, R.L. Histocompatibility antigens on murine tumors. Science 1985, 230, 777–783. [Google Scholar] [CrossRef]
- Mimori, K.; Shiraishi, T.; Mashino, K.; Sonoda, H.; Yamashita, K.; Yoshinaga, K.; Masuda, T.; Utsunomiya, T.; Alonso, M.A.; Inoue, H.; et al. MAL gene expression in esophageal cancer suppresses motility, invasion and tumorigenicity and enhances apoptosis through the Fas pathway. Oncogene 2003, 22, 3463–3471. [Google Scholar] [CrossRef] [Green Version]
- Kurashige, J.; Sawada, G.; Takahashi, Y.; Eguchi, H.; Sudo, T.; Ikegami, T.; Yoshizumi, T.; Soejima, Y.; Ikeda, T.; Kawanaka, H.; et al. Suppression of MAL gene expression in gastric cancer correlates with metastasis and mortality. Fukuoka Igaku Zasshi 2013, 104, 344–349. [Google Scholar]
- Fidler, I.J.; Radinsky, R. Search for genes that suppress cancer metastasis. J. Natl. Cancer Inst. 1996, 88, 1700–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Shen, S.S.; Hoshida, Y.; Subramanian, A.; Ross, K.; Brunet, J.P.; Wagner, S.N.; Ramaswamy, S.; Mesirov, J.P.; Hynes, R.O. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol. Cancer Res. 2008, 6, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O.; Yamada, K.M. Fibronectins: Multifunctional modular glycoproteins. J. Cell Biol. 1982, 95, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, T.; Saito, D.; Atsuta, Y.; Uchiyama, C.; Ueda, S.; Sekiguchi, K.; Takahashi, Y. Interepithelial signaling with nephric duct is required for the formation of overlying coelomic epithelial cell sheet. Proc. Natl. Acad. Sci. USA 2014, 111, 6660–6665. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Zhang, H.; Cui, X.; Ma, C.; Wang, L.; Liu, W. Runx3 Induces a Cell Shape Change and Suppresses Migration and Metastasis of Melanoma Cells by Altering a Transcriptional Profile. Int. J. Mol. Sci. 2021, 22, 2219. https://doi.org/10.3390/ijms22042219
Wang N, Zhang H, Cui X, Ma C, Wang L, Liu W. Runx3 Induces a Cell Shape Change and Suppresses Migration and Metastasis of Melanoma Cells by Altering a Transcriptional Profile. International Journal of Molecular Sciences. 2021; 22(4):2219. https://doi.org/10.3390/ijms22042219
Chicago/Turabian StyleWang, Ning, Haiying Zhang, Xiulin Cui, Chao Ma, Linghui Wang, and Wenguang Liu. 2021. "Runx3 Induces a Cell Shape Change and Suppresses Migration and Metastasis of Melanoma Cells by Altering a Transcriptional Profile" International Journal of Molecular Sciences 22, no. 4: 2219. https://doi.org/10.3390/ijms22042219
APA StyleWang, N., Zhang, H., Cui, X., Ma, C., Wang, L., & Liu, W. (2021). Runx3 Induces a Cell Shape Change and Suppresses Migration and Metastasis of Melanoma Cells by Altering a Transcriptional Profile. International Journal of Molecular Sciences, 22(4), 2219. https://doi.org/10.3390/ijms22042219