
New 3-Aryl-2-(2-thienyl)acrylonitriles with High Activity Against Hepatoma Cells

 , ,
, ,  , and
, and 
Abstract
:
1. Introduction
2. Results
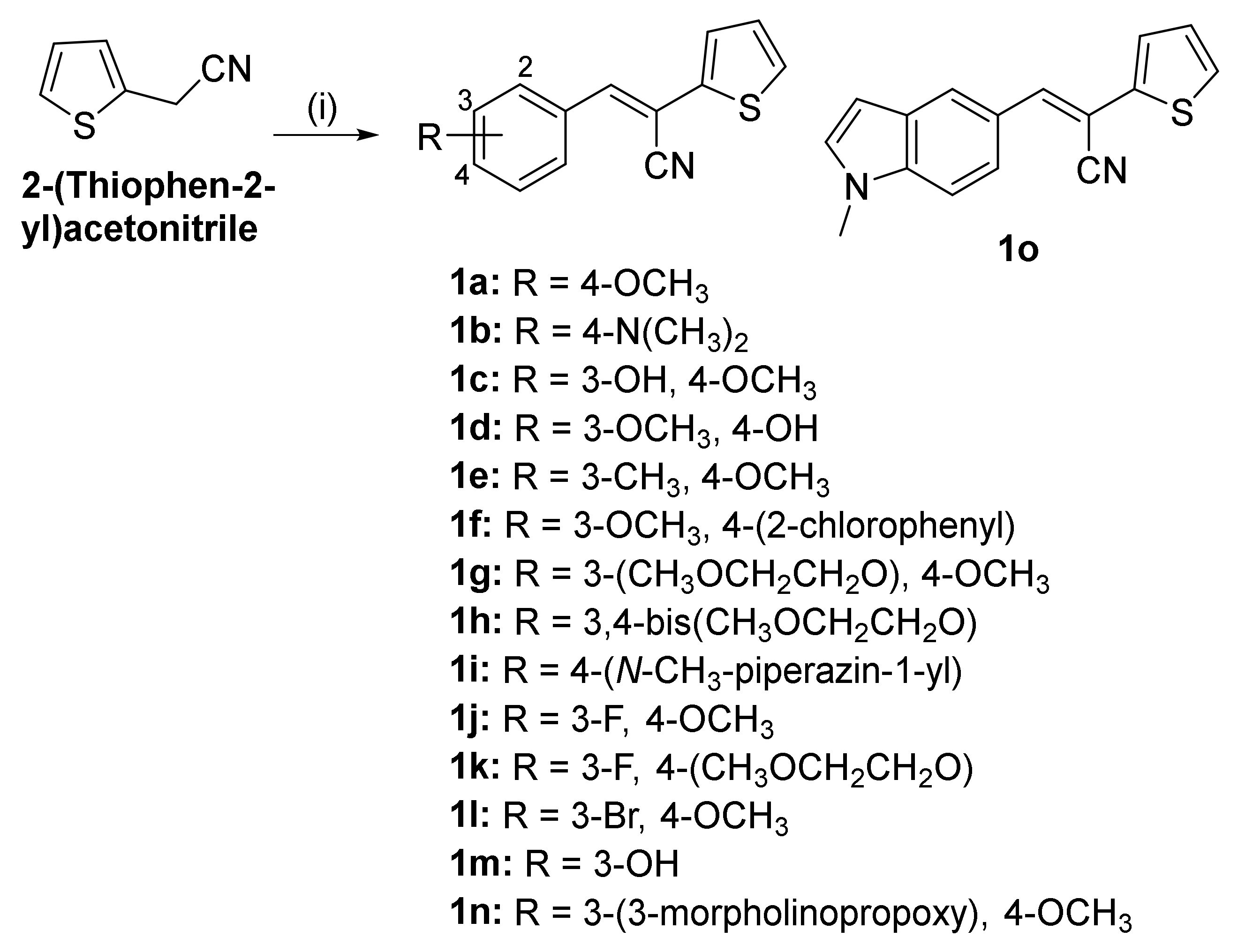
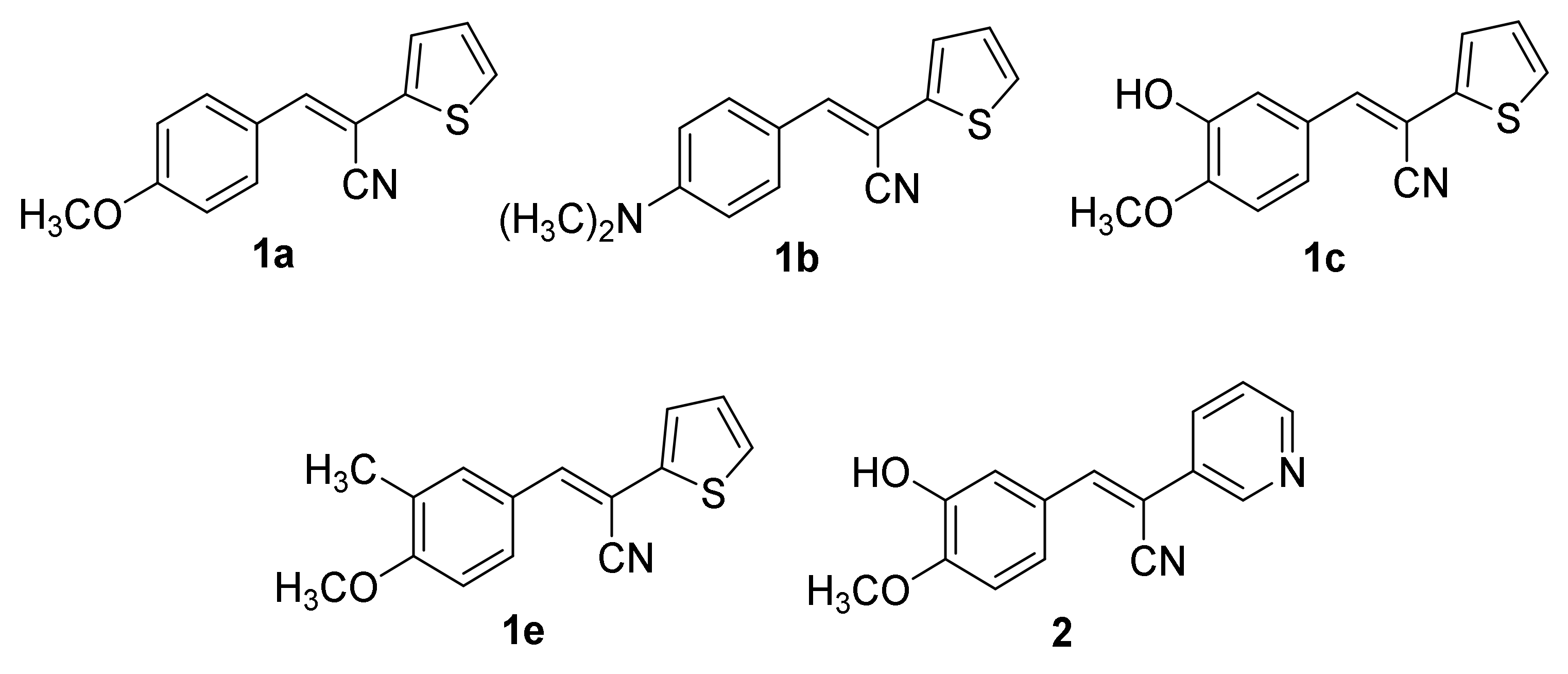
2.1. Chemistry
2.2. Biological Evaluation
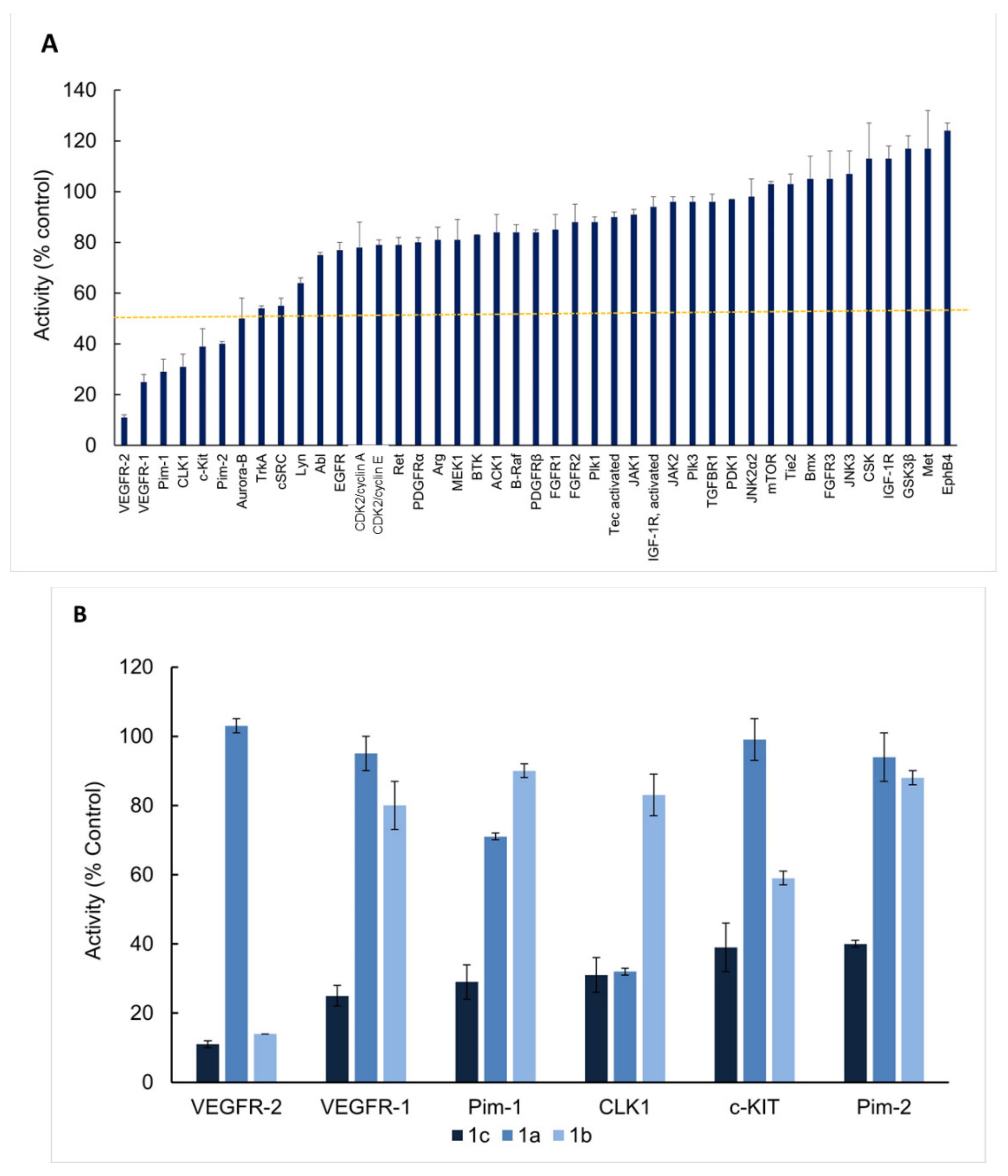
2.3. Enzymatic Kinase Assays
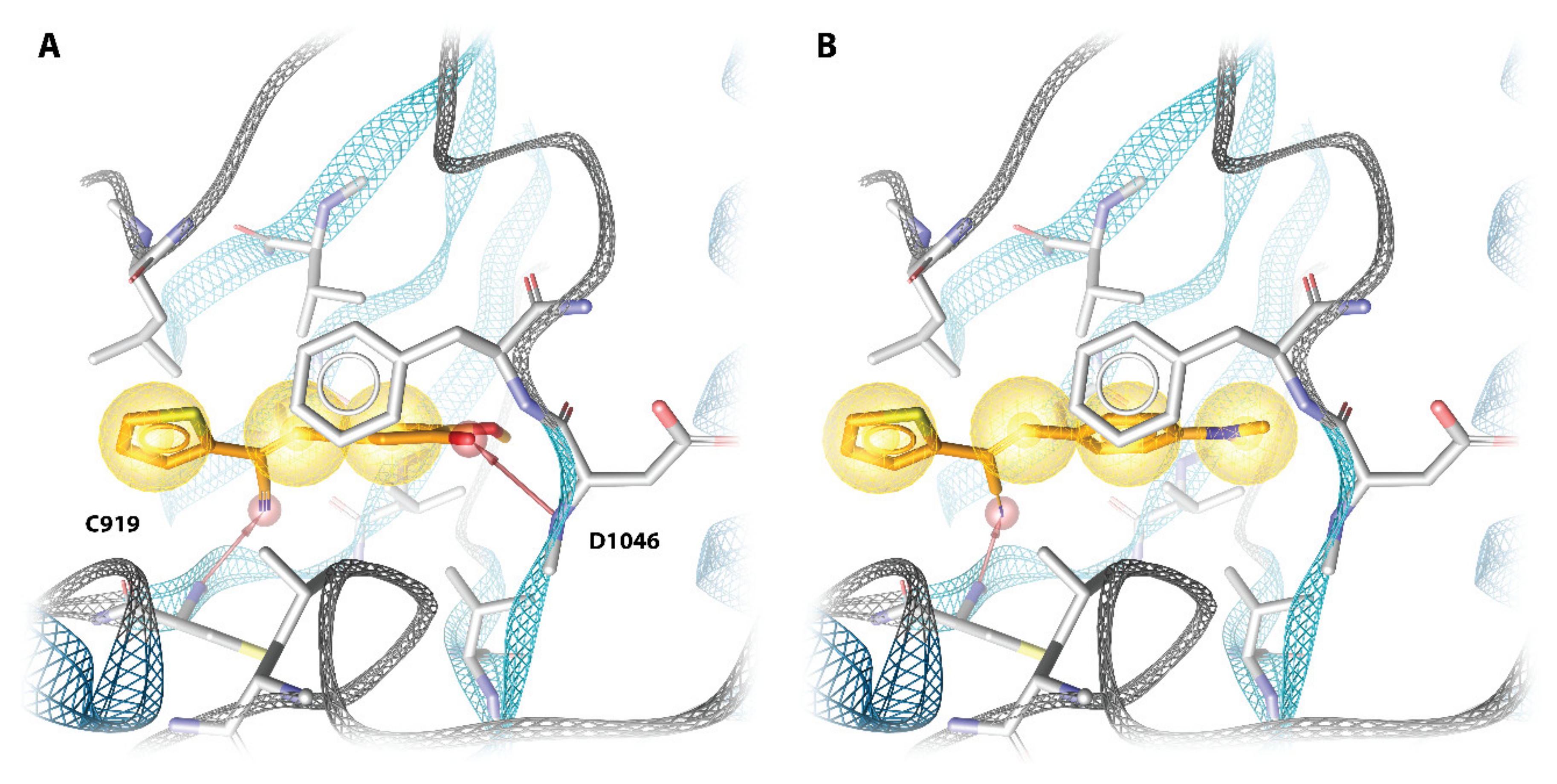
2.4. Molecular Modeling
3. Discussion
4. Materials and Methods
4.1. General Procedures
4.2. Materials
4.3. Synthesis of Compounds 1c–o
4.4. Biological Evaluations
4.4.1. Cell Lines and Culture Conditions
4.4.2. Determination of Growth Inhibition
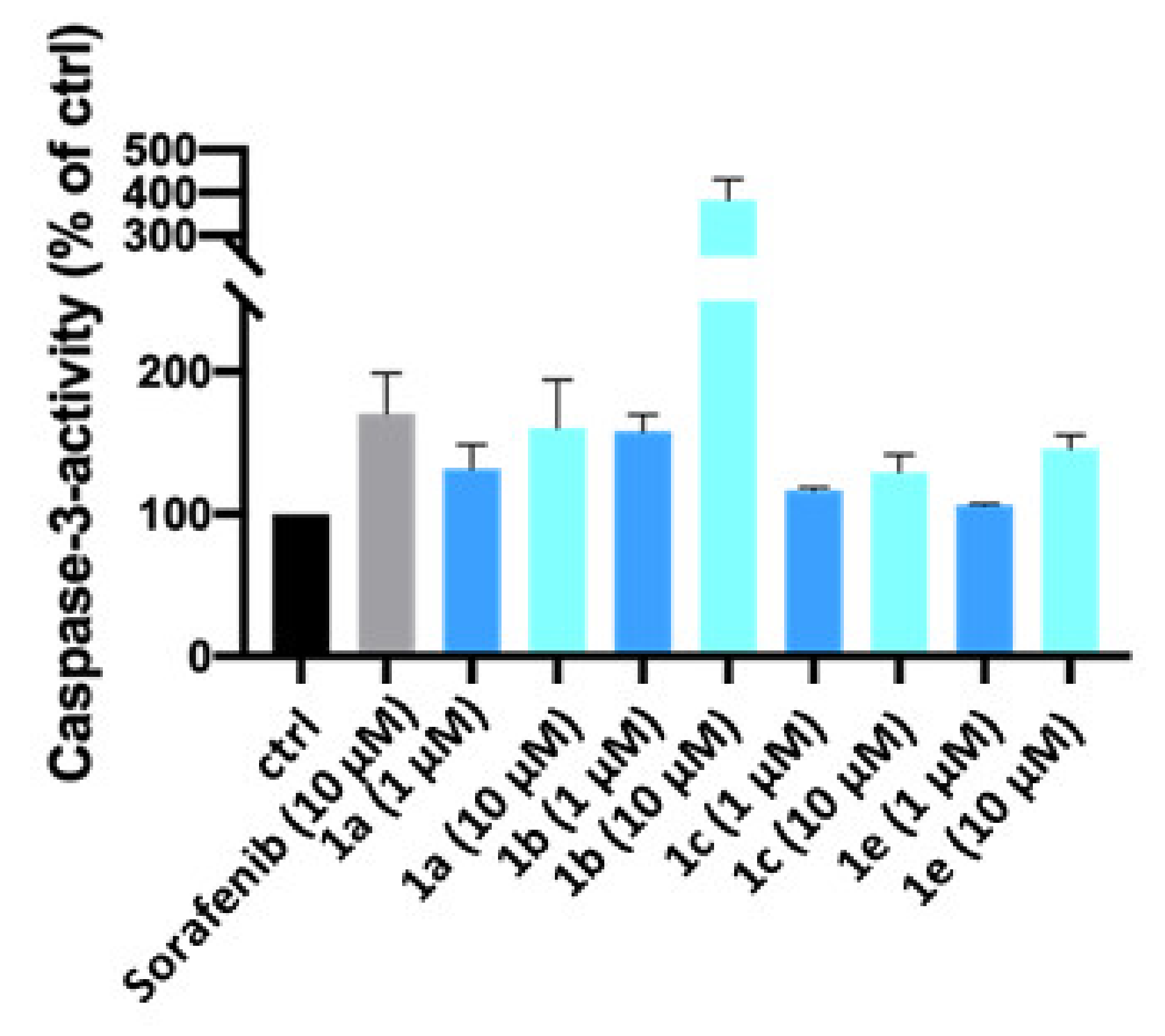
4.4.3. Measurement of Apoptosis-Specific Caspase-3 Activity
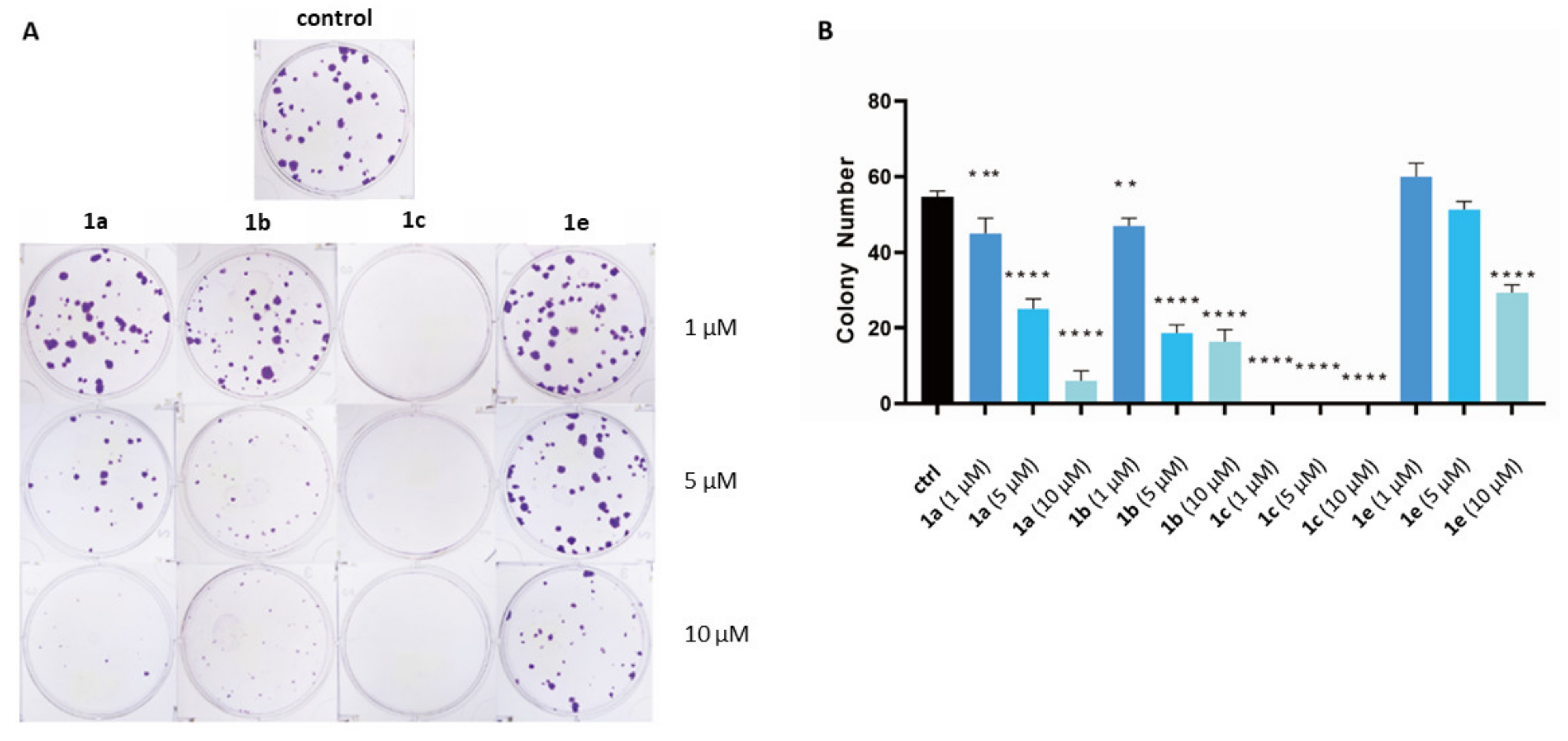
4.4.4. Colony-Formation Assay
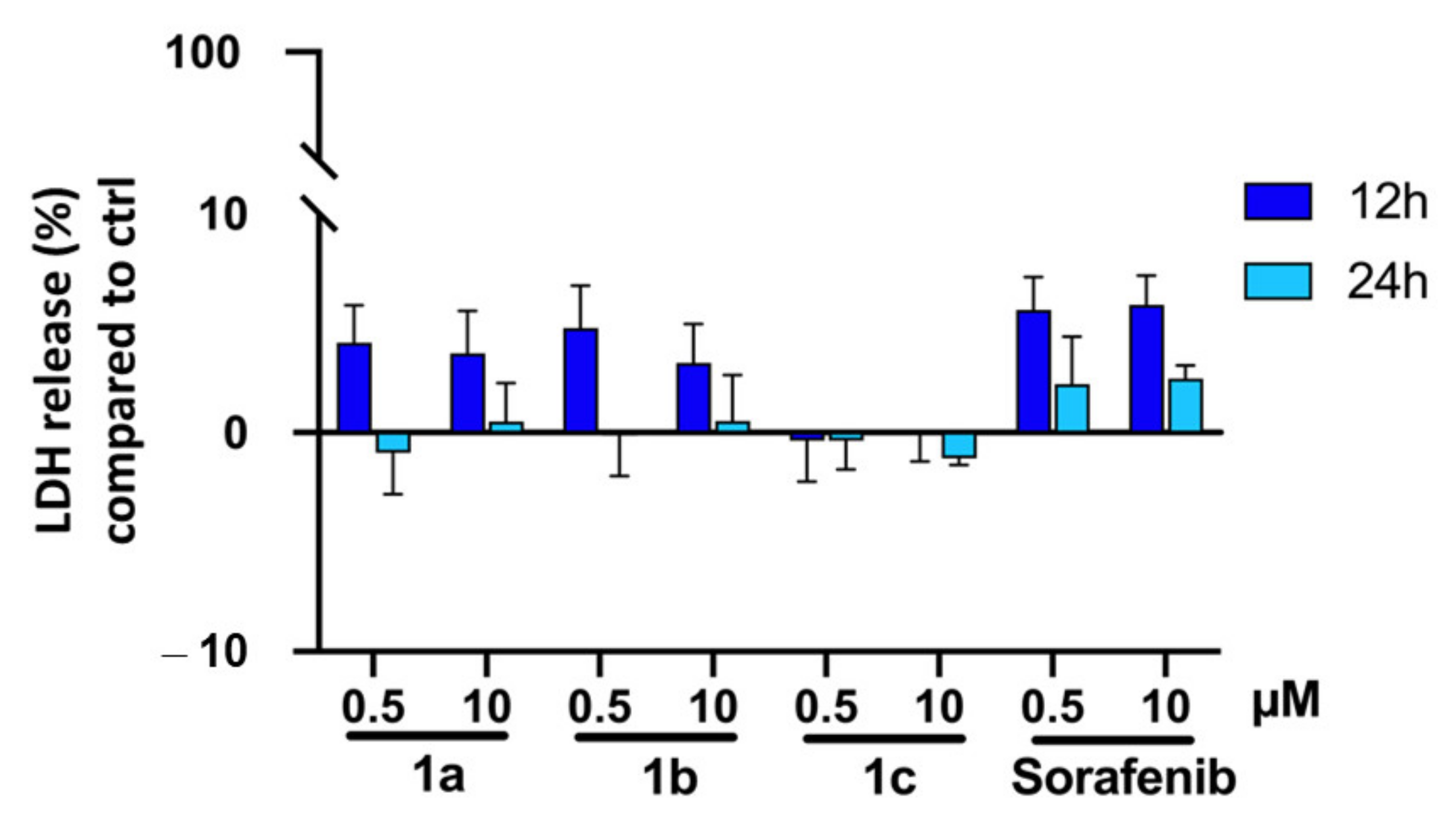
4.4.5. LDH Assay
4.4.6. Enzymatic Kinase Assay
4.4.7. In Vivo Evaluation of Antineoplastic Effects Using the Chorioallantoic Membrane (CAM) Assay
4.5. Computational Evaluation
4.5.1. Structure Selection
4.5.2. Molecular Docking
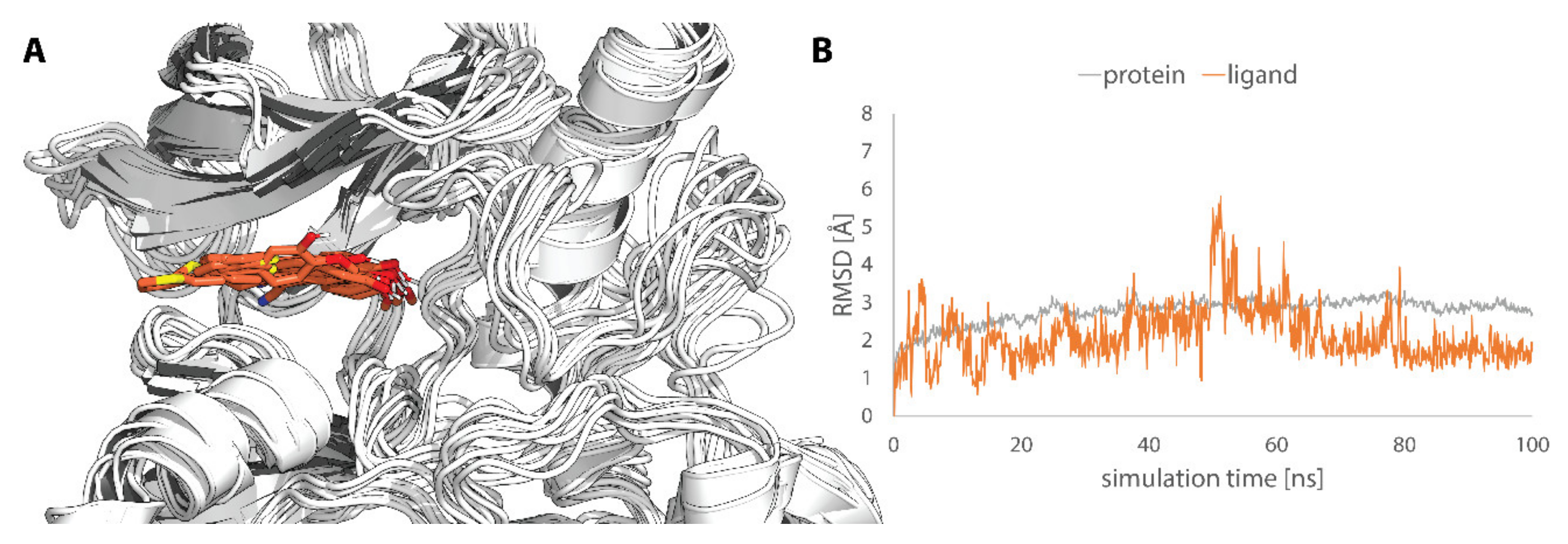
4.5.3. Molecular Dynamics Simulation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAM | Chorioallantoic membrane |
| CLK1 | Cdc2-like kinase-1 |
| EGFR | Epidermal growth factor receptor |
| FGFR | Fibroblast growth factor receptor |
| GIST | Gastrointestinal stromal tumor |
| HCC | Hepatocellular carcinoma |
| IGF-1R | Insulin-like growth factor 1-receptor |
| LDH | Lactate dehydrogenase |
| PDGFR | Platelet-derived growth factor receptor |
| Pim | Proviral integration site for Moloney murine leukemia virus |
| RCC | Renal cell carcinoma |
| VEGFR | Vascular endothelial growth factor receptor |
References
- López-Terrada, D.; Cheung, S.W.; Finegold, M.J.; Knowles, B.B. Hep G2 is a hepatoblastoma-derived cell line. Hum. Pathol. 2009, 40, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Thomas, M.B.; Zhu, A.X. Hepatocellular carcinoma: The need for progress. J. Clin. Oncol. 2005, 23, 2892–2899. [Google Scholar] [CrossRef]
- Cidon, E.U. Systemic treatment of hepatocellular carcinoma: Past, present and future. World J. Hepatol. 2017, 9, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Ganten, T.M.; Stauber, R.E.; Schott, E.; Malfertheiner, P.; Buder, R.; Galle, P.R.; Gohler, T.; Walther, M.; Koschny, R.; Gerken, G. Sorafenib in patients with hepatocellular carcinoma—Results of the observational INSIGHT study. Clin. Cancer Res. 2017, 23, 5720–5728. [Google Scholar] [CrossRef] [Green Version]
- Frenette, C. The role of regorafenib in hepatocellular carcinoma. Gastroenterol. Hepatol. 2017, 13, 122–124. [Google Scholar] [PubMed]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; Román-Gil, M.S.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine kinase receptors in oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef] [PubMed]
- Giannini, E.; Farinati, F.; Ciccarese, F.; Pecorelli, A.; Rapaccini, G.; Di Marco, M.; Benvegnu, L.; Caturelli, E.; Zoli, M.; Borzio, F.; et al. Prognosis of untreated hepatocellular carcinoma. Hepatology 2015, 61, 184–190. [Google Scholar] [CrossRef]
- Dua, R.; Shrivastava, S.; Sonwane, S.K.; Srivastava, S.K. Pharmacological significance of synthetic heterocycles scaffold: A review. Adv. Biol. Res. 2011, 5, 120–144. [Google Scholar]
- Mishra, R.; Jha, K.K.; Kumar, S.; Tomer, I. Synthesis, properties and biological activity of thiophene: A review. Der Pharma Chem. 2011, 3, 38–54. [Google Scholar]
- Karunakaran, J.; Moorthy, N.D.; Chowdhury, S.R.; Iqbal, S.; Majumder, H.K.; Gunasekaran, K.; Vellaichamy, E.; Mohanakrishnan, A.K. Divergent synthesis and evaluation of the in vitro cytotoxicity profiles of 3,4-ethylenedioxythiophenyl-2-propen-1-one analogues. ChemMedChem 2019, 14, 1418–1430. [Google Scholar] [CrossRef]
- Amawi, H.; Karthikeyan, C.; Pathak, R.; Hussein, N.; Christman, R.; Robey, R.; Ashby Jr., C. R.; Trivedi, P.; Malhotra, A.; Tiwari, A.K. Thienopyrimidine derivatives exert their anticancer efficacy via apoptosis induction, oxidative stress and mitotic catastrophe. Eur. J. Med. Chem. 2017, 138, 1053–1065. [Google Scholar] [CrossRef]
- Gururaja, T.L.; Goff, D.; Kinoshita, T.; Goldstein, E.; Yung, S.; McLaughlin, J.; Pali, E.; Huang, J.; Singh, R.; Daniel-Issakani, S.; et al. R-253 disrupts microtubule networks in multiple tumor cell lines. Clin. Cancer Res. 2006, 12, 3831–3842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, S.U.F.; Siddiqui, H.L.; Nisar, M.; Khan, N.; Khan, I. Discovery and molecular docking of quinolyl-thienyl chalcones as anti-angiogenic agents targeting VEGFR-2 tyrosine kinase. Bioorg. Med. Chem. Lett. 2012, 22, 942–944. [Google Scholar] [CrossRef]
- Khare, R.; Pandey, J.; Smriti; Rupanwar, R. The importance and applications of Knoevenagel reaction. Orient. J. Chem. 2019, 35, 423–429. [Google Scholar] [CrossRef]
- Loupy, A.; Pellet, M.; Petit, A.; Vo-Thanh, G. Solvent-free condensation of phenylacetonitrile and nonanenitrile with 4-methoxybenzaldehyde: Optimization and mechanistic studies. Org. Biomol. Chem. 2005, 3, 1534–1540. [Google Scholar] [CrossRef]
- Yoneda, T.; Lyall, R.M.; Alsina, M.M.; Persons, P.E.; Spada, A.P.; Levitzki, A.; Zilberstein, A.; Mundy, G.R. The antiproliferative effects of tyrosine kinase inhibitors tyrphostins on a human squamous cell carcinoma in vitro and in nude mice. Cancer Res. 1991, 51, 4430–4435. [Google Scholar] [PubMed]
- Biersack, B.; Zoldakova, M.; Effenberger, K.; Schobert, R. (Arene)Ru(II) complexes of epidermal growth factor receptor inhibiting tyrphostins with enhanced selectivity and cytotoxicity in cancer cells. Eur. J. Med. Chem. 2010, 45, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Nam, Y.-J.; Lee, D.-U. Synthesis and evaluation of (Z)-2,3-diphenylacrylonitrile analogs as anti-cancer and anti-microbial agents. Eur. J. Med. Chem. 2013, 69, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Tarleton, M.; Gilbert, J.; Sakoff, J.A.; McCluskey, A. Cytotoxic 2-phenyacrylnitriles, the importance of the cyanide moiety and discovery of potent broad spectrum cytotoxic agents. Eur. J. Med. Chem. 2012, 57, 65–73. [Google Scholar] [CrossRef]
- Madadi, N.R.; Ketkar, A.; Penthala, N.R.; Bostian, A.C.L.; Eoff, R.L.; Crooks, P.A. Dioxol and dihydrodioxin analogs of 2- and 3-phenylacetonitriles as potent anti-cancer agents with nanomolar activity against a variety of human cancer cells. Bioorg. Med. Chem. Lett. 2016, 26, 2164–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tcherniuk, S.; Skoufias, D.A.; Labriere, C.; Rath, O.; Gueritte, F.; Guillou, C.; Kozielski, F. Relocation of aurora B and survivin from centromeres to the central spindle impaired by a kinesin-specific MKLP-2 inhibitor. Angew. Chem. Int. Ed. 2010, 49, 8228–8231. [Google Scholar] [CrossRef] [PubMed]
- Penthala, N.R.; Janganati, V.; Bommagani, S.; Crooks, P.A. Synthesis and evaluation of a series of quinolinyl trans-cyanostilbene analogs as anticancer agents. Med. Chem. Commun. 2014, 5, 886–890. [Google Scholar] [CrossRef]
- Tarleton, M.; Gilbert, J.; Robertson, M.J.; McCluskey, A.; Sakoff, J.A. Library synthesis and cytotoxicity of a family of 2-phenylacrylonitriles and discovery of an estrogen dependent breast cancer lead compound. Med. Chem. Commun. 2011, 2, 31–37. [Google Scholar] [CrossRef]
- Carta, A.; Briguglio, I.; Piras, S.; Boatto, G.; La Colla, P.; Loddo, R.; Tolomeno, M.; Grimaudo, S.; Di Cristina, A.; Pipitone, R.M.; et al. 3-Aryl-2-[1H-benzotriazol-1-yl]acrylonitriles: A novel class of potent tubulin inhibitors. Eur. J. Med. Chem. 2011, 46, 4151–4167. [Google Scholar] [CrossRef]
- Buu-Hoi, N.P.; Hoan, N.; Lavit, D. Thiophen derivatives of potential biological interest. Part I. Thiophen analogues of stilbene and of related compounds. J. Chem. Soc. 1951, 251–255. [Google Scholar] [CrossRef]
- Yamazaki, R.; Nishiyama, Y.; Furuta, T.; Hatano, H.; Igarashi, Y.; Asakawa, N.; Kodaira, H.; Takahashi, H.; Aiyama, R.; Matsuzaki, T.; et al. Novel acrylonitrile derivatives, YHO-13177 and YHO-13351, reverse BCRP/ABCG2-mediated drug resistance in vitro and in vivo. Mol. Cancer Ther. 2011, 10, 1252–1263. [Google Scholar] [CrossRef] [Green Version]
- Quiroga, J.; Cobo, D.; Insuasty, B.; Abonía, R.; Nogueras, M.; Cobo, J.; Vásquez, Y.; Gupta, M.; Derita, M.; Zacchino, S. Synthesis and evaluation of novel E-2-(2-thienyl)- and Z-2-(3-thienyl)-3-arylacrylonitriles as antifungal and anticancer agents. Arch. Pharm. Chem. Life Sci. 2007, 340, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Potter, G.A.; Patterson, L.H.; Burke, M.D.; Butler, P.C. Hydroxylation Activated Prodrugs. WO 99/40056, 12 August 1999. [Google Scholar]
- Li, L.; Wang, L.; Song, P.; Geng, X.; Liang, X.; Zhou, M.; Wang, Y.; Chen, C.; Jia, J.; Zeng, J. Critical role of histone demethylase RBP2 in human gastric cancer angiogenesis. Mol. Cancer 2014, 13, 81. [Google Scholar] [CrossRef] [Green Version]
- Modi, V.; Dunbrack, L., Jr. Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. USA 2019, 116, 6818–6827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, J.L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef] [Green Version]
- Oguro, Y.; Miyamoto, N.; Okada, K.; Takagi, T.; Iwata, H.; Awazu, Y.; Miki, H.; Hori, A.; Kamiyama, K.; Imamura, S. Design, synthesis, and evaluation of 5-methyl-4-phenoxy-5H-pyrrolo[3,2-d]pyrimidine derivatives: Novel VEGFR2 kinase inhibitors binding to inactive kinase conformation. Bioorg. Med. Chem. 2010, 18, 7260–7273. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. JCC 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. Ligand Scout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Höpfner, M.; Sutter, A.P.; Huether, A.; Schuppan, D.; Zeitz, M.; Scherübl, H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J. Hepatol. 2004, 41, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Fernando, J.; Sancho, P.; Fernández-Rodriguez, C.M.; Lledó, J.L.; Caja, L.; Campbell, J.S.; Fausto, N.; Fabregat, I. Sorafenib sensitizes hepatocellular carcinoma cells to physiological apoptotic stimuli. J. Cell. Physiol. 2012, 227, 1319–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivy, S.P.; Wick, J.Y.; Kaufman, B.M. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 2009, 6, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, L.-J.; Jiang, B.; Wu, N.; Li, X.; Liu, S.; Luo, J.; Shi, D. Anti-angiogenic properties of BDDPM, a bromophenol from marine red alga Rhodomela confervoides, with multi receptor tyrosine kinase inhibition effects. Int. J. Mol. Sci. 2015, 16, 13548–13560. [Google Scholar] [CrossRef] [Green Version]
- Sangande, F.; Julianti, E.; Tjahjono, D.H. Ligand-based pharmacophore modeling, molecular docking, and molecular dynamic studies of dual tyrosine kinase inhibitor of EGFR and VEGFR2. Int. J. Mol. Sci. 2020, 21, 779. [Google Scholar] [CrossRef]
- El-Adil, K.; Ibrahim, M.-K.; Khedr, F.; Abulkhair, H.S.; Eissa, I.H. N-Substituted-4-phenylphthalazin-1-amine-derived VEGFR-2 inhibitors: Design, synthesis, molecular docking, and anticancer evaluation studies. Arch. Pharm. 2020, e2000219. [Google Scholar] [CrossRef]
- Yan, W.; Zhu, Z.; Pan, F.; Huang, A.; Dai, G. Overexpression of c-kit(CD117), relevant with microvessel density, is an independent survival prognostic factor for patients with HBV-related hepatocellular carcinoma. OncoTargets Ther. 2018, 11, 1285–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tursynbay, Y.; Zhang, J.; Li, Z.; Tokay, T.; Zhumadilov, Z.; Wu, D.; Xie, Y. Pim-1 kinase as cancer drug target: An update. Biomed. Rep. 2016, 4, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Boss, C.; Fischli, W.; Meyer, S.; Richard-Bildstein, S.; Weller, T. Substituted Alkyldiamines. US20040067927A1, 8 April 2001. [Google Scholar]
- Boobalan, R.; Liu, K.-K.; Chao, J.-I.; Chen, C. Synthesis and biological assay of erlotinib analogues and BSA-conjugated erlotinib analogue. Bioorg. Med. Chem. Lett. 2017, 27, 1784–1788. [Google Scholar] [CrossRef] [PubMed]
- Saglik, B.N.; Ilgin, S.; Özkay, Y. Synthesis of new donepezil analogues and investigation of their effects on cholinesterase enzymes. Eur. J. Med. Chem. 2016, 124, 1026–1040. [Google Scholar] [CrossRef]
- Epifano, F.; Sosa, S.; Tubaro, A.; Marcotullio, M.C.; Curini, M.; Genovese, S. Topical anti-inflammatory activity of boropinic acid and its natural and semi-synthetic derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Chandregowda, V.; Rao, G.V.; Reddy, G.C. One-pot conversion 2-nitrobenzonitriles to quinazolin-4(3H)-ones and synthesis of gefitinib and erlotinib hydrochloride. Heterocycles 2007, 71, 39–48. [Google Scholar] [CrossRef]
- Höpfner, M.; Huether, A.; Sutter, A.P.; Baradari, V.; Schuppan, D.; Scherübl, H. Blockade of IGF-1 receptor tyrosine kinase has antineoplastic effects in hepatocellular carcinoma cells. Biochem. Pharmacol. 2006, 71, 1435–1448. [Google Scholar] [CrossRef]
- Steinemann, G.; Dittmer, A.; Kuzyniak, W.; Hoffmann, B.; Schrader, M.; Schobert, R.; Biersack, B.; Nitzsche, B.; Höpfner, M. Animacroxam, a novel dual-mode compound targeting histone deacetylases and cytoskeletal integrity of testicular germ cell cancer cells. Mol. Cancer Ther. 2017, 16, 2364–2374. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.; Kuzyniak, W.; Berkholz, J.; Steinemann, G.; Ogbodu, R.; Hoffmann, B.; Nouailles, G.; Gürek, A.; Nitzsche, B.; Höpfner, M. Novel zinc- and silicon-phthalocyanines as photosensitizers for photodynamic therapy of cholangiocarcinoma. Int. J. Mol. Med. 2018, 42, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Schomburg, K.T.; Bietz, S.; Briem, H.; Henzler, A.M.; Urbaczek, S.; Rarey, M. Facing the Challenges of Structure-Based Target Prediction by Inverse Virtual Screening. J. Chem. Inf. Model. 2014, 54, 1676–1686. [Google Scholar] [CrossRef]
- Pons, J.; Labesse, G. @TOME-2: A new pipeline for comparative modeling of protein-ligand complexes. Nucleic Acids Res. 2009, 37, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eurofins Kinase & Profiling Service. Available online: https://www.eurofinsdiscoveryservices.com/services/in-vitro-assays/kinases/screening-profiling-services/ (accessed on 28 January 2021).
- Gloesenkamp, C.; Nitzsche, B.; Lim, A.R.; Normant, E.; Vosburgh, E.; Schrader, M.; Ocker, M.; Scherübl, H.; Höpfner, M. Heat shock protein 90 is a promising target for effective growth inhibition of gastrointestinal neuroendocrine tumors. Int. J. Oncol. 2012, 40, 1659–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanev, G.K.; de Graaf, C.; Westerman, B.A.; de Esch, I.J.P.; Kooistra, A.J. KLIFS: An overhaul after the first 5 years of supporting kinase research. Nucleic Acids Res. 2021, 49, D562–D569. [Google Scholar] [CrossRef]
- Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey, M.A. A consistent description of HYdrogen bond and DEhydration energies in protein-ligand complexes: Methods behind the HYDE scoring function. J. Comput. Aided Mol. Des. 2013, 27, 15–29. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | HepG2 | Huh-7 | AML-12 |
|---|---|---|---|
| 1a | 1.46 ± 0.21 | 1.2 ± 0.42 | 6.79 ± 0.32 |
| 1b | 0.72 ± 0.09 | 0.67 ± 0.13 | 2.72 ± 0.78 |
| 1c | 0.55 ± 0.26 | 0.32 ± 0.15 | 1.81 ± 0.31 |
| 1d | >20 | >20 | - |
| 1e | 1.30 ± 0.17 | 1.85 ± 0.21 | 14.78 ± 2.39 |
| 1f | >20 | >20 | - |
| 1g | >20 | >20 | - |
| 1h | >20 | >20 | - |
| 1i | 9.30 ± 0.82 | 11.79 ± 0.56 | 20.53 ± 1.25 |
| 1j | 4.90 ± 1.54 | 5.70 ± 1.20 | 9.42 ± 0.91 |
| 1k | >20 | >20 | - |
| 1l | 13.87 ± 1.16 | - | - |
| 1m | >20 | >20 | - |
| 1n | >20 | >20 | - |
| 1o | >20 | >20 | - |
| 2 | 6.26 ± 1.18 | - | - |
| Sorafenib | 2.73 ± 0.76 | 2.50 ± 0.14 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaller, E.; Ma, A.; Gosch, L.C.; Klefenz, A.; Schaller, D.; Goehringer, N.; Kaps, L.; Schuppan, D.; Volkamer, A.; Schobert, R.; et al. New 3-Aryl-2-(2-thienyl)acrylonitriles with High Activity Against Hepatoma Cells. Int. J. Mol. Sci. 2021, 22, 2243. https://doi.org/10.3390/ijms22052243
Schaller E, Ma A, Gosch LC, Klefenz A, Schaller D, Goehringer N, Kaps L, Schuppan D, Volkamer A, Schobert R, et al. New 3-Aryl-2-(2-thienyl)acrylonitriles with High Activity Against Hepatoma Cells. International Journal of Molecular Sciences. 2021; 22(5):2243. https://doi.org/10.3390/ijms22052243
Chicago/Turabian StyleSchaller, Eva, Andi Ma, Lisa Chiara Gosch, Adrian Klefenz, David Schaller, Nils Goehringer, Leonard Kaps, Detlef Schuppan, Andrea Volkamer, Rainer Schobert, and et al. 2021. "New 3-Aryl-2-(2-thienyl)acrylonitriles with High Activity Against Hepatoma Cells" International Journal of Molecular Sciences 22, no. 5: 2243. https://doi.org/10.3390/ijms22052243
APA StyleSchaller, E., Ma, A., Gosch, L. C., Klefenz, A., Schaller, D., Goehringer, N., Kaps, L., Schuppan, D., Volkamer, A., Schobert, R., Biersack, B., Nitzsche, B., & Höpfner, M. (2021). New 3-Aryl-2-(2-thienyl)acrylonitriles with High Activity Against Hepatoma Cells. International Journal of Molecular Sciences, 22(5), 2243. https://doi.org/10.3390/ijms22052243