
Selective Cardiomyocyte Oxidative Stress Leads to Bystander Senescence of Cardiac Stromal Cells

 , ,
, ,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
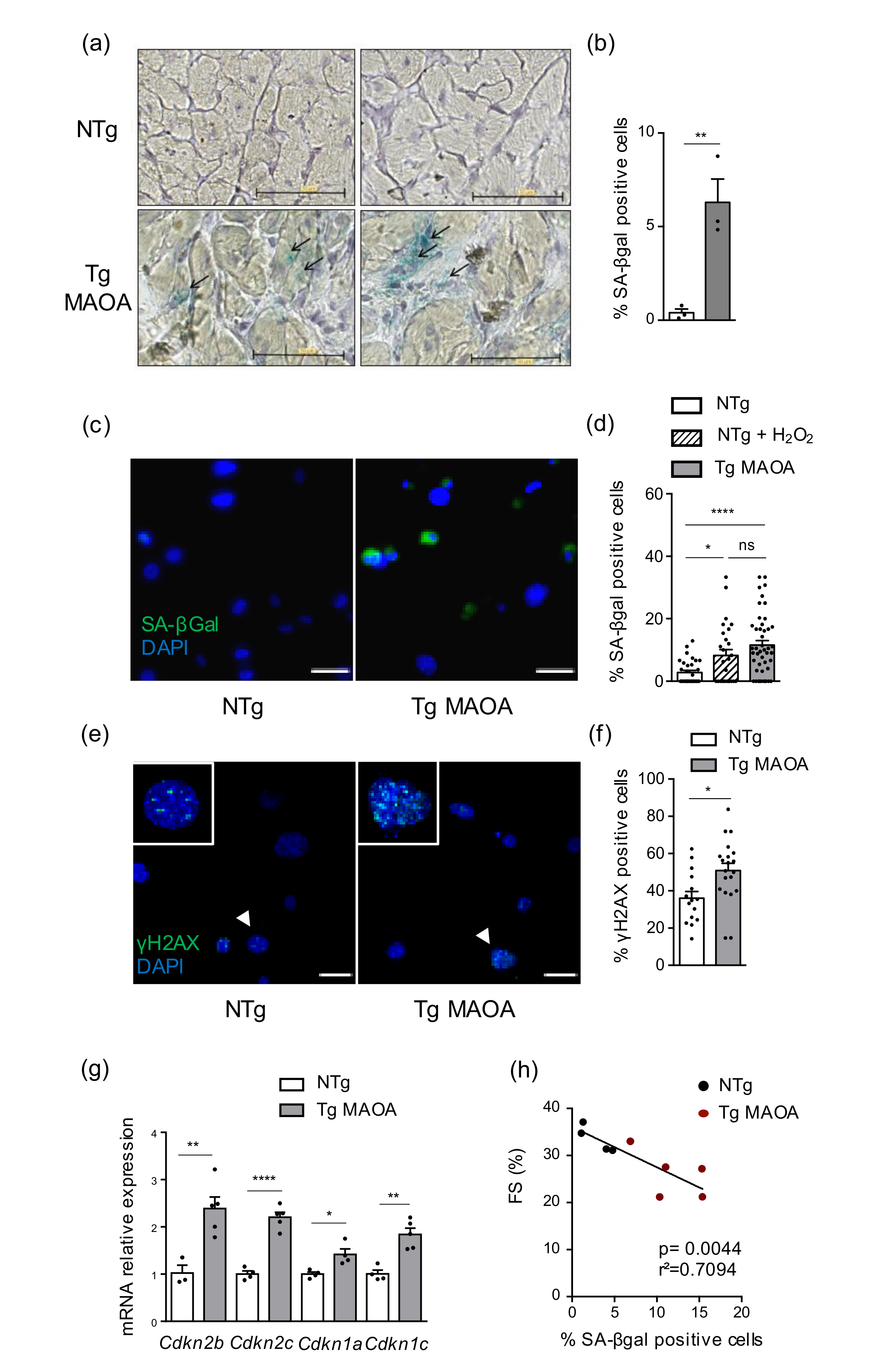
2.1. MAOA Dependent Oxidative Stress in Cardiomyocytes Triggers Stress-Induced Premature Senescence in Cardiac Stromal Cells
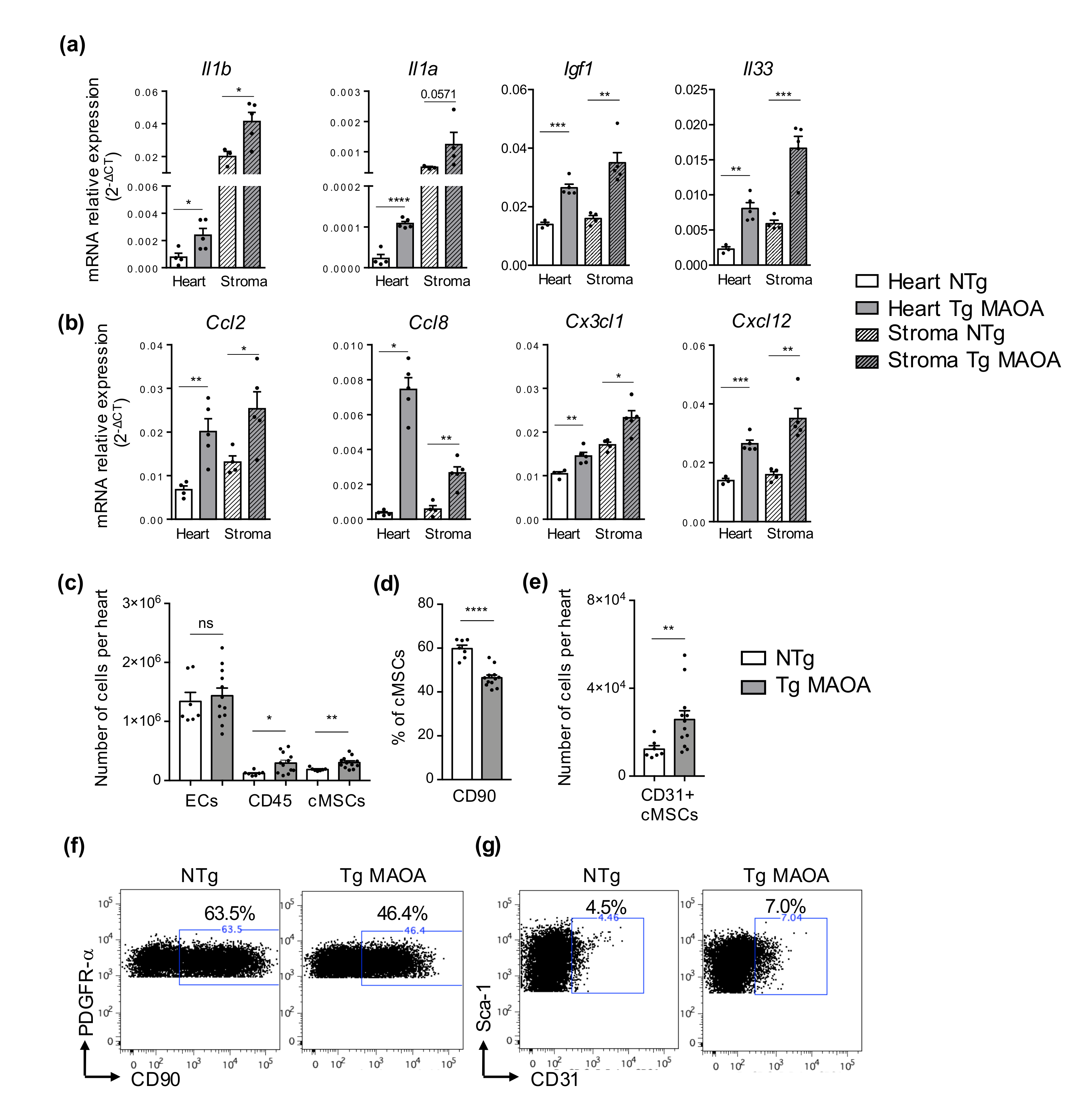
2.2. SIPS of Cardiac Stromal Cells in Tg MAOA Mice Increased Expression of SASP Factors and Is Associated to Modification of Stromal Cell Subsets
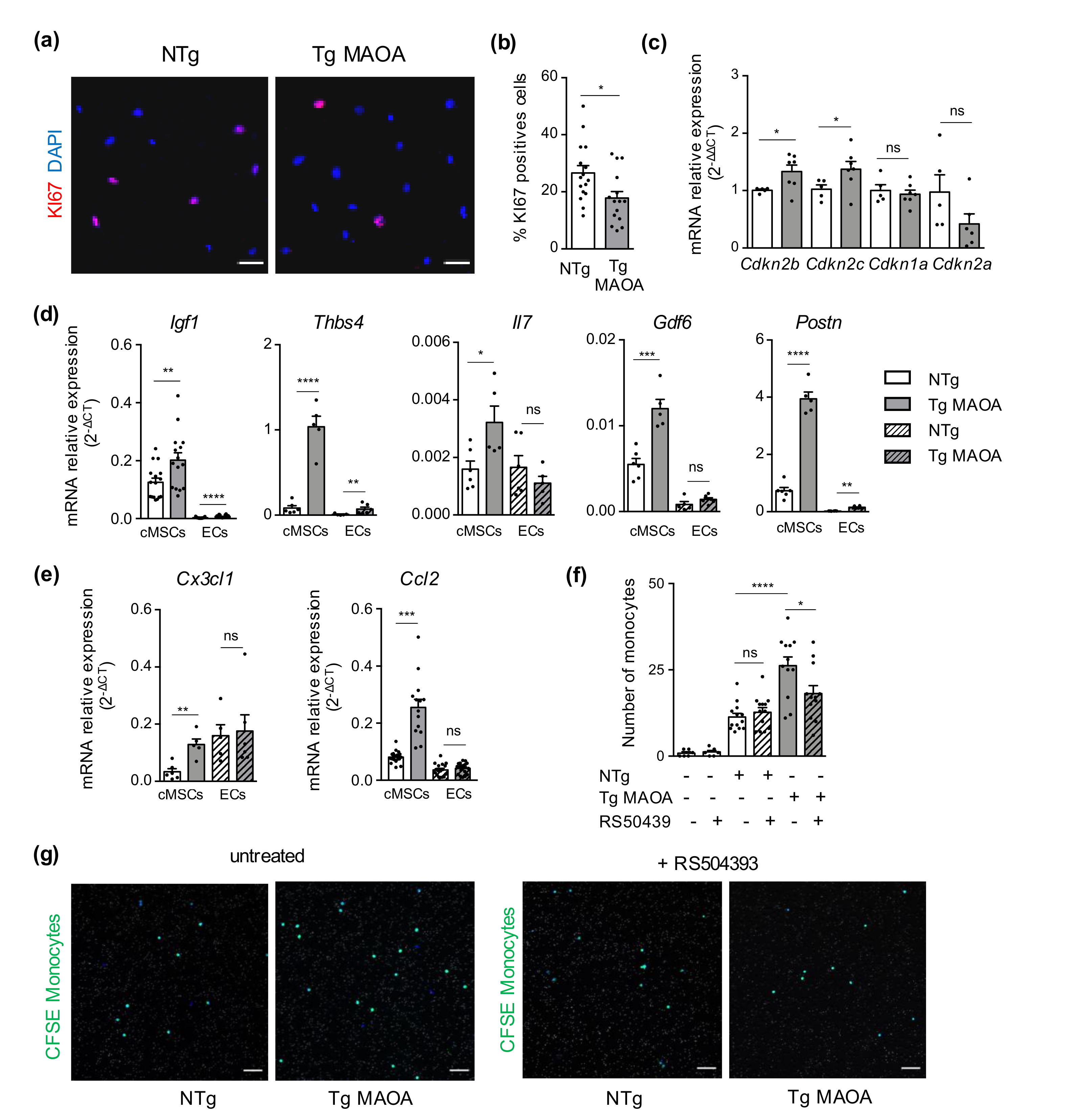
2.3. Oxidative Stress-Induced Premature Senescence of Cardiac Mesenchymal Stromal Cells Shares Some Common Features with Physiological Aging-Associated Senescence
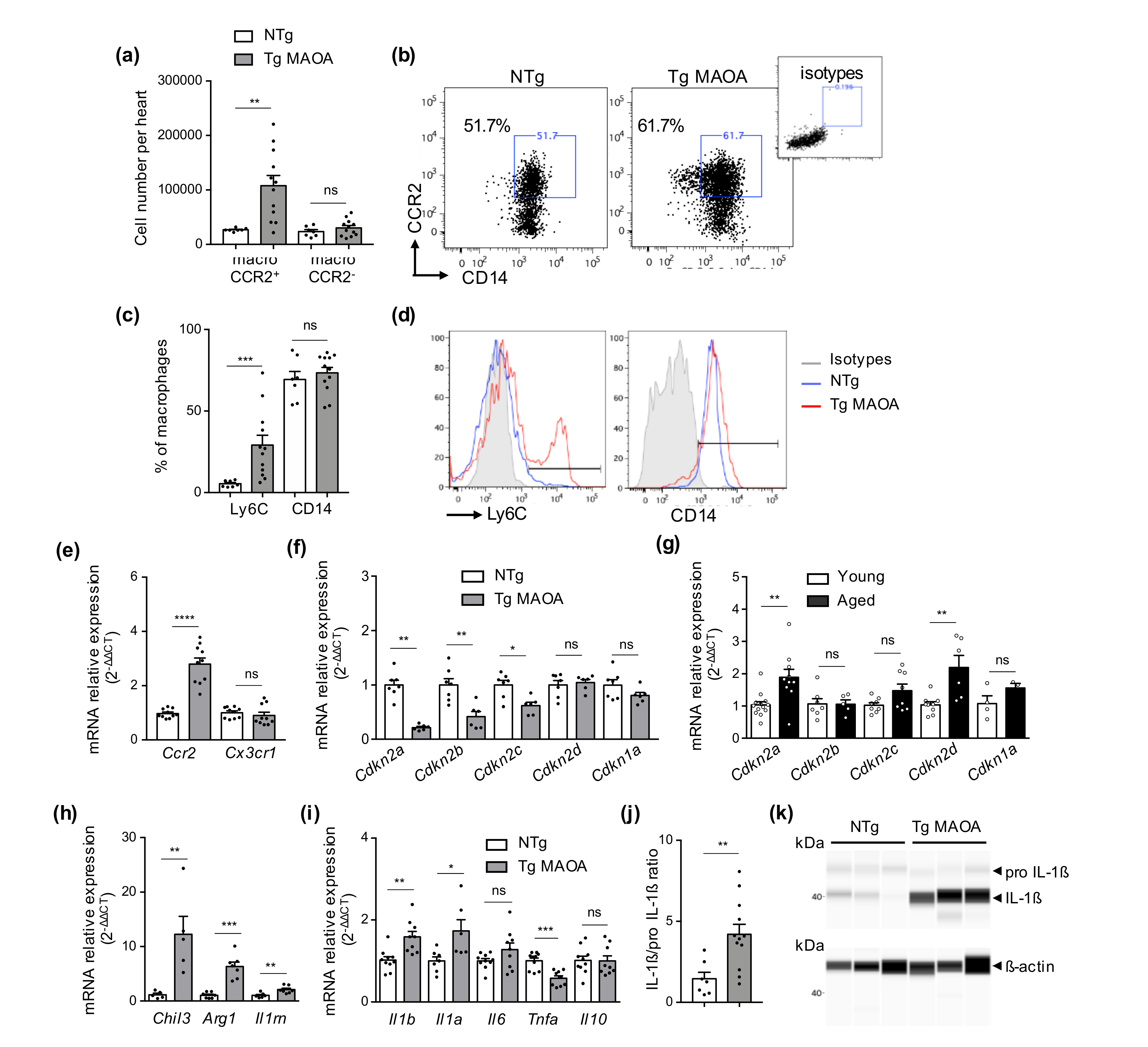
2.4. Oxidative Stress-Derived Cardiomyocytes and SIPS of Cardiac Stromal Cells Promote CCR2+ Cardiac Macrophages and Expression of Cytokines of the IL-1 Family
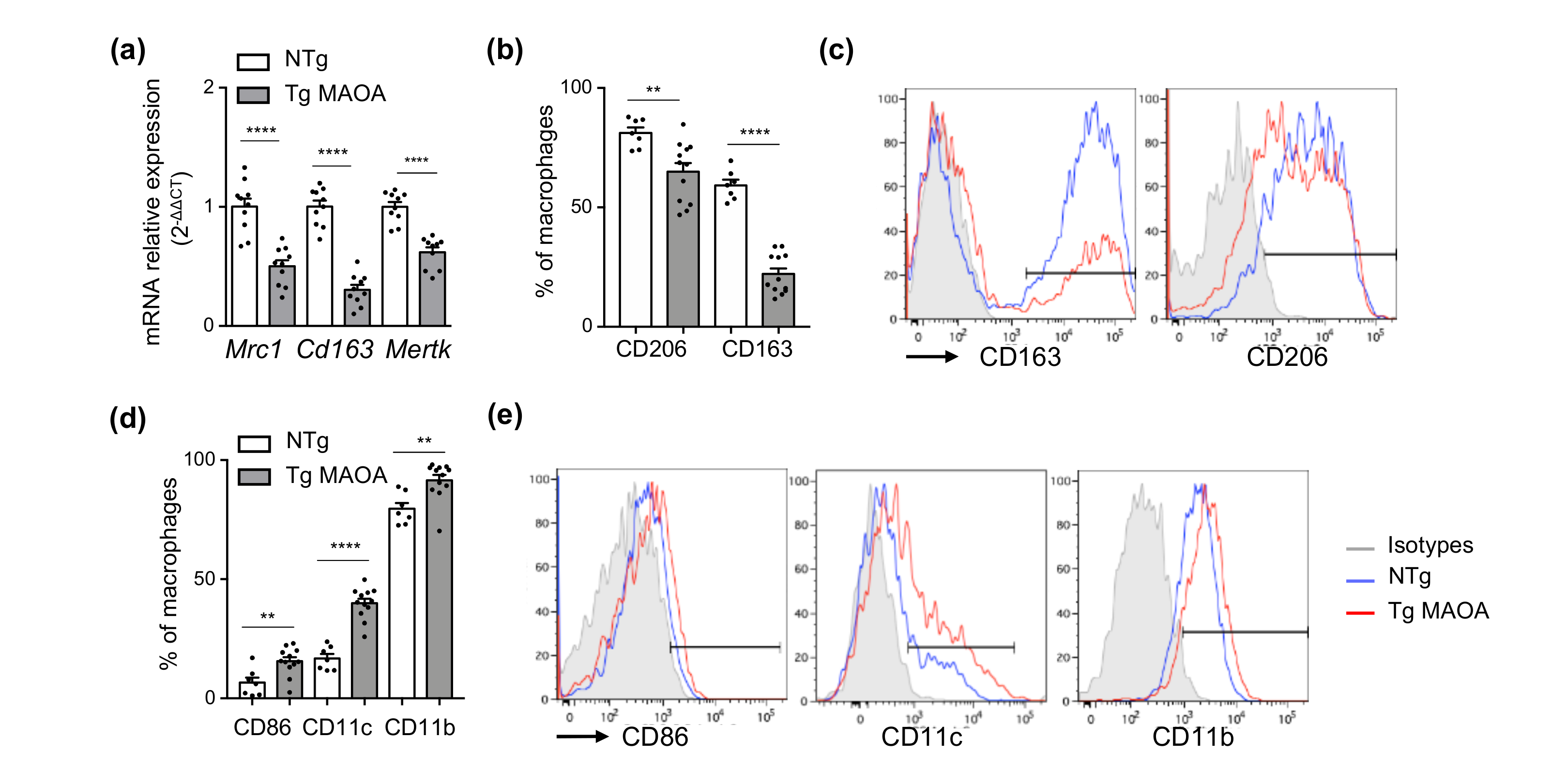
2.5. Macrophages Repressed Expression of Phagocytic Receptors in the Cardiac Microenvironment of Tg MAOA Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Echocardiography
4.3. Preparation of Cardiac Stromal Cell Suspensions
4.4. Protein Quantification by Capillary-Based Western Blot
4.5. Cardiac Stromal Cell Staining for Flow Cytometry
4.6. Cell Immunofluorescence Assays and Confocal Microscopy
4.7. SA-β Galactosidase Activity
4.8. Chemotaxis Assays
4.9. RT-PCR
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| cMSC | Cardiac Mesenchymal Stromal Cells |
| MAOA | Mono Amine Oxidase |
| MP | Macrophage |
| NTg | Non Transgenic |
| Tg | Transgenic |
References
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschenes-Simard, X.; Ferbeyre, G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell. Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef]
- Steinberg, S.F. Oxidative stress and sarcomeric proteins. Circ. Res. 2013, 112, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox. Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Shah, A.M. ROS signalling between endothelial cells and cardiac cells. Cardiovasc. Res. 2014, 102, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Santin, Y.; Sicard, P.; Vigneron, F.; Guilbeau-Frugier, C.; Dutaur, M.; Lairez, O.; Couderc, B.; Manni, D.; Korolchuk, V.I.; Lezoualc’h, F.; et al. Oxidative Stress by Monoamine Oxidase-A Impairs Transcription Factor EB Activation and Autophagosome Clearance, Leading to Cardiomyocyte Necrosis and Heart Failure. Antioxid. Redox. Signal. 2016, 25, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, C.; Guilbeau-Frugier, C.; Sicard, P.; Lairez, O.; Ordener, C.; Duparc, T.; De Paulis, D.; Couderc, B.; Spreux-Varoquaux, O.; Tortosa, F.; et al. p53-PGC-1alpha pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: Role in chronic left ventricular dysfunction in mice. Antioxid. Redox. Signal. 2013, 18, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Maurel, A.; Hernandez, C.; Kunduzova, O.; Bompart, G.; Cambon, C.; Parini, A.; Frances, B. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1460–H1467. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Manzella, N.; Santin, Y.; Maggiorani, D.; Martini, H.; Douin-Echinard, V.; Passos, J.F.; Lezoualc’h, F.; Binda, C.; Parini, A.; Mialet-Perez, J. Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging Cell 2018, 17, e12811. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.E4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef] [Green Version]
- Martini, H.; Iacovoni, J.S.; Maggiorani, D.; Dutaur, M.; Marsal, D.J.; Roncalli, J.; Itier, R.; Dambrin, C.; Pizzinat, N.; Mialet-Perez, J.; et al. Aging induces cardiac mesenchymal stromal cell senescence and promotes endothelial cell fate of the CD90 + subset. Aging Cell 2019, 18, e13015. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Abernethy, A.; Raza, S.; Sun, J.L.; Anstrom, K.J.; Tracy, R.; Steiner, J.; VanBuren, P.; LeWinter, M.M. Pro-Inflammatory Biomarkers in Stable Versus Acutely Decompensated Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Collier, P.; Watson, C.J.; Voon, V.; Phelan, D.; Jan, A.; Mak, G.; Martos, R.; Baugh, J.A.; Ledwidge, M.T.; McDonald, K.M. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur. J. Heart Fail. 2011, 13, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Lefevre, L.; Iacovoni, J.S.; Martini, H.; Belliere, J.; Maggiorani, D.; Dutaur, M.; Marsal, D.J.; Decaunes, P.; Pizzinat, N.; Mialet-Perez, J.; et al. Kidney inflammaging is promoted by CCR2(+) macrophages and tissue-derived micro-environmental factors. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mogilenko, D.A.; Shpynov, O.; Andhey, P.S.; Arthur, L.; Swain, A.; Esaulova, E.; Brioschi, S.; Shchukina, I.; Kerndl, M.; Bambouskova, M.; et al. Comprehensive Profiling of an Aging Immune System Reveals Clonal GZMK(+) CD8(+) T Cells as Conserved Hallmark of Inflammaging. Immunity 2021, 54, 99–115.e12. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martinez, L.; Sanchez-Diaz, M.; Diaz-Garcia, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef]
- Youm, Y.H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Marin-Aguilar, F.; Lechuga-Vieco, A.V.; Alcocer-Gomez, E.; Castejon-Vega, B.; Lucas, J.; Garrido, C.; Peralta-Garcia, A.; Perez-Pulido, A.J.; Varela-Lopez, A.; Quiles, J.L.; et al. NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell 2020, 19, e13050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabal, M.; Calleja, D.J.; Simpson, D.S.; Lawlor, K.E. Stressing out the mitochondria: Mechanistic insights into NLRP3 inflammasome activation. J. Leukoc. Biol. 2019, 105, 377–399. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rodriguez, R.; Munari, F.; Angioni, R.; Venegas, F.; Agnellini, A.; Castro-Gil, M.P.; Castegna, A.; Luisetto, R.; Viola, A.; Canton, M. Targeting monoamine oxidase to dampen NLRP3 inflammasome activation in inflammation. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Laroumanie, F.; Douin-Echinard, V.; Pozzo, J.; Lairez, O.; Tortosa, F.; Vinel, C.; Delage, C.; Calise, D.; Dutaur, M.; Parini, A.; et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 2014, 129, 2111–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martini, H.; Lefevre, L.; Sayir, S.; Itier, R.; Maggiorani, D.; Dutaur, M.; Marsal, D.J.; Roncalli, J.; Pizzinat, N.; Cussac, D.; et al. Selective Cardiomyocyte Oxidative Stress Leads to Bystander Senescence of Cardiac Stromal Cells. Int. J. Mol. Sci. 2021, 22, 2245. https://doi.org/10.3390/ijms22052245
Martini H, Lefevre L, Sayir S, Itier R, Maggiorani D, Dutaur M, Marsal DJ, Roncalli J, Pizzinat N, Cussac D, et al. Selective Cardiomyocyte Oxidative Stress Leads to Bystander Senescence of Cardiac Stromal Cells. International Journal of Molecular Sciences. 2021; 22(5):2245. https://doi.org/10.3390/ijms22052245
Chicago/Turabian StyleMartini, Hélène, Lise Lefevre, Sylvain Sayir, Romain Itier, Damien Maggiorani, Marianne Dutaur, Dimitri J. Marsal, Jérôme Roncalli, Nathalie Pizzinat, Daniel Cussac, and et al. 2021. "Selective Cardiomyocyte Oxidative Stress Leads to Bystander Senescence of Cardiac Stromal Cells" International Journal of Molecular Sciences 22, no. 5: 2245. https://doi.org/10.3390/ijms22052245
APA StyleMartini, H., Lefevre, L., Sayir, S., Itier, R., Maggiorani, D., Dutaur, M., Marsal, D. J., Roncalli, J., Pizzinat, N., Cussac, D., Parini, A., Mialet-Perez, J., & Douin-Echinard, V. (2021). Selective Cardiomyocyte Oxidative Stress Leads to Bystander Senescence of Cardiac Stromal Cells. International Journal of Molecular Sciences, 22(5), 2245. https://doi.org/10.3390/ijms22052245