
Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the MiR156, MiR5488 and MiR399 Are Involved in the Regulation of Male Sterility in PTGMS Rice


Abstract
:1. Introduction
2. Results
2.1. Abnormal Anther Development in PA64S under High-Temperature Treatment
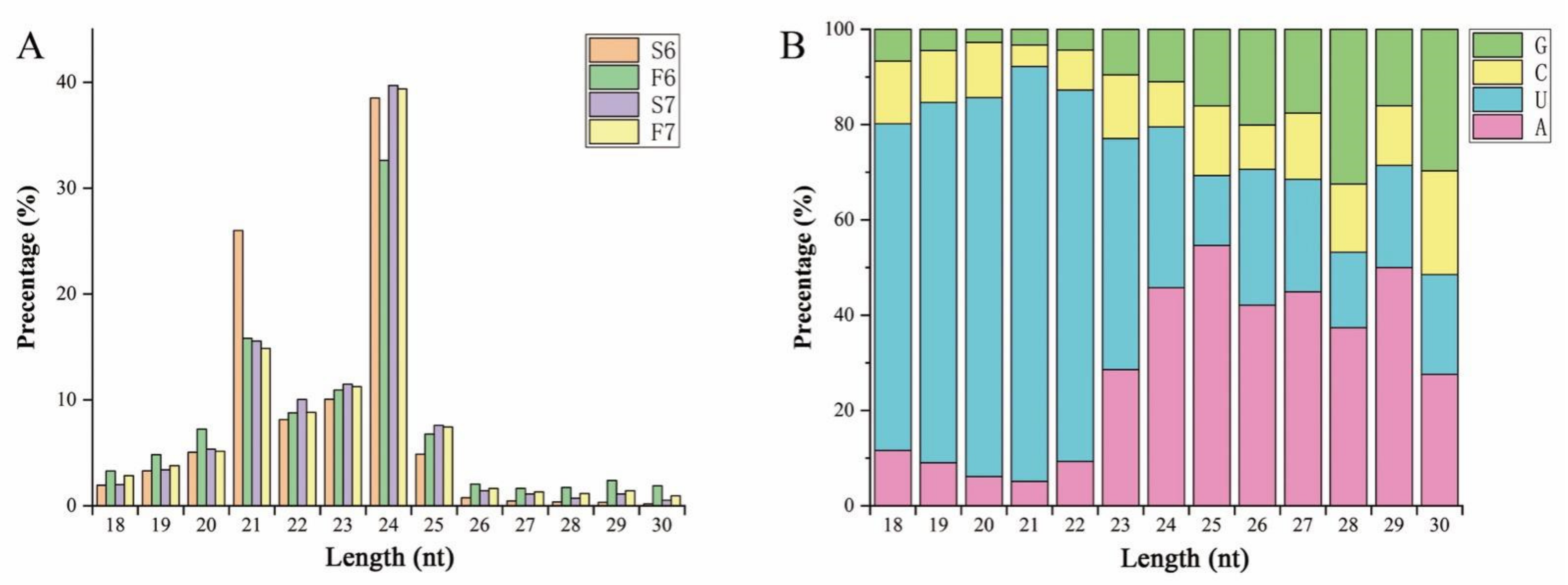
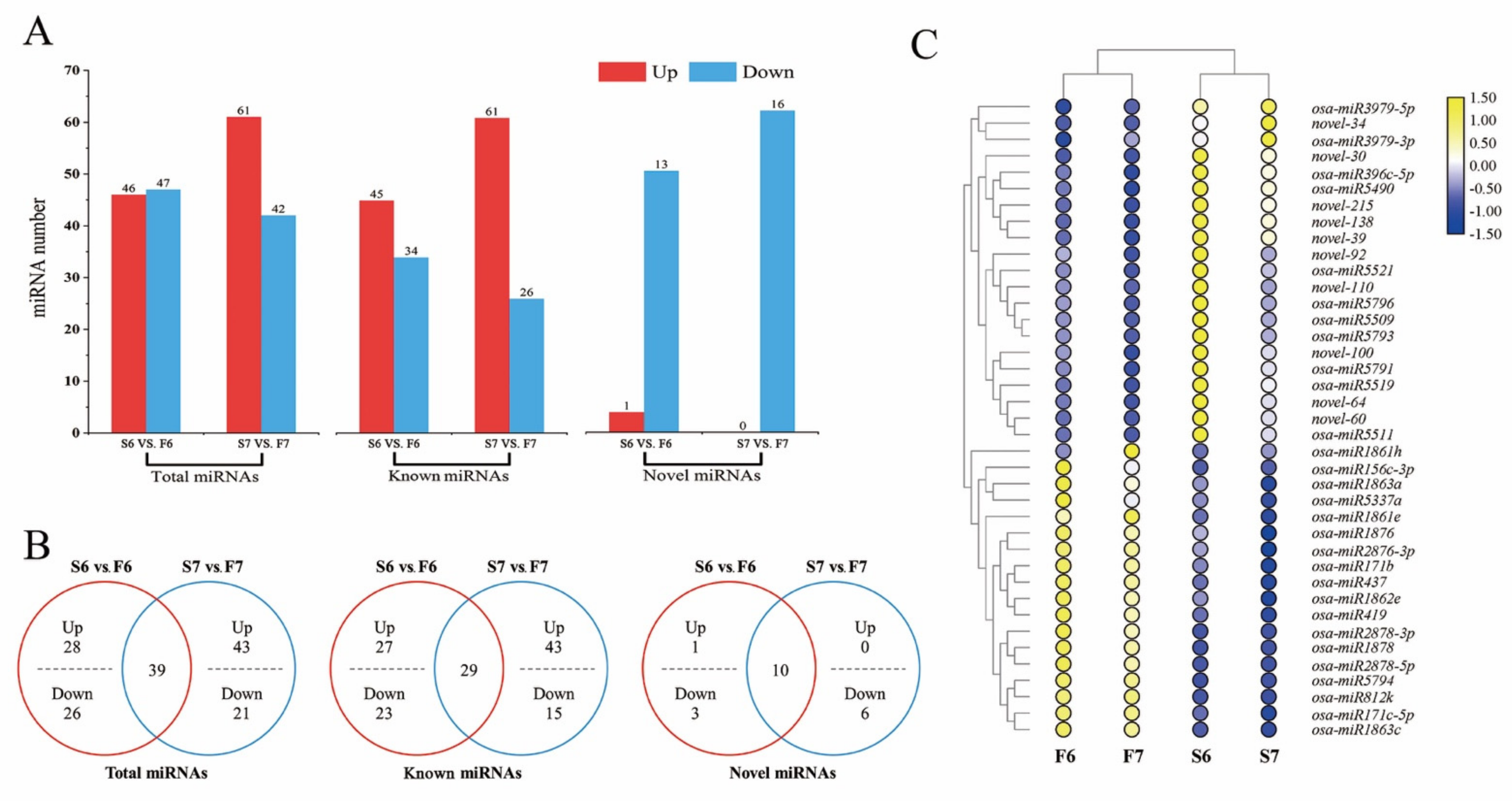
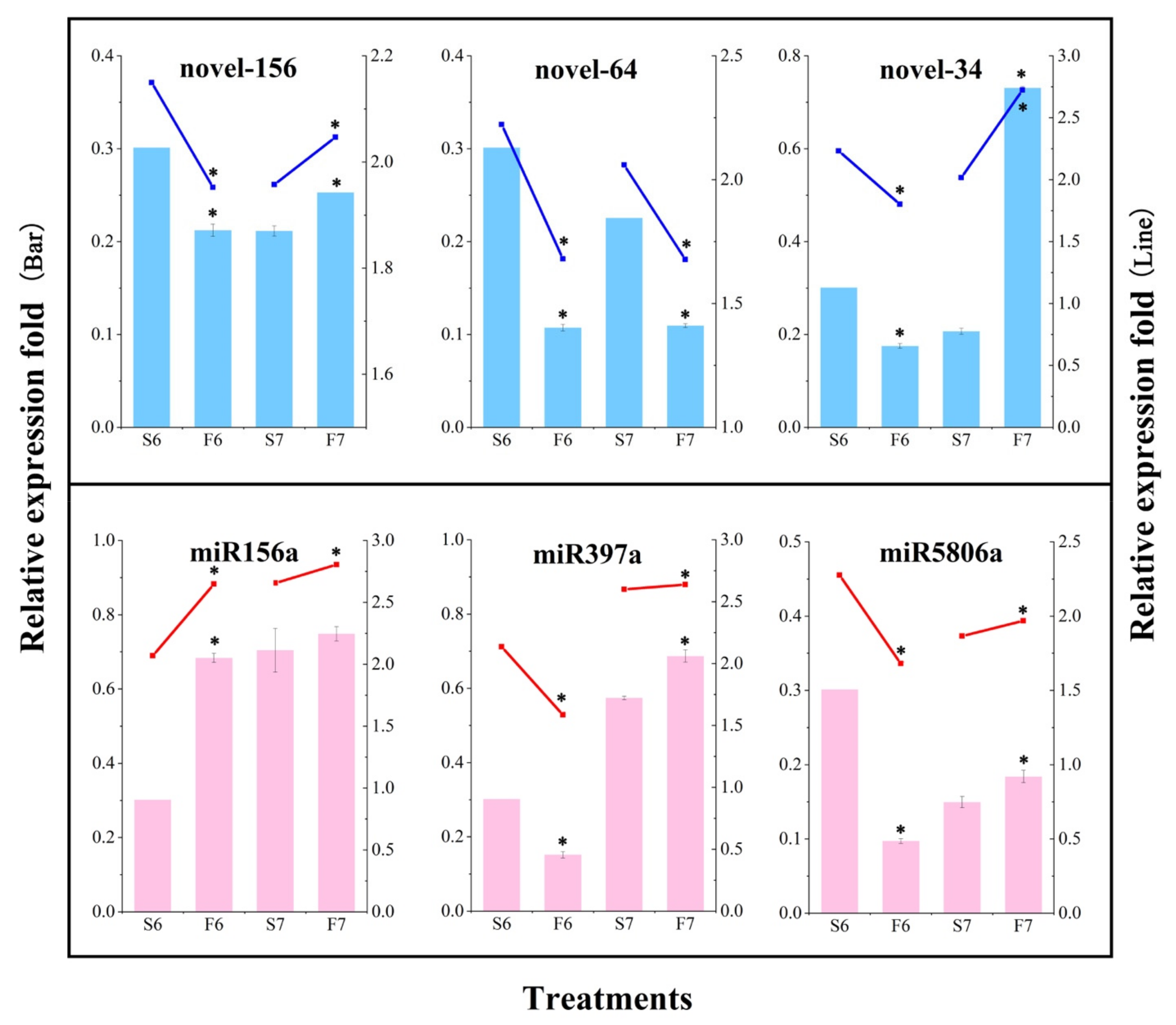
2.2. Expression Changes of miRNAs Are Involved in the Response to Temperature Variation in PA64S
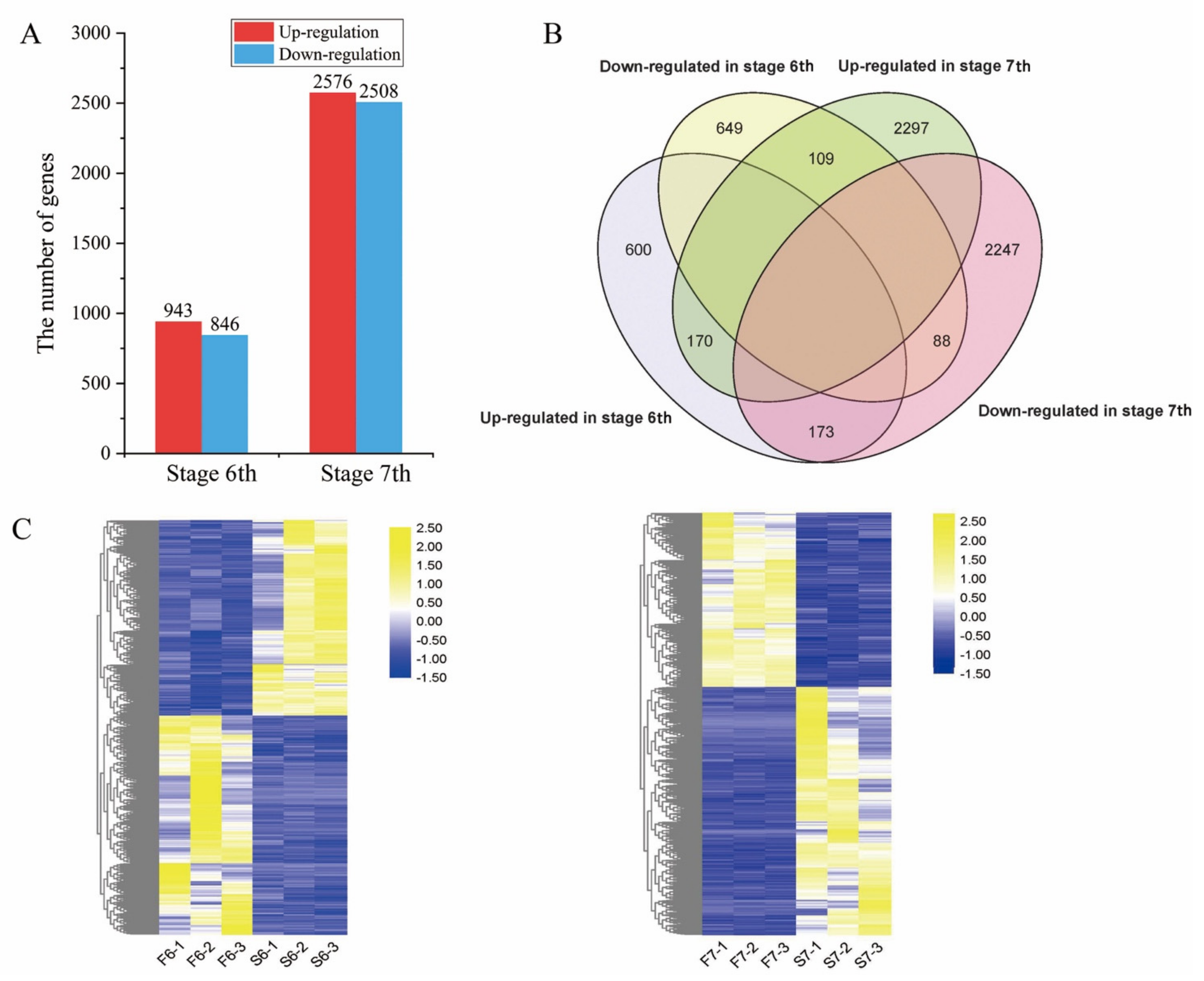
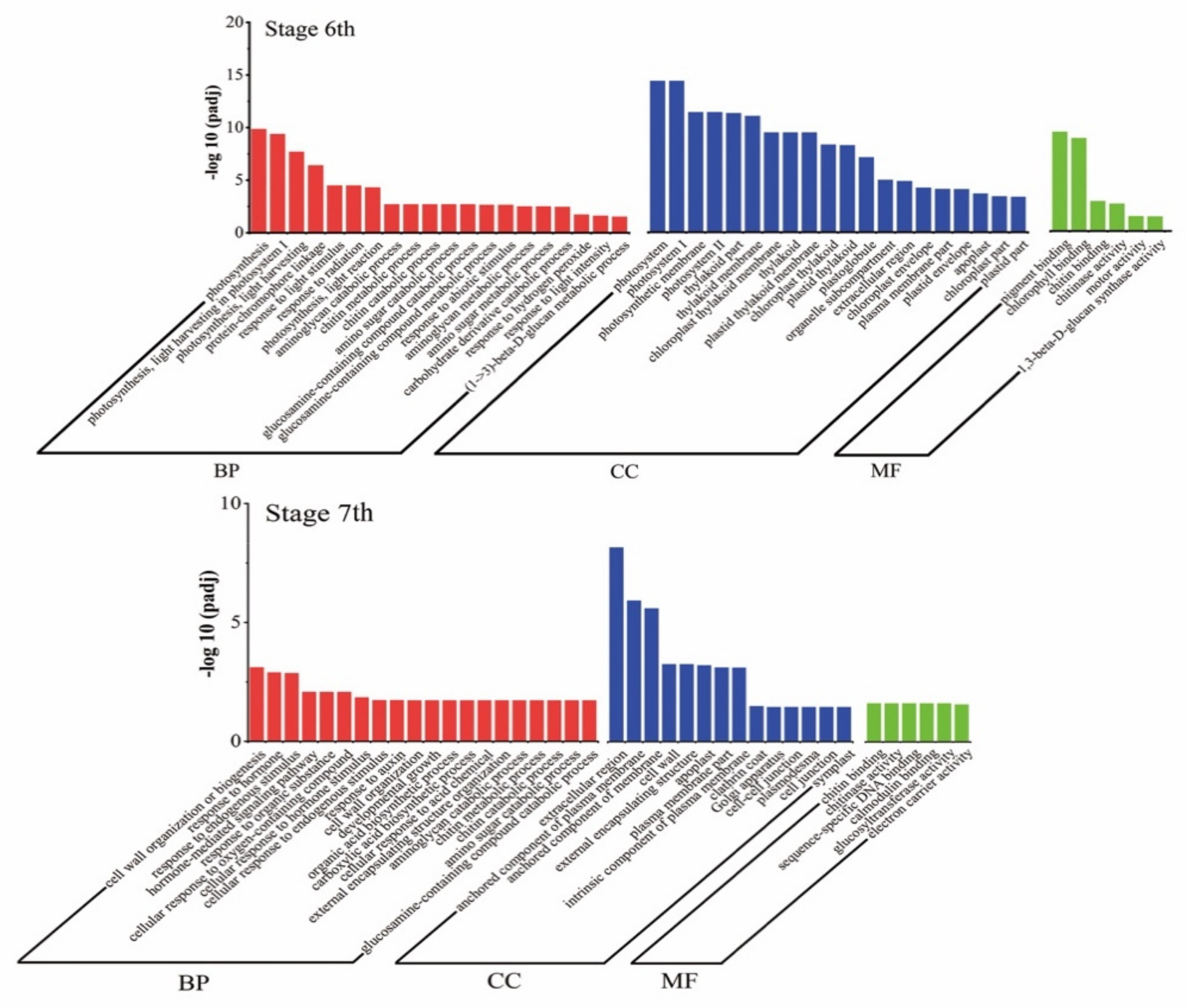
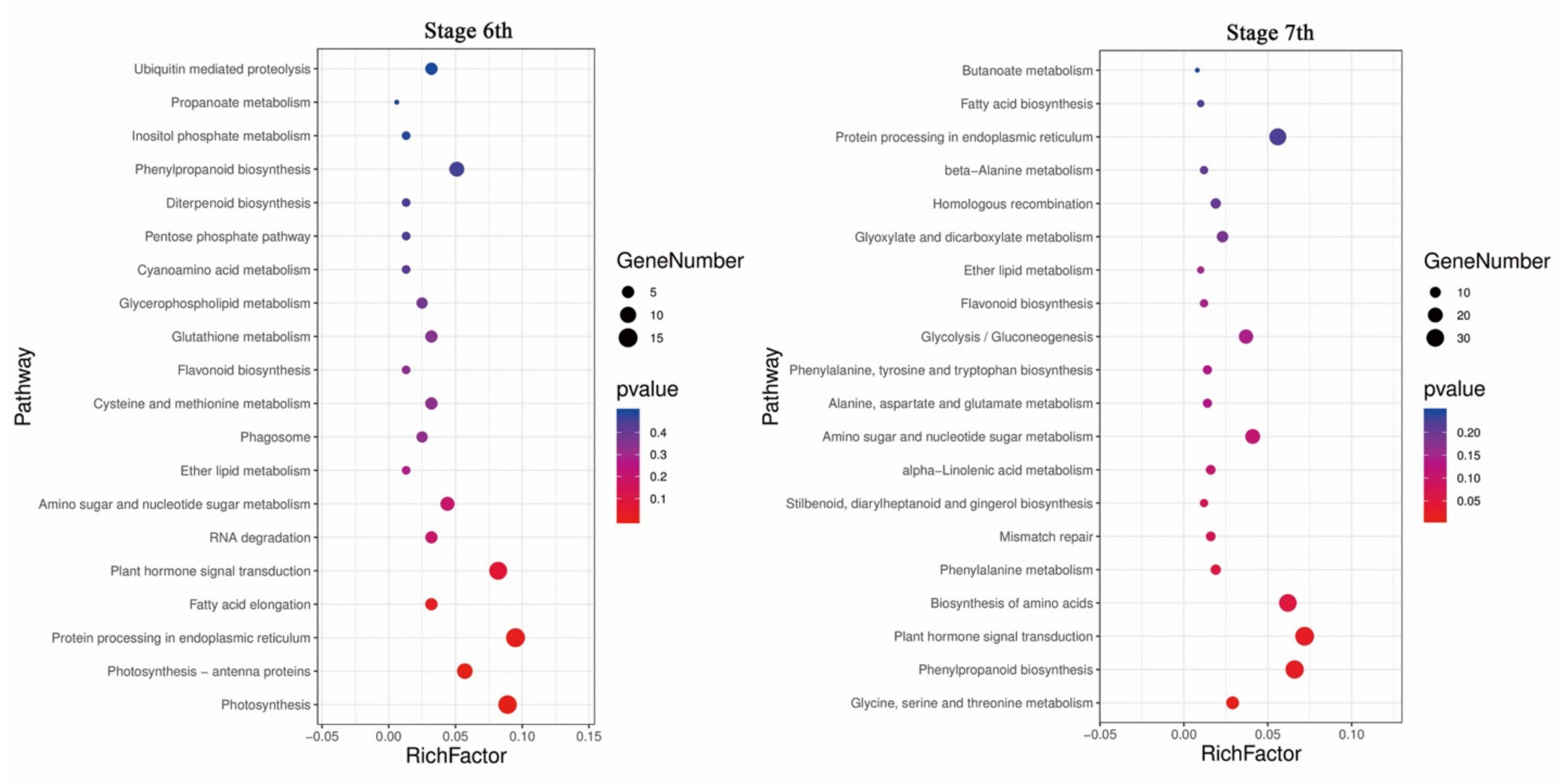
2.3. Changes in Gene Transcript Levels Are Involved in the Response to Temperature Variation in PA64S
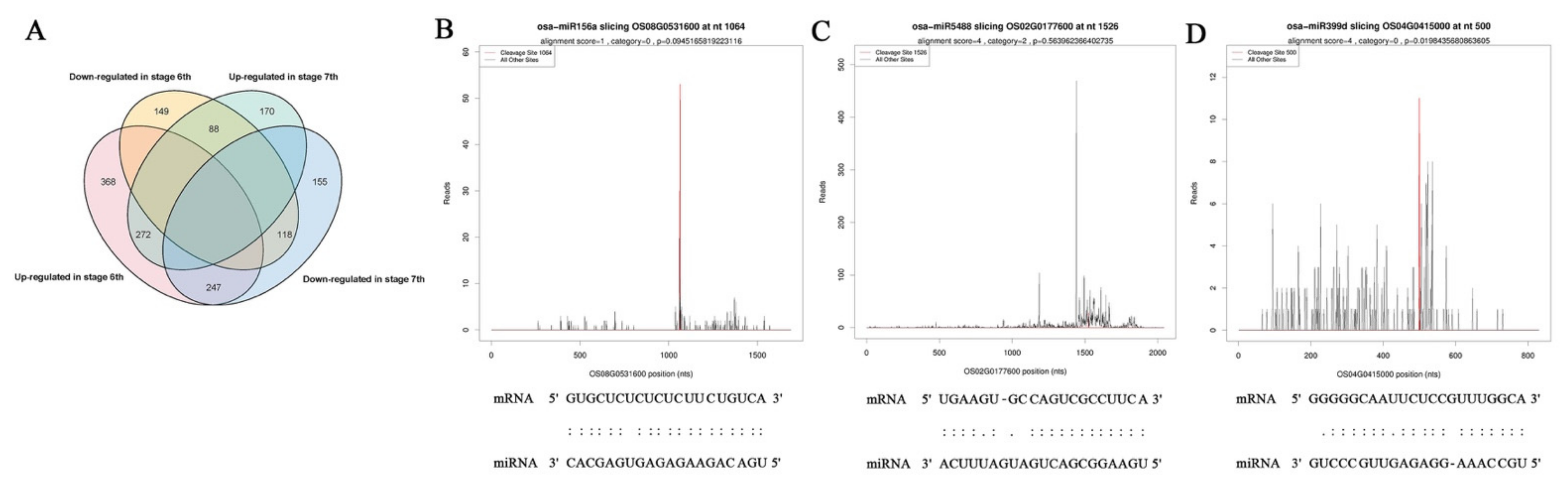
2.4. Target Identification of miRNAs by Degradome Analysis
2.5. Comprehensive Analysis of miRNA Expression Profiles and Target Genes in Response to Temperature Variation in PA64S
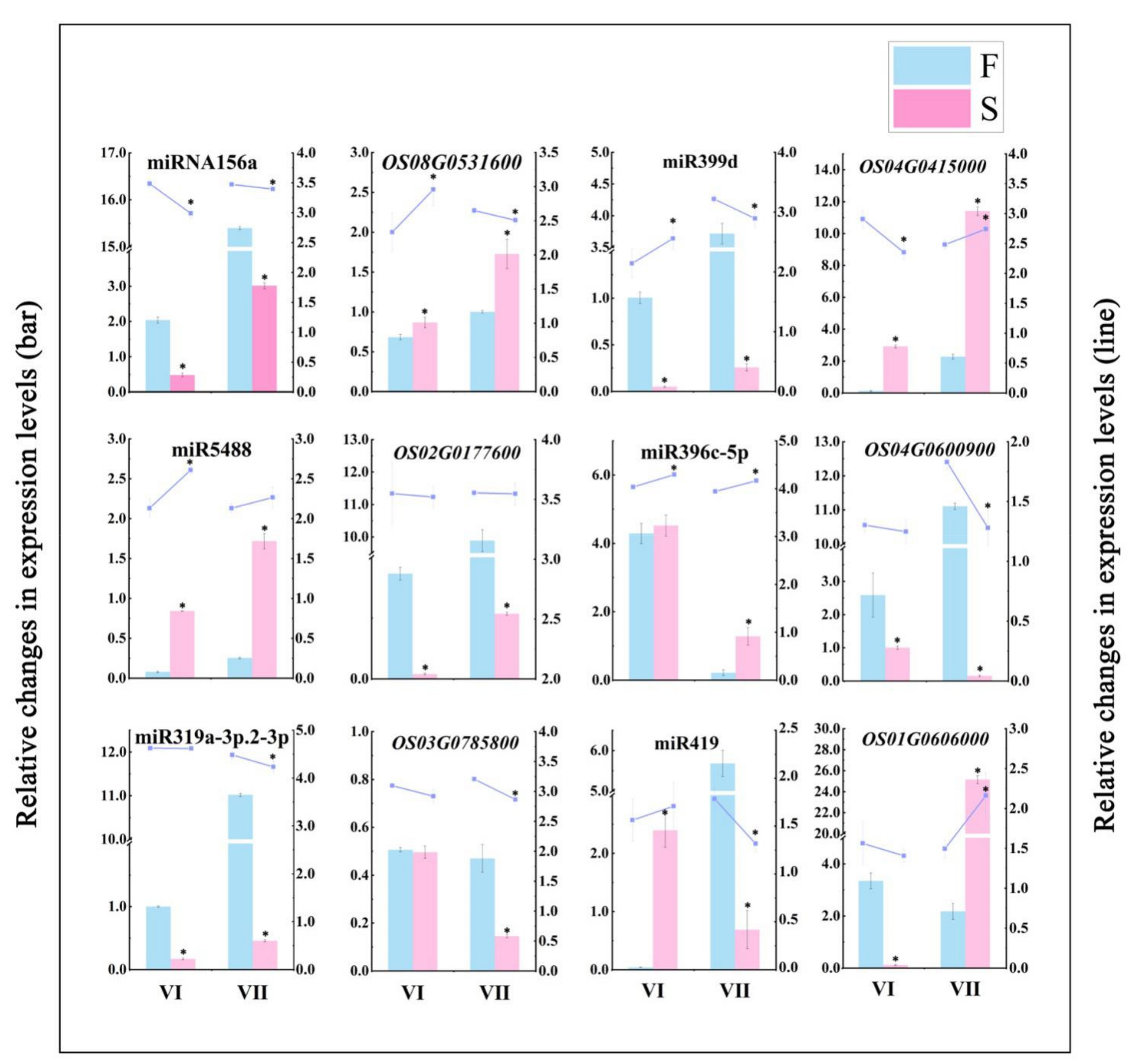
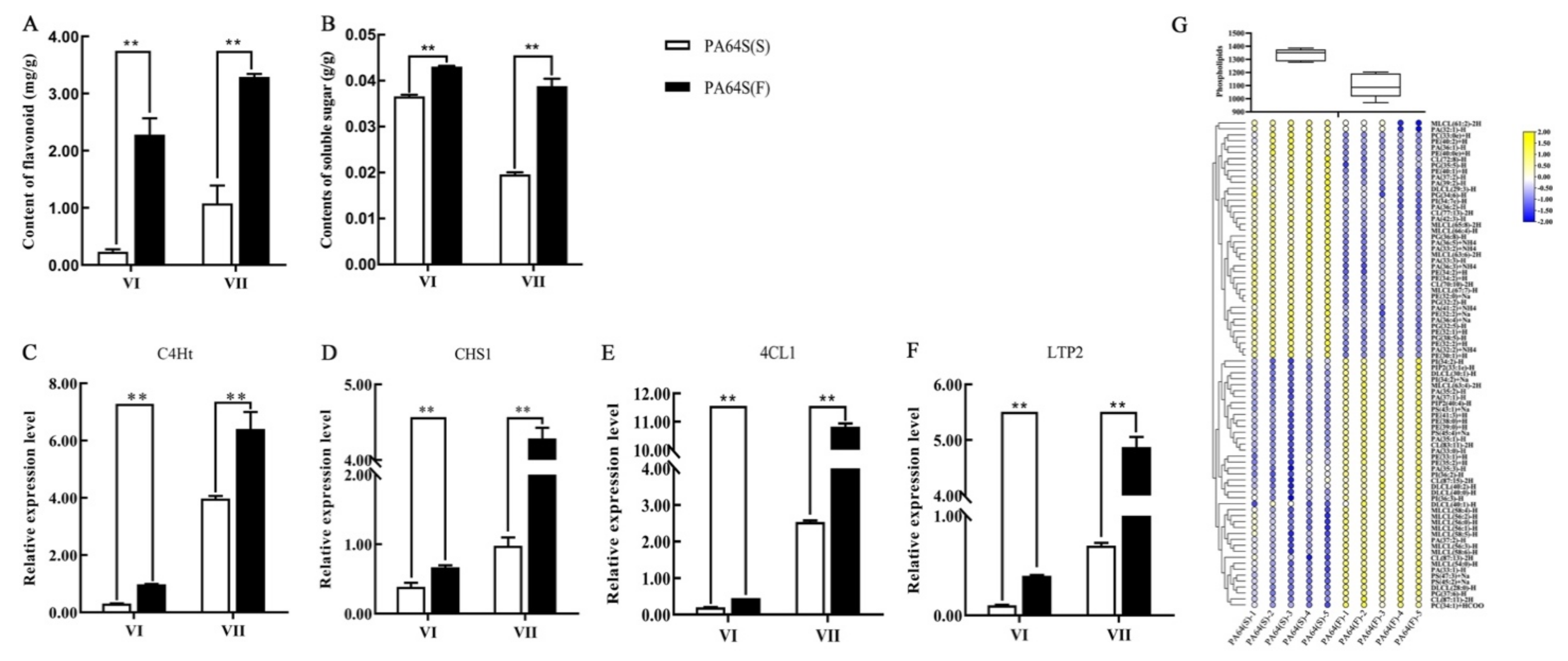
2.6. The Content of Metabolites and Relative Expression Level of Related Genes Involved in the Regulation of Male Fertility Processes
3. Discussion
3.1. Abnormal Tapetum Causes Male Sterility under High Temperature in PA64S
3.2. MiRNAs Are Involved in the Regulation of Male Fertility in PA64S in Response to Temperature Changes
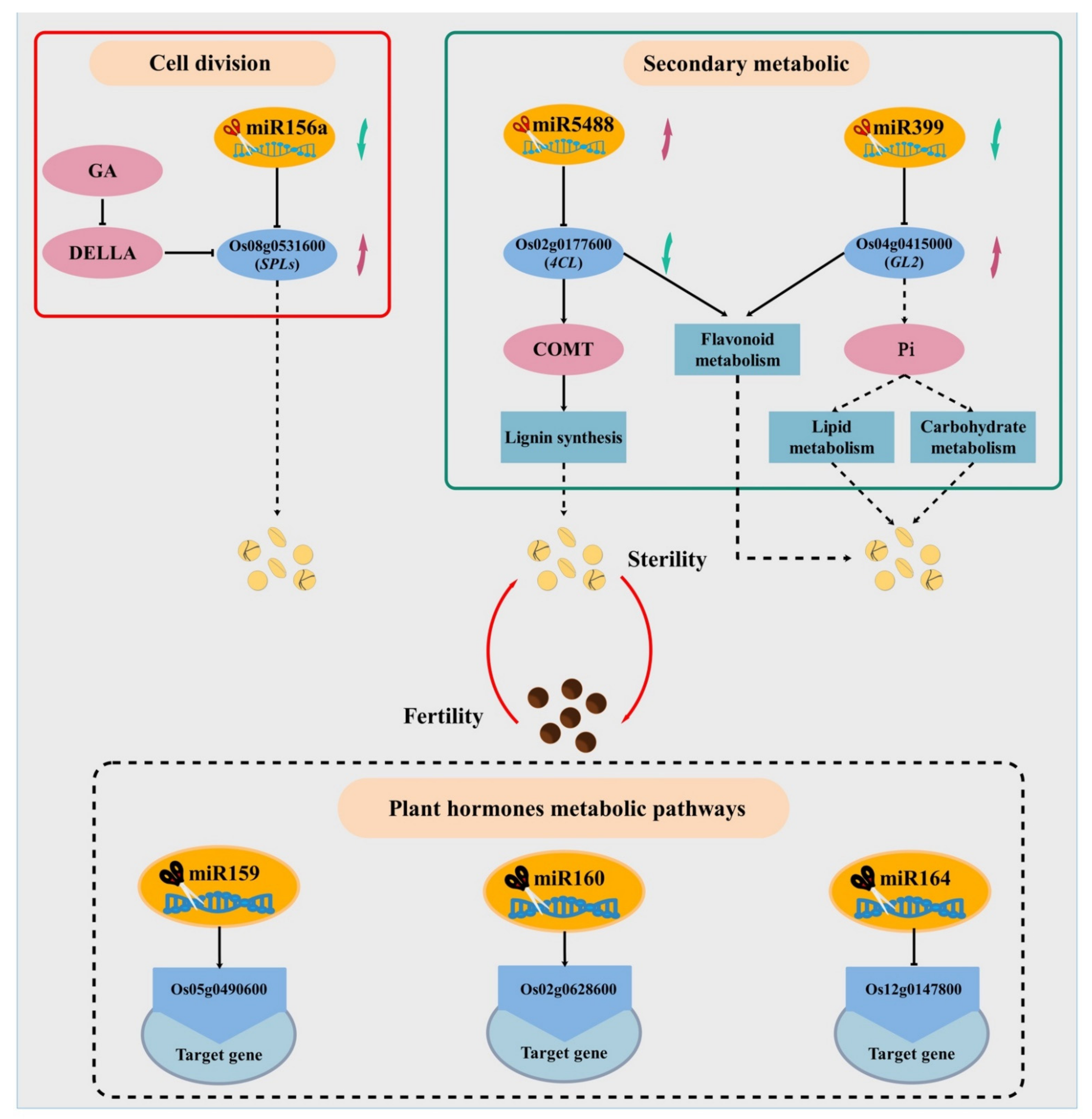
3.3. MIR156, MIR5488, and MIR399 Regulated Male Fertility in Rice by Responding to Temperature Change
4. Materials and Methods
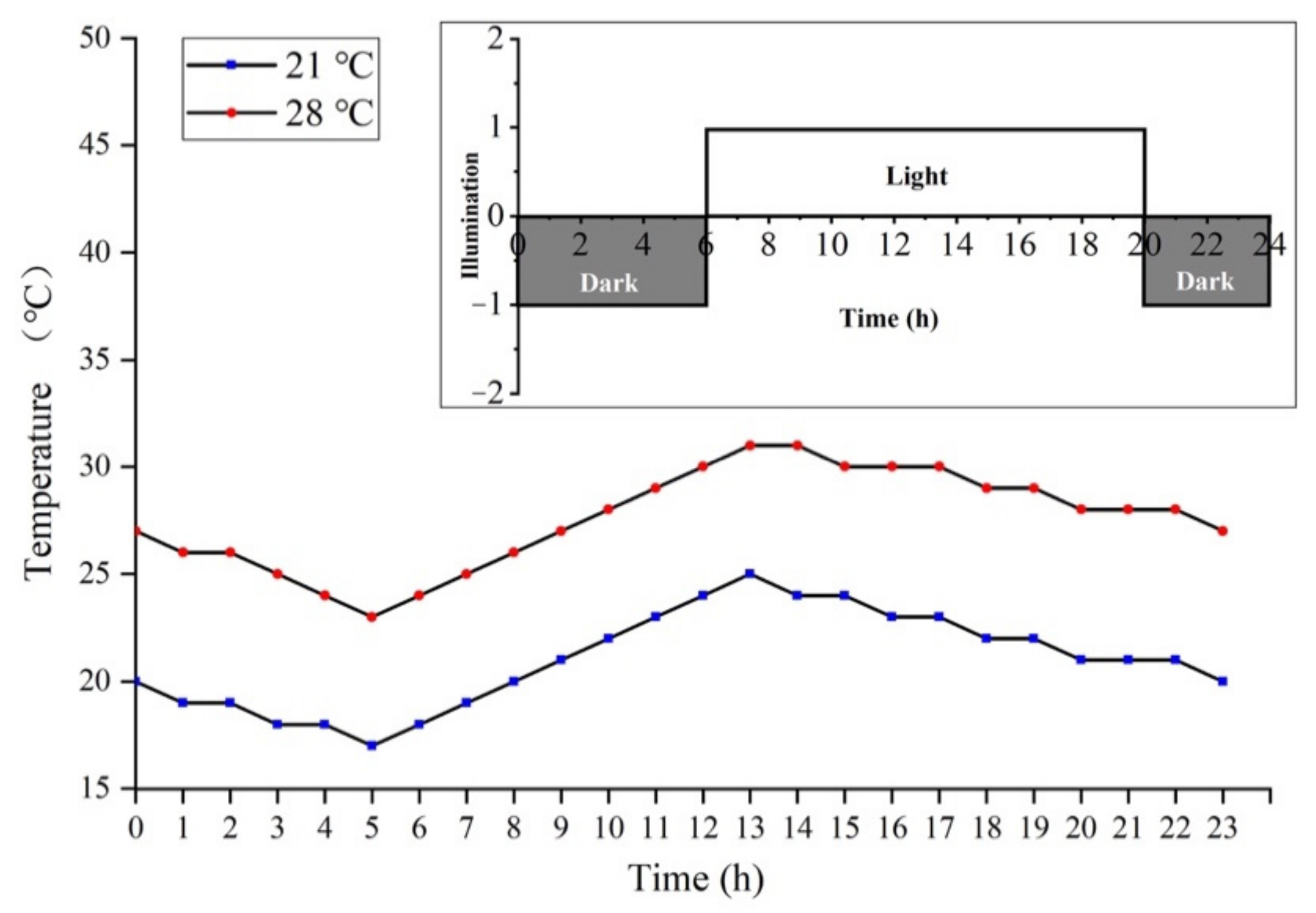
4.1. Plant Materials and Experimental Treatment
4.2. Phenotype Analysis of PA64S
4.3. Small RNA and RNA Sequencing
4.3.1. Small RNA Sequencing and Data Analysis
4.3.2. RNA Sequencing
4.4. Degradome Sequencing and Data Analysis
4.5. qPCR Validation
4.6. Determination of Metabolite Content
4.6.1. Flavonoids and Soluble Sugar Content
4.6.2. Lipid Type and Content
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Huang, X.; Yang, S.; Gong, J.; Zhao, Q.; Feng, Q.; Zhan, Q.; Zhao, Y.; Li, W.; Cheng, B.; Xia, J. Genomic architecture of heterosis for yield traits in rice. Nature 2016. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, C.; He, Y.; Zong, J.; Yang, X.; Si, H.; Sun, Z.; Hu, J.; Liang, W.; Zhang, O. Mutation in CSA creates a new photoperiod-sensitive genic male sterile line applicable for hybrid rice seed production. Proc. Natl. Acad. Sci. USA 2012, 110, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, Y.G. Male Sterility and Fertility Restoration in Crops. Annu. Rev. Plant. Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef]
- Wang, H.; Deng, X.W. Development of the “Third-Generation” Hybrid Rice in China. Genom. Proteom. Bioinform. 2018, 16, 19–22. [Google Scholar] [CrossRef]
- Yuan, L.P. Purification and Production of Foundation Seed of Rice PGMS and TGMS Lines. Hybrid Rice 1994. [Google Scholar] [CrossRef]
- Xu, X.M.; Zhang, S.G.; Liang, K.J. Progress and Discussion in Breeding of Indica Rice CMS Lines in China. Chin. Agric. Sci. Bull. 2007, 23, 176–180. [Google Scholar] [CrossRef]
- Chen, L.Y.; Lei, D.Y.; Tang, W.B.; Xiao, Y.H. Thoughts and Practice on Some Problems about Research and Application of Two-Line Hybrid Rice. Rice Sci. 2011, 18, 79–85. [Google Scholar] [CrossRef]
- Chang, Z.; Chen, Z.; Wang, N.; Xie, G.; Lu, J.; Yan, W.; Zhou, J.; Tang, X.; Deng, X.W. Construction of a male sterility system for hybrid rice breeding and seed production using a nuclear male sterility gene. Proc. Natl. Acad. Sci. USA 2016, 14145. [Google Scholar] [CrossRef] [Green Version]
- Shi, M. The discovery and preliminary studies of the photoperiod-sensitive recessive male-sterile rice (Oryza sativa L. subsp. Japonica). Sci. Agric. Sin. 1985, 2, 44–48. [Google Scholar]
- Sun, Z.X. Identification of the Temperature Sensitive Male-sterile Rice. Chin. J. Rice Sci. 1989, 2, 49–55. [Google Scholar] [CrossRef]
- Wang, Z.; Fan, W.U.; Yi, T.; Qian, L.I.; De, Z.; Huang, X. Selection of the dual-purpose genic male sterile line Mian 9S, A New germplasm of indica rice. S. China J. Agric. Sci. 1999, 12, 11–14. [Google Scholar] [CrossRef]
- Li, S.; Xiong, G.; Gao, Y. Discovery and Exploitation of the Genetic Male Sterility Induced by Short Daylength in Rice. Hybrid Rice 2006, 10–13. [Google Scholar] [CrossRef]
- Chen, L.B.; Zhou, G.Q.; Huang, Y.X. Effects of Temperature and Photoperiod on Fertility and Physiological Activities of Rice Annong S-1 and Hengnong S-1. J. Integr. Plant. Biol. 1994, 36, 119–123. [Google Scholar]
- Yang, Y.; Tang, P.; Yang, W.; Liu, A.; Chen, Y.; Ling, W.; Shi, T. Breeding and Utilization of TGMS Line Zhu 1 S in Rice. Hybrid Rice 2000, 2, 6–9. [Google Scholar] [CrossRef]
- Dong, N.V.; Subudhi, P.K.; Luong, P.N.; Quang, V.D.; Quy, T.D.; Zheng, H.G.; Wang, B.; Nguyen, H.T. Molecular mapping of a rice gene conditioning thermosensitive genic male sterility using AFLP, RFLP and SSR techniques. Theor. Appl. Genet. 2000, 100, 727–734. [Google Scholar] [CrossRef]
- Lee, D.S.; Chen, L.J.; Suh, H.S. Genetic characterization and fine mapping of a novel thermo-sensitive genic male-sterile gene tms6 in rice (Oryza sativa L.). Theor. Appl. Genet. 2005, 111, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shen, B.Z.; Dai, X.K.; Mei, M.H.; Saghai Maroof, M.A.; Li, Z.B. Using bulked extremes and recessive class to map genes for photoperiod-sensitive genic male sterility in rice. Proc. Natl. Acad. Sci. USA 1994, 91, 8675–8679. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Xu, W.W.; Wang, J.Z.; Wu, W.; Zheng, H.G.; Yang, Z.Y.; Ray, J.D.; Nguyen, H.T. Tagging and mapping the thermo-sensitive genic male-sterile gene in rice (Oryza sativa L.) with molecular markers. Theor. Appl. Genet. 1995, 91, 1111–1114. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Lkeda, R.; Hirasawa, H.; Minami, M.; Ujihara, A. Linkage Analysis of Thermosensitive Genic Male Sterility Gene, tms-2 in Rice (Oryza sativa L.). Jpn. J. Breed. 1997, 47, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, B.; Wu, Y.; Du, P.; Wang, J.; Wang, M.; Yi, C.; Gu, M.; Liang, G. Fine mapping and candidate gene analysis of ptgms2–1, the photoperiod-thermo-sensitive genic male sterile gene in rice (Oryza sativa L.). Theor. Appl. Genet. 2011, 122, 365–372. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, Q.; Li, J.; Jiang, D.; Zhou, L.; Wu, P.; Lu, S.; Li, F.; Zhu, L.; Liu, Z.; et al. Photoperiod- and thermo-sensitive genic male sterility in rice are caused by a point mutation in a novel noncoding RNA that produces a small RNA. Cell Res. 2012, 22, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.; Wei, X.; Shao, G.; Chen, M.; Song, J.; Tang, S.; Luo, J.; Hu, Y.; Hu, P.; Chen, L. Genetic analysis and fine mapping of tms9, a novel thermosensitive genic male-sterile gene in rice (Oryza sativa L.). Plant Breed. 2013, 132, 159–164. [Google Scholar] [CrossRef]
- Li, C.; Zhang, B. MicroRNAs in Control of Plant Development. J. Cell. Physiol. 2016, 231, 303–313. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. Micrornas And Their Regulatory Roles in Plants. Annu. Rev. Plant. Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef]
- Jin, Q.Y.; Peng, H.Z.; Lin, E.-P.; Li, N.; Huang, D.N.; Xu, Y.L.; Hua, X.Q.; Wang, K.H.; Zhu, T.J. Identification and characterization of differentially expressed miRNAs between bamboo shoot and rhizome shoot. J. Plant Biol. 2016. [Google Scholar] [CrossRef]
- Sharma, D.; Tiwari, M.; Pandey, A.; Bhatia, C.; Sharma, A.; Trivedi, P.K. MicroRNA858 is a potential regulator of phenylpropanoid pathway and plant development in Arabidopsis. Plant. Physiol. 2016, 944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curaba, J.; Spriggs, A.; Taylor, J.; Li, Z.; Helliwell, C. miRNA regulation in the early development of barley seed. BMC Plant Biol. 2012, 12, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.Y.; Xu, Y.P.; Zhao, L.; Li, S.S.; Cai, X.Z. Tight regulation of the interaction between Brassica napus and Sclerotinia sclerotiorum at the microRNA level. Plant Mol. Biol. 2016, 92, 39–55. [Google Scholar] [CrossRef]
- Candar-Cakir, B.; Arican, E.; Zhang, B. Small RNA and degradome deep sequencing reveals drought-and tissue-specific micrornas and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol. J. 2016, 14, 1727–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanemian, M.; Barlet, X.; Sorin, C.; Yadeta, K.A.; Keller, H.; Favery, B.; Simon, R.; Thomma, B.P.H.J.; Hartmann, C.; Crespi, M. Arabidopsis CLAVATA1 and CLAVATA2 receptors contribute to Ralstonia solanacearum pathogenicity through a miR169-dependent pathway. New Phytol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Omidvar, V.; Mohorianu, I.; Dalmay, T.; Fellner, M. Identification of miRNAs with potential roles in regulation of anther development and male-sterility in 7B-1 male-sterile tomato mutant. BMC Genomics 2015, 16, 878. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhang, X.; Liu, G.; Guo, L.; Qi, T.; Zhang, M.; Li, X.; Wang, H.; Tang, H.; Qiao, X.; et al. A combined small RNA and transcriptome sequencing analysis reveal regulatory roles of miRNAs during anther development of Upland cotton carrying cytoplasmic male sterile Gossypium harknessii (D2) cytoplasm. BMC Plant Biol. 2018, 18, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Xie, Y.; Xu, L.; Wang, Y.; Zhu, X.; Wang, R.; Zhang, Y.; Muleke, E.M.; Liu, L. Identification of microRNAs and Their Target Genes Explores miRNA-Mediated Regulatory Network of Cytoplasmic Male Sterility Occurrence during Anther Development in Radish (Raphanus sativus L.). Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Wang, Y.; Jin, F.-w.; Xing, L.-j.; Fang, Y.; Zhang, Z.-y.; Zou, J.-j.; Wang, L.; Xu, M.-y. Differentially expressed miRNAs in anthers may contribute to the fertility of a novel Brassica napus genic male sterile line CN12A. J. Integr. Agric. 2020, 19, 1731–1742. [Google Scholar] [CrossRef]
- Chen, J.; Pan, A.; He, S.; Su, P.; Yuan, X.; Zhu, S.; Liu, Z. Different MicroRNA Families Involved in Regulating High Temperature Stress Response during Cotton (Gossypium hirsutum L.) Anther Development. Int. J. Mol. Sci. 2020, 21, 1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Chen, Z.; Li, H.; Sun, Y.; Wang, L.; Zeng, H.; He, Y. Lipid metabolism is involved in male fertility regulation of the photoperiod- and thermo sensitive genic male sterile rice line Peiai 64S. Plant Sci. 2020, 299, 110581. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Zhang, D. Developing Near Isogenic Lines of Different Critical Male Sterile Temperature of Thermo-photoperiod Sensitive Male Sterile Rice Peiai 64S. Acta Agron. Ica Sin. 2001, 27, 351–355. [Google Scholar] [CrossRef]
- Ma, H. Molecular genetic analyses of microsporogenesis and microgametogenesis in flowering plants. Annu. Rev. Plant Biol. 2005, 56, 393–434. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Khurana, R.; Malik, N.; Badoni, S.; Parida, S.K.; Kapoor, S.; Tyagi, A.K. bHLH142 regulates various metabolic pathway-related genes to affect pollen development and anther dehiscence in rice. Sci. Rep. 2017, 7, 43397. [Google Scholar] [CrossRef] [Green Version]
- Wilson, Z.A.; Song, J.; Taylor, B.; Yang, C. The final split: The regulation of anther dehiscence. J. Exp. Bot. 2011, 62, 1633–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Yang, C.; Yuan, Z.; Zhang, D.; Gondwe, M.Y.; Ding, Z.; Liang, W.; Zhang, D.; Wilson, Z.A. The ABORTED MICROSPORES Regulatory Network Is Required for Postmeiotic Male Reproductive Development in Arabidopsis thaliana. Plant Cell 2010, 22, 91–107. [Google Scholar] [CrossRef] [Green Version]
- Yong, S.S.; Ying, C.; Lu, L.; Benjamin, S.Y.H.; Zao, M.C.; Bo, G.Y.; Hao, Y. OsFTIP7 determines auxin-mediated anther dehiscence in rice. Nat. Plants 2018. [Google Scholar] [CrossRef]
- Dechkrong, P.; Sreewongchai, T.; Paopun, Y.; Sripichitt, P.; Worede, F. Cytological observation of anther development of cytoplasmic male sterility and thermosensitive genic male sterility systems in rice. Agric. Nat. Resour. 2019, 53, 114–119. [Google Scholar]
- Xiang, X.J.; Sun, L.P.; Yu, P.; Yang, Z.F.; Zhang, P.P.; Zhang, Y.X.; Wu, W.X.; Chen, D.B.; Zhan, X.D.; Khan, R.M.; et al. The MYB transcription factor Baymax1 plays a critical role in rice male fertility. Theor. Appl. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Q. MicroRNA-Based Biotechnology for Plant Improvement. J. Cell. Physiol. 2015, 230, 1–15. [Google Scholar] [CrossRef]
- Gong, S.; Ding, Y.; Huang, S.; Zhu, C. Identification of miRNAs and Their Target Genes Associated with Sweet Corn Seed Vigor by Combined Small RNA and Degradome Sequencing. J. Agric. Food Chem. 2015, 63, 5485–5491. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, S.; Tan, J.; Chen, X.; Zhang, M. MiRNAs profiling and degradome sequencing between the CMS-line N816S and its maintainer line Ning5m during anther development in pepper (Capsicum annuum L.). bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Li, T.; Zhao, Y.; Zhang, B.; Li, A.; Zhao, S.; Hou, L.; Xia, H.; Fan, S.; Qiu, J.; et al. Integrated small RNA and mRNA expression profiles reveal miRNAs and their target genes in response to Aspergillus flavus growth in peanut seeds. BMC Plant Biol. 2020, 20, 215. [Google Scholar] [CrossRef]
- Fu, Y.; Mason, A.S.; Zhang, Y.; Lin, B.; Xiao, M.; Fu, D.; Yu, H. MicroRNA-mRNA expression profiles and their potential role in cadmium stress response in Brassica napus. BMC Plant Biol. 2019, 19, 570. [Google Scholar] [CrossRef]
- Bai, J.F.; Wang, Y.K.; Wang, P.; Duan, W.J.; Yuan, S.H.; Sun, H.; Yuan, G.L.; Ma, J.-X.; Wang, N.; Zhang, F.T.; et al. Uncovering Male Fertility Transition Responsive miRNA in a Wheat Photo-Thermosensitive Genic Male Sterile Line by Deep Sequencing and Degradome Analysis. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Nie, H.; Wang, Y.; Su, Y.; Hua, J. Exploration of miRNAs and target genes of cytoplasmic male sterility line in cotton during flower bud development. Funct. Integr. Genom. 2018, 18, 457–476. [Google Scholar] [CrossRef]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and Functional Diversification of MIRNA Genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Axtell, M.J. Classification and Comparison of Small RNAs from Plants. Annu. Rev. Plant Biol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Jones-Rhoades, M.W. Conservation and divergence in plant microRNAs. Plant Mol. Biol. 2012, 80, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Zhang, L.; Li, W.W.; Hu, X.L.; Wang, M.B.; Fan, Y.L.; Zhang, C.Y.; Wang, L. Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J. Exp. Bot. 2014, 65, 89–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Park, M.Y.; Conway, S.R. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Ma, Y.; Liu, N.; Xu, J.; Hu, Q. microRNAs involved in auxin signalling modulate male sterility under high-temperature stress in cotton (Gossypium hirsutum). Plant J. 2017. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Liu, C.; Feng, M.; Liu, Y.; Wu, X.; Guo, W. miR156-SPL modules regulate induction of somatic embryogenesis in citrus callus. J. Exp. Bot. 2018, 69, 2979–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, K.; Wu, C.; Xiong, L. Genomic Organization, Differential Expression, and Interaction of SQUAMOSA Promoter-Binding-Like Transcription Factors and microRNA156 in Rice. Plant Physiol. 2006, 142, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.Q.; Sun, G.L.; Shi, C.X.; Sun, D.F. Transcriptome analysis reveals new microRNAs-mediated pathway involved in anther development in male sterile wheat. BMC Genomics 2018, 19, 333. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Fang, Y.N.; Wu, X.M.; Qing, M.; Li, C.C.; Xie, K.D.; Deng, X.X.; Guo, W.W. The miR399-CsUBC24 Module Regulates Reproductive Development and Male Fertility in Citrus. Plant Physiol. 2020, 183, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, J.; Sang, Y.; Xing, S.; Wu, Q. Identification and Characterization of MicroRNAs in Ginkgo biloba var. epiphylla Mak. PLoS ONE 2015. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, J.N.; Chand, S.K.; Joshi, R.K. Multiple microRNAs Regulate the Floral Development and Sex Differentiation in the Dioecious Cucurbit Coccinia grandis (L.) Voigt. Plant Mol. Biol. Report. 2019, 37, 111–128. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, L.; Zhao, Y.; Wang, Z.; Zhou, S. Comparative analysis and functional identification of temperature-sensitive miRNA in Arabidopsis anthers. Biochem. Biophys. Res. Commun. 2020, 532, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, H.; Weigel, D. Small RNAs in flower development. Eur. J. Cell Biol. 2010, 89, 250–257. [Google Scholar] [CrossRef]
- Cui, L.G.; Shan, J.X.; Shi, M.; Gao, J.P.; Lin, H.X. The miR156-SPL 9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014, 80, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Gou, J.-Y.; Felippes, F.F.; Liu, C.-J.; Weigel, D.; Wang, J.-W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef] [Green Version]
- Yu, N.; Niu, Q.W.; Ng, K.H.; Chua, N.H. The role of miR156/SPL s modules in Arabidopsis lateral root development. Plant J. 2015, 83, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Wu, M.-F.; Yang, L.; Wu, G.; Poethig, R.S.; Wagner, D. The MicroRNA-Regulated SBP-Box Transcription Factor SPL3 Is a Direct Upstream Activator of LEAFY, FRUITFULL, and APETALA1. Dev. Cell 2009, 17, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Ye, M.; Sang, M.; Wu, R. A Regulatory Network for miR156-SPL Module in Arabidopsis thaliana. Int. J. Mol. Sci. 2019, 20, 6166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, S.; Salinas, M.; Höhmann, S.; Berndtgen, R.; Huijser, P. miR156-Targeted and Nontargeted SBP-Box Transcription Factors Act in Concert to Secure Male Fertility in Arabidopsis. Plant Cell 2010, 22, 3935–3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hongyuan, Z.; Jihong, H.; Qian, Q.; Hao, C.; Jing, J.; Yi, D. Small RNA Profiles of the Rice PTGMS Line Wuxiang S Reveal miRNAs Involved in Fertility Transition. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Yoo, S.J.; Lee, J.H.; Kim, W.; Yoo, S.K.; Fitzgerald, H.; Carrington, J.C.; Ahn, J.H. Genetic framework for flowering-time regulation by ambient temperature-responsive miRNAs in Arabidopsis. Nucleic Acids Res. 2010, 38, 3081–3093. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Galvão, V.C.; Zhang, Y.-C.; Horrer, D.; Zhang, T.-Q.; Hao, Y.-H.; Feng, Y.-Q.; Wang, S.; Schmid, M.; Wang, J.-W. Gibberellin Regulates the Arabidopsis Floral Transition through miR156-Targeted SQUAMOSA PROMOTER BINDING–LIKE Transcription Factors. Plant Cell 2012, 24, 3320–3332. [Google Scholar] [CrossRef] [Green Version]
- Austin, M.B.; Noel, J.P. The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 2003, 20, 79–110. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.J.; Harding, S.A.; Lung, J.; Popko, J.L.; Ralph, J.; Stokke, D.D.; Tsai, C.J.; Chiang, V.L. Repression of lignin biosynthesis promotes cellulose accumulation and growth in transgenic trees. Nat. Biotechnol. 1999, 17, 808–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajita, S.; Hishiyama, S.; Tomimura, Y.; Katayama, Y.; Omori, S. Structural Characterization of Modified Lignin in Transgenic Tobacco Plants in Which the Activity of 4-Coumarate:Coenzyme A Ligase Is Depressed. Plant Physiol. 1997, 114, 871–879. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Meyer, K.; Chapple, C.; Douglas, C.J. Antisense suppression of 4-coumarate:coenzyme A ligase activity in Arabidopsis leads to altered lignin subunit composition. Plant Cell 1997, 9, 1985–1998. [Google Scholar] [CrossRef] [Green Version]
- Gui, J.; Shen, J.; Li, L. Functional Characterization of Evolutionarily Divergent 4-Coumarate:Coenzyme A Ligases in Rice. Plant Physiol. 2011, 157, 574–586. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.-S.; Zhang, B.; Zhan, H.; Lv, Y.-L.; Jia, X.-L.; Wang, T.; Yang, N.-Y.; Lou, Y.-X.; Zhang, Z.-B.; Hu, W.-J.; et al. Phenylpropanoid Derivatives Are Essential Components of Sporopollenin in Vascular Plants. Mol. Plant 2020, 13, 1644–1653. [Google Scholar] [CrossRef]
- Hu, B.; Zhu, C.; Li, F.; Tang, J.; Wang, Y.; Lin, A.; Liu, L.; Che, R.; Chu, C. LEAF TIP NECROSIS1 Plays a Pivotal Role in the Regulation of Multiple Phosphate Starvation Responses in Rice. Plant Physiol. 2011, 156, 1101–1115. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.G.; Wang, K.; Zou, C.; Xu, C.; Li, W.X. The PILNCR1-miR399 Regulatory Module Is Important for Low Phosphate Tolerance in Maize. Plant Physiol. 2018, 1743–1753. [Google Scholar] [CrossRef] [Green Version]
- Hiroaki, F.; Jen, C.T.; Len, S.I.; Kyaw, A.; Kang, Z.J. A miRNA Involved in Phosphate-Starvation Response in Arabidopsis. Curr. Biol. 2005, 15, 2038–2043. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Wang, J.; Zhao, J.; Tian, J.; Liao, H. Control of phosphate homeostasis through gene regulation in crops. Curr. Opin. Plant Biol. 2014, 21, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Misson, J.; Raghothama, K.G.; Jain, A.; Jouhet, J.; Block, M.A.; Bligny, R.; Ortet, P.; Creff, A.; Somerville, S.; Rolland, N.; et al. A genome-wide transcriptional analysis using Arabidopsis thaliana Affymetrix gene chips determined plant responses to phosphate deprivation. Proc. Natl. Acad. Sci. USA 2005, 102, 11934–11939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasaki, J.; Yonetani, R.; Kuroda, S.; Shinano, T.; Yazaki, J.; Fujii, F.; Shimbo, K.; Yamamoto, K.; Sakata, K.; Sasaki, T.; et al. Transcriptomic analysis of metabolic changes by phosphorus stress in rice plant roots. Plant Cell Environ. 2003, 26, 1515–1523. [Google Scholar] [CrossRef]
- Zhang, K.; Song, Q.; Wei, Q.; Wang, C.; Su, Z. Down-regulation of OsSPX1 caused semi-male sterility, resulting in reduction of grain yield in rice. Plant Biotechnol. J. 2016, 14, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Sui, X.; Lucas, W.J.; Li, Y.; Feng, S.; Ma, S.; Fan, J.; Gao, L.; Zhang, Z. Down-regulation of the Sucrose Transporter CsSUT1 Causes Male Sterility by Altering Carbohydrate Supply. Plant Physiol. 2019, 180, 986–997. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, X.; Hu, Q.; Dai, X.; Tian, H.; Zheng, K.; Wang, X.; Mao, T.; Chen, J.-G.; Wang, S. Characterization of an activation-tagged mutant uncovers a role of GLABRA2 in anthocyanin biosynthesis in Arabidopsis. Plant J. 2015, 83, 300–311. [Google Scholar] [CrossRef]
- Chen, S.; Wang, S. GLABRA2, a Common Regulator for Epidermal Cell Fate Determination and Anthocyanin Biosynthesis in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 4997. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, J.; Chen, S.; Heng, Y.; Chen, Z.; Yang, J.; Zhou, K.; Pei, J.; He, H.; Deng, X.W.; et al. Poaceae-specific MS1 encodes a phospholipid-binding protein for male fertility in bread wheat. Proc. Natl. Acad. Sci. USA 2017, 114, 12614–12619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhao, G.; Li, Y.; Mo, N.; Zhang, J.; Liang, Y. Transcriptomic Analysis Implies That GA Regulates Sex Expression via Ethylene-Dependent and Ethylene-Independent Pathways in Cucumber (Cucumis sativus L.). Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Tanurdzic, M.; Banks, J.A. Sex-Determining Mechanisms in Land Plants. Plant Cell 2004, 16, S61–S71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Q.Q.; Wang, S.G. Judgment of young panicle differentiation stage of hybrid Rice. Crop Res. 1997, 11, 14–16. [Google Scholar] [CrossRef]
- Deng, Q.Y.; OU, A.H.; Fun, Z.Q.; Zhu, Q.R. A preliminary study on the method for identifying the practival photo-and thermo-sensitive genic male sterile rice. Hybrid Rice 1996, 2, 23–27. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2011, 40, 37–52. [Google Scholar] [CrossRef]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.J.; Ma, Y.K.; Chen, T.; Wang, M.; Wang, X.J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2008, 25, 130–131. [Google Scholar] [CrossRef]
- Sumczynski, D.; Bubelova, Z.; Sneyd, J.; Erb-Weber, S.; Mlcek, J. Total phenolics, flavonoids, antioxidant activity, crude fibre and digestibility in non-traditional wheat flakes and muesli. Food Chem. 2015, 174, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.-F.; Song, J.; Xiong, J.; Li, Y.-R.; Chen, H.-Z.; Le, M.-K.; Tao, L.-X. Changes of Oxidative Stress and Soluble Sugar in Anthers Involve in Rice Pollen Abortion Under Drought Stress. Agric. Sci. China 2011, 10, 1016–1025. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Treatment | Pollen Fertility (%) | Seed Setting Rate (%) |
|---|---|---|---|
| 2018 | PA64S(S) | 0.00 | 0.00 |
| PA64S(F) | 41.35 ± 1.76 ** | 34.27 ± 5.99 ** | |
| 2019 | PA64S(S) | 0.00 | 0.00 |
| PA64S(F) | 30.72 ± 2.69 ** | 31.40 ± 7.87 ** |
| miRNA Family | Stage 6 | Stage 7 | ||||
|---|---|---|---|---|---|---|
| miRNA Number | Up | Down | miRNA Number | Up | Down | |
| MIR156 | 1 | 1 | 0 | - | - | - |
| MIR164 | 1 | 0 | 1 | - | - | - |
| MIR396_2 | 1 | 1 | 0 | - | - | - |
| MIR397 | 2 | 0 | 2 | - | - | - |
| MIR5079 | 1 | 1 | 0 | - | - | - |
| MIR5143 | 1 | 1 | 0 | - | - | - |
| MIR171_1 | 1 | 1 | 0 | 1 | 1 | 0 |
| MIR1861 | 2 | 2 | 0 | 4 | 4 | 0 |
| MIR1862 | 1 | 1 | 0 | 2 | 2 | 0 |
| MIR1863 | 1 | 1 | 0 | 1 | 1 | 0 |
| MIR1878 | 1 | 1 | 0 | 1 | 1 | 0 |
| MIR2118 | 2 | 0 | 2 | 1 | 1 | 0 |
| MIR2863 | 1 | 1 | 1 | 1 | 1 | 0 |
| MIR437 | 1 | 1 | 0 | 1 | 1 | 0 |
| MIR812 | 3 | 1 | 2 | 2 | 1 | 1 |
| MIR169_1 | - | - | - | 1 | 1 | 0 |
| MIR169_2 | - | - | - | 2 | 2 | 0 |
| MIR169_4 | - | - | - | 1 | 1 | 0 |
| MIR1883 | - | - | - | 1 | 1 | 0 |
| MIR2275 | - | - | - | 2 | 1 | 1 |
| MIR395 | - | - | - | 4 | 4 | 0 |
| MIR399 | - | - | - | 3 | 3 | 0 |
| MIR444 | - | - | - | 1 | 0 | 1 |
| MIR5160 | - | - | - | 1 | 0 | 1 |
| MIR529 | - | - | - | 1 | 0 | 1 |
| MIR820 | - | - | - | 2 | 0 | 2 |
| MIR827 | - | - | - | 1 | 1 | 0 |
| Degradome Category Type | Cleavages Events | Genes | miRNAs |
|---|---|---|---|
| Category 0 | 633 | 287 | 89 |
| Category 1 | 99 | 68 | 59 |
| Category 2 | 1440 | 960 | 262 |
| Category 3 | 195 | 161 | 75 |
| Category 4 | 851 | 776 | 233 |
| miRNA | Target Genes | Relative Expression Level in Response to Fertility | Alignment Range | Cleavage Site | Category | ||||
|---|---|---|---|---|---|---|---|---|---|
| miRNA log2 | miRNA | Target log2 | Target | ||||||
| osa-miR156a | OS08G0531600 | −1.81 | down | 2.04 | up | 1053–1073 | 1064 | 2 | |
| osa-miR164a | OS06G0675600 | 0.32 | up | −2.58 | down | 954–974 | 965 | 0 | |
| osa-miR528-5p | OS09G0365900 | 1.05 | up | −2.41 | down | 164–183 | 174 | 2 | |
| OS12G0552300 | 1.05 | up | 1.83 | up | 2790–2809 | 2800 | 4 | ||
| osa-miR5488 | OS02G0177600 | 1.22 | up | −2.14 | down | 1516–1535 | 1526 | 2 | |
| osa-miR171b | OS10G0551200 | −0.83 | down | −1.16 | down | 549–569 | 560 | 2 | |
| OS05G0417100 | −0.83 | down | 0.87 | up | 1957–1977 | 1968 | 2 | ||
| osa-miR319a-3p.2-3p | OS01G0755500 | −0.97 | down | −1.09 | down | 1236–1254 | 1245 | 0 | |
| OS03G0785800 | −0.97 | down | −1.14 | down | 1182–1201 | 1192 | 0 | ||
| osa-miR396c-5p | OS04G0600900 | 0.52 | up | −1.78 | down | 404–425 | 415 | 0 | |
| OS02G0776900 | 0.52 | up | −1.26 | down | 570–590 | 581 | 0 | ||
| osa-miR156l-5p | OS01G0922600 | −1.81 | down | −0.70 | down | 615–635 | 626 | 0 | |
| osa-miR172d-5p | OS02G0582400 | 1.21 | up | −0.75 | down | 671–689 | 681 | 2 | |
| osa-miR399a | OS04G0415000 | −1.36 | down | 0.90 | up | 489–510 | 500 | 2 | |
| osa-miR399d | OS04G0415000 | −1.14 | down | 0.90 | up | 489–510 | 500 | 2 | |
| osa-miR399j | OS04G0415000 | −1.10 | down | 0.90 | up | 489–510 | 500 | 2 | |
| osa-miR419 | OS01G0606000 | −1.86 | down | 2.44 | up | 108–127 | 119 | 4 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Xiong, X.; Wang, Q.; Zhu, L.; Wang, L.; He, Y.; Zeng, H. Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the MiR156, MiR5488 and MiR399 Are Involved in the Regulation of Male Sterility in PTGMS Rice. Int. J. Mol. Sci. 2021, 22, 2260. https://doi.org/10.3390/ijms22052260
Sun Y, Xiong X, Wang Q, Zhu L, Wang L, He Y, Zeng H. Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the MiR156, MiR5488 and MiR399 Are Involved in the Regulation of Male Sterility in PTGMS Rice. International Journal of Molecular Sciences. 2021; 22(5):2260. https://doi.org/10.3390/ijms22052260
Chicago/Turabian StyleSun, Yujun, Xinguo Xiong, Qian Wang, Lan Zhu, Lei Wang, Ying He, and Hanlai Zeng. 2021. "Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the MiR156, MiR5488 and MiR399 Are Involved in the Regulation of Male Sterility in PTGMS Rice" International Journal of Molecular Sciences 22, no. 5: 2260. https://doi.org/10.3390/ijms22052260
APA StyleSun, Y., Xiong, X., Wang, Q., Zhu, L., Wang, L., He, Y., & Zeng, H. (2021). Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the MiR156, MiR5488 and MiR399 Are Involved in the Regulation of Male Sterility in PTGMS Rice. International Journal of Molecular Sciences, 22(5), 2260. https://doi.org/10.3390/ijms22052260