
Mitochondrial Calcium Signaling in Pancreatic β-Cell


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pancreatic β-Cell Signal Transduction and Ca2+ Homeostasis
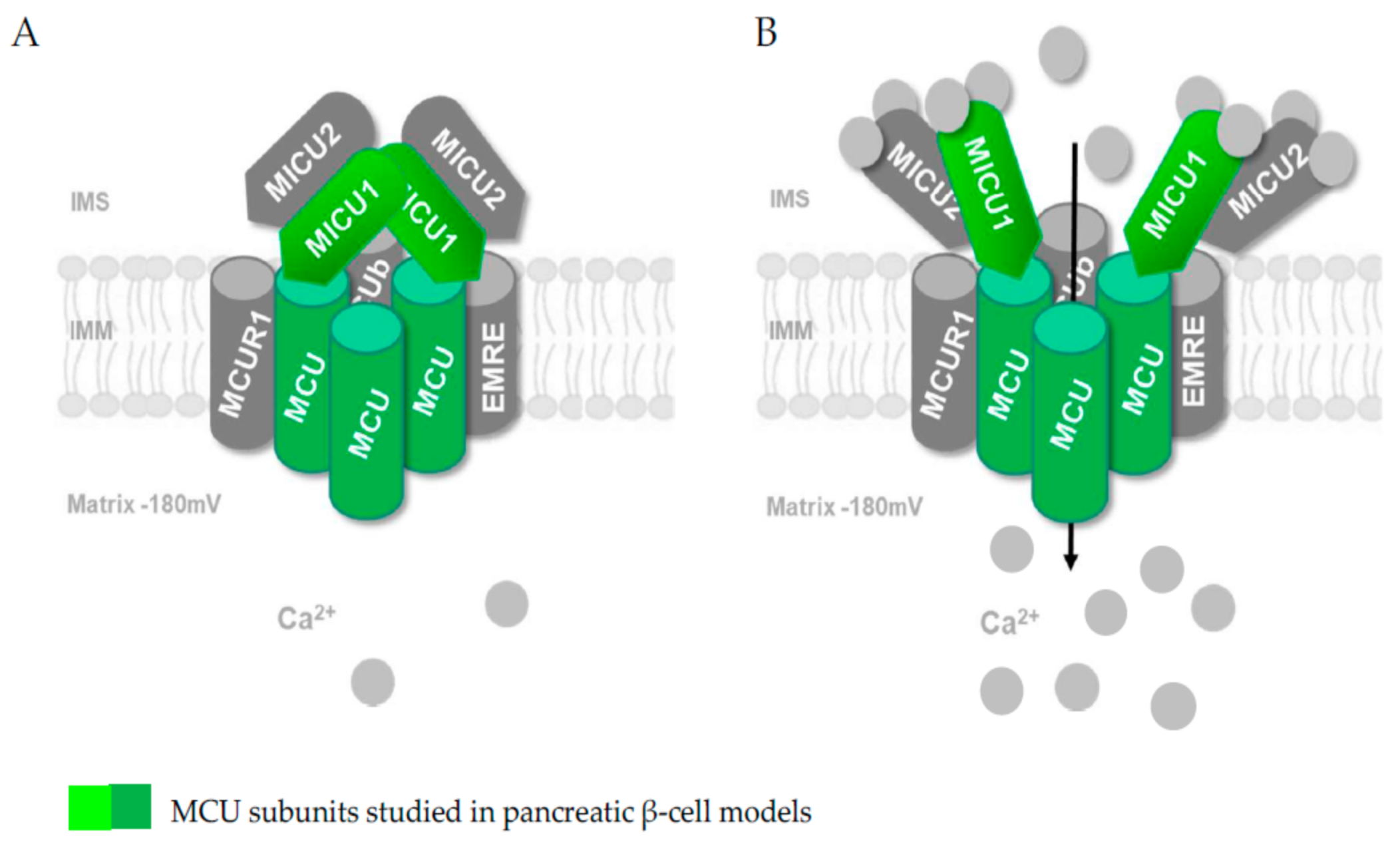
3. The Mitochondrial Calcium Uniporter and Its Existence in Pancreatic β-Cells
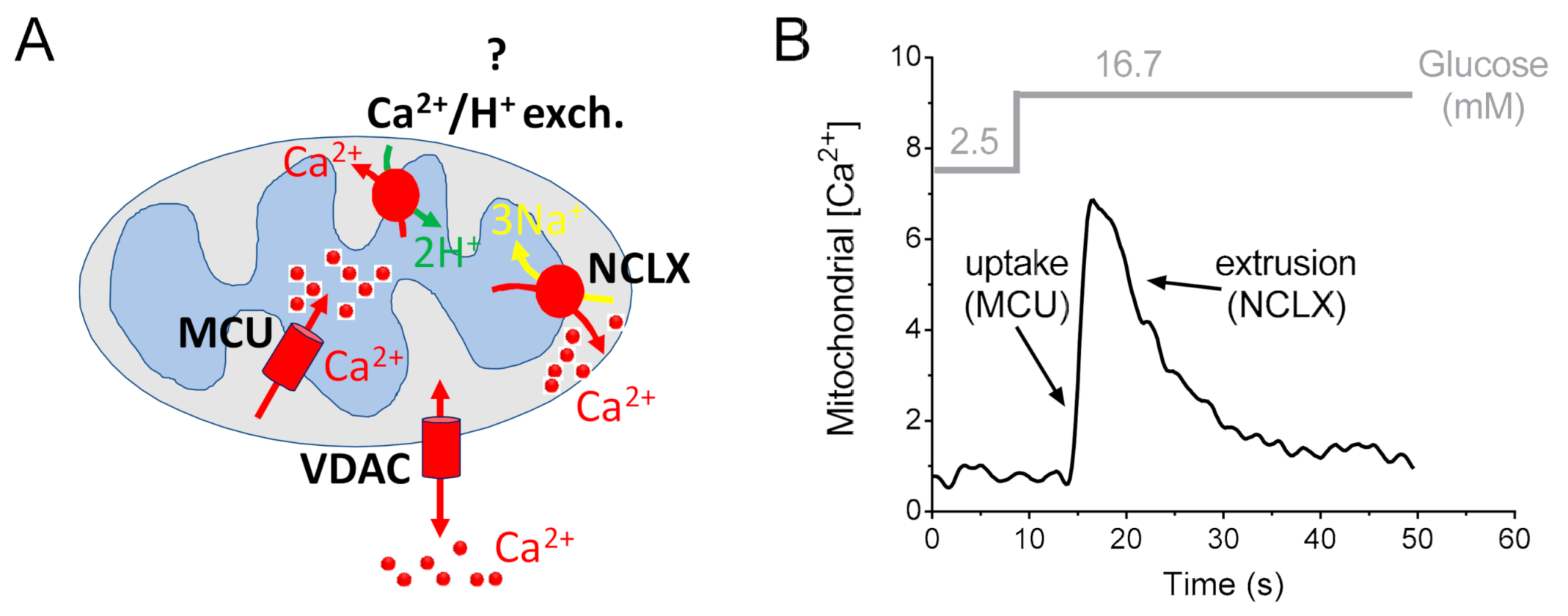
4. Role of Mitochondrial Ca2+ Uptake in the Pancreatic β-Cell
5. Mitochondrial Ca2+ Extrusion in the Pancreatic β-Cell
6. Mitochondrial Ca2+-Targeted Intervention Strategies to Modulate Pancreatic β-Cell Function
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahier, J.; Goebbels, R.M.; Henquin, J.-C. Cellular composition of the human diabetic pancreas. Diabetologia 1983, 24, 366–371. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef] [Green Version]
- Weir, G.C.; Bonner-Weir, S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004, 53, S16–S21. [Google Scholar] [CrossRef] [Green Version]
- Vetere, A.; Choudhary, A.; Burns, S.M.; Wagner, B.K. Targeting the pancreatic beta-cell to treat diabetes. Nat. Rev. Drug. Discov. 2014, 13, 278–289. [Google Scholar] [CrossRef]
- Gilon, P.; Chae, H.Y.; Rutter, G.A.; Ravier, M.A. Calcium signaling in pancreatic beta-cells in health and in Type 2 diabetes. Cell Calcium 2014, 56, 340–361. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Ashcroft, F.M. Pancreatic beta-cell electrical activity and insulin secretion: Of mice and men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Kanekura, K.; Lavagnino, Z.; Spears, L.D.; Abreu, D.; Mahadevan, J.; Yagi, T.; Semenkovich, C.F.; Piston, D.W.; Uranoet, F. Targeting cellular calcium homeostasis to prevent cytokine-mediated beta cell death. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, A.; Wollheim, C.B. Mitochondrial signals drive insulin secretion in the pancreatic beta-cell. Mol. Cell Endocrinol. 2012, 353, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic beta-cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef]
- Santo-Domingo, J.; Wiederkehr, A.; De Marchi, U. Modulation of the matrix redox signaling by mitochondrial Ca2+. World J. Biol. Chem. 2015, 6, 310–323. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, A.; Szanda, G.; Akhmedov, D.; Mataki, C.; Heizmann, C.W.; Schoonjans, K.; Pozzan, T.; Spat, A.; Wollheim, C.B. Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab. 2011, 13, 601–611. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Alam, M.R.; Groschner, L.N.; Parichatikanond, W.; Kuo, L.; Bondarenko, A.I.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic beta-cells. J. Biol. Chem. 2012, 287, 34445–34454. [Google Scholar] [CrossRef] [Green Version]
- Quan, X.; Nguyen, T.T.; Choi, S.K.; Xu, S.; Das, R.; Cha, S.K.; Kim, N.; Han, J.; Wiederkehr, A.; Wollheim, C.B.; et al. Essential role of mitochondrial Ca2+ uniporter in the generation of mitochondrial pH gradient and metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 2015, 290, 4086–4096. [Google Scholar] [CrossRef] [Green Version]
- Tarasov, A.I.; Semplici, F.; Ravier, M.A.; Bellomo, E.A.; Pullen, T.J.; Gilon, P.; Sekler, I.; Rizzuto, R.; Rutter, G.A. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic beta-cells. PLoS ONE 2012, 7, e39722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederkehr, A.; Wollheim, C.B. Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic beta-cell. Cell Calcium 2008, 44, 64–76. [Google Scholar] [CrossRef]
- Nicholls, D.G. The Pancreatic beta-cell: A bioenergetic perspective. Physiol. Rev. 2016, 96, 1385–1447. [Google Scholar] [CrossRef] [Green Version]
- Georgiadou, E.; Rutter, G.A. Control by Ca(2+) of mitochondrial structure and function in pancreatic beta-cells. Cell Calcium 2020, 91, 102282. [Google Scholar] [CrossRef]
- De Marchi, U.; Fernandez-Martinez, S.; de la Fuente, S.; Wiederkehr, A.; Santo-Domingo, J. Mitochondrial ion channels in pancreatic beta-cells: Novel pharmacological targets for the treatment of Type 2 diabetes. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maechler, P. Mitochondrial function and insulin secretion. Mol. Cell. Endocrinol. 2013, 379, 12–18. [Google Scholar] [CrossRef]
- Henquin, J.-C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000, 49, 1751–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, B.R.; Wollheim, C.B. Synaptotagmins bind calcium to release insulin. Am. J. Physiol. Metab. 2008, 295, E1279–E1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rorsman, P.; Braun, M.; Zhang, Q. Regulation of calcium in pancreatic alpha- and beta-cells in health and disease. Cell Calcium 2012, 51, 300–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idevall-Hagren, O.; Tengholm, A. Metabolic regulation of calcium signaling in beta cells. Semin. Cell Dev. Biol. 2020, 103, 20–30. [Google Scholar] [CrossRef]
- Klec, C.; Ziomek, G.; Pichler, M.; Malli, R.; Graier, W.F. Calcium signaling in ß-cell physiology and pathology: A revisit. Int. J. Mol. Sci. 2019, 20, 6110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P. Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 2008, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabourin, J.; Allagnat, F. Store-operated Ca2+ entry: A key component of the insulin secretion machinery. J. Mol. Endocrinol. 2016, 57, F35–F39. [Google Scholar] [CrossRef]
- Miura, Y.; Henquin, J.C.; Gilon, P. Emptying of intracellular Ca2+ stores stimulates Ca2+ entry in mouse pancreatic beta-cells by both direct and indirect mechanisms. J. Physiol. 1997, 503, 387–398. [Google Scholar] [CrossRef]
- Sabourin, J.; Le Gal, L.; Saurwein, L.; Haefliger, J.A.; Raddatz, E.; Allagnat, F. Store-operated Ca2+ entry mediated by orai1 and TRPC1 participates to insulin secretion in rat beta. Cells J. Biol. Chem. 2015, 290, 30530–30539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentki, M.; Janjic, D.; Biden, T.J.; Blondel, B.; Wollheim, C.B. Regulation of Ca2+ transport by isolated organelles of a rat insulinoma: Studies with endoplasmic reticulum and secretory granules. J. Biol. Chem. 1984, 259, 10118–10123. [Google Scholar] [CrossRef]
- Santulli, G.; Pagano, G.; Sardu, C.; Xie, W.; Reiken, S.; D’Ascia, S.L.; Cannone, M.; Marziliano, N.; Trimarco, B.; Guise, T.A.; et al. Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J. Clin. Investig. 2015, 125, 1968–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klec, C.; Madreiter-Sokolowski, C.T.; Ziomek, G.; Stryeck, S.; Sachdev, V.; Duta-Mare, M.; Gottschalk, B.; Depaoli, M.R.; Rost, R.; Hay, J.; et al. Presenilin-1 established ER-Ca(2+) leak: A Follow up on its importance for the initial insulin secretion in pancreatic islets and beta-cells upon elevated glucose. Cell. Physiol. Biochem. 2019, 53, 573–586. [Google Scholar] [PubMed] [Green Version]
- Klec, C.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Sachdev, V.; Duta-Mare, M.; Gottschalk, B.; Depaoli, M.R.; Rost, R.; Hay, J.; Waldeck-Weiermair, M.; et al. Glycogen synthase kinase 3 beta controls presenilin-1-mediated endoplasmic reticulum Ca2+ leak directed to mitochondria in pancreatic islets and beta-cells. Cell. Physiol. Biochem. 2019, 52, 57–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouriou, Y.; Demaurex, N.; Bijlenga, P.; De Marchi, U. Mitochondrial calcium handling during ischemia-induced cell death in neurons. Biochimie 2011, 93, 2060–2067. [Google Scholar] [CrossRef] [Green Version]
- Gembal, M.; Detimary, P.; Gilon, P.; Gao, Z.Y.; Henquin, J.C. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J. Clin. Investig. 1993, 91, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Jonas, J.C.; Li, G.; Palmer, M.; Weller, U.; Wollheim, C.B. Dynamics of Ca2+ and guanosine 5′-[gamma-thio]triphosphate action on insulin secretion from alpha-toxin-permeabilized HIT-T15 cells. Biochem. J. 1994, 301, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.; Theler, J.M.; Murgia, M.; Wollheim, C.B.; Pozzan, T.; Rizzuto, R. Stimulated Ca2+ influx raises mitochondrial free Ca2+ to supramicromolar levels in a pancreatic beta-cell line: Possible role in glucose and agonist-induced insulin secretion. J. Biol. Chem. 1993, 268, 22385–22390. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nat. Cell Biol. 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Patron, M.; Rizzuto, R. Structure and function of the mitochondrial calcium uniporter complex. Biochim. Biophys. Acta Bioenerg. 2015, 1853, 2006–2011. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Jhun, B.S.; Hurst, S.; O-Uchi, J.; Csordás, G.; Sheu, S.-S. The mitochondrial Ca2+ uniporter: Structure, function, and pharmacology. Handb. Exp. Pharmacol. 2017, 240, 129–156. [Google Scholar]
- Mallilankaraman, K.; Doonan, P.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foskett, J.K.; Madesh, M. Regulation of the mitochondrial Ca2+ uniporter by MICU1 and micubiochem. Biophys. Res. Commun. 2014, 449, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Bermont, F.; Hermant, A.; Benninga, R.; Chabert, C.; Jacot, G.; Santo-Domingo, J.; Kraus, M.R.; Feige, J.N.; De Marchi, U. Targeting mitochondrial calcium uptake with the natural flavonol kaempferol, to promote metabolism/secretion coupling in pancreatic β-cells. Nutrients 2020, 12, 538. [Google Scholar] [CrossRef] [Green Version]
- Pitter, J.; Maechler, P.; Wollheim, C.; Spät, A. Mitochondria respond to Ca2+ already in the submicromolar range: Correlation with redox state. Cell Calcium 2002, 31, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Golenar, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; de la Fuente Perez, S.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesada, I.; Villalobos, C.; Nunez, L.; Chamero, P.; Alonso, M.T.; Nadal, A.; Garcia-Sancho, J. Glucose induces synchronous mitochondrial calcium oscillations in intact pancreatic islets. Cell Calcium 2008, 43, 39–47. [Google Scholar] [CrossRef]
- Kennedy, E.D.; Rizzuto, R.; Theler, J.M.; Pralong, W.F.; Bastianutto, C.; Pozzan, T.; Wollheim, C.B. Glucose-stimulated insulin secretion correlates with changes in mitochondrial and cytosolic Ca2+ in aequorin-expressing INS-1 cells. J. Clin. Investig. 1996, 98, 2524–2538. [Google Scholar] [CrossRef]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [Green Version]
- Fan, M. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Kamer, K.J.; Jiang, W.; Kaushik, V.K.; Mootha, V.K.; Grabarek, Z. Crystal structure of MICU2 and comparison with MICU1 reveal insights into the uniporter gating mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 3546–3555. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.B.; Tsai, C.W.; Tsai, M.F. The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. eLife 2019, 8. [Google Scholar] [CrossRef]
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulović, S.; Eroglu, E.; Sattler, W.; et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat. Commun. 2019, 10, 3732. [Google Scholar] [CrossRef] [Green Version]
- Georgiadou, E.; Haythorne, E.; Dickerson, M.T.; Lopez-Noriega, L.; Pullen, T.J.; da Silva Xavier, G.; Davis, S.P.X.; Martinez-Sanchez, A.; Semplici, F.; Rizzuto, R.; et al. The pore-forming subunit MCU of the mitochondrial Ca(2+) uniporter is required for normal glucose-stimulated insulin secretion in vitro and in vivo in mice. Diabetologia 2020, 63, 1368–1381. [Google Scholar] [CrossRef] [PubMed]
- Harrington, J.L.; Murphy, E. The mitochondrial calcium uniporter: Mice can live and die without it. J. Mol. Cell. Cardiol. 2015, 78, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Pallafacchina, G.; Zanin, S.; Rizzuto, R. Recent advances in the molecular mechanism of mitochondrial calcium uptake. F1000Res 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Grabarek, Z.; Mootha, V.K. High-affinity cooperative Ca(2+) binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep. 2017, 18, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Csordás, G.; Huang, K.-T.; Várnai, P.; Joseph, S.K.; Hajnóczky, G. MICU1 interacts with the D-ring of the MCU pore to control its Ca(2+) flux and sensitivity to Ru. Mol. Cell 2018, 72, 778–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.-F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.-W.; Wu, Y.; Willliams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. eLife 2016, 5, e15545. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Sancak, Y.; Fomina, Y.; Meisel, J.D.; Chaudhuri, D.; Grabarek, Z.; Mootha, V.K. MICU1 imparts the mitochondrial uniporter with the ability to discriminate between Ca2+ and Mn2+. Proc. Natl. Acad. Sci. USA 2018, 115, E7960–E7969. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.-X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 serves as a molecular gatekeeper to prevent In Vivo mitochondrial calcium overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- De la Fuente, S.; Matesanz-Isabel, J.; Fonteriz, R.I.; Montero, M.; Alvarez, J. Dynamics of mitochondrial Ca2+ uptake in MICU1-knockdown cells. Biochem. J. 2014, 458, 33–40. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 2014, 289, 20377–20385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Min, C.K.; Kim, T.G.; Song, H.K.; Lim, Y.; Kim, D.; Shin, K.; Kang, M.; Kang, J.Y.; Youn, H.S.; et al. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep. 2015, 16, 1318–1333. [Google Scholar] [CrossRef]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef] [Green Version]
- Enslow, B.T.; Madaris, T.; Ravichandran, J.; Jacob, R.; Srikantan, S.; Stathopulos, P.; Muniswamy, M. Identification of critical MCUR1 domains in the mitochondrial calcium uniporter complex that regulates cellular metabolism. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Paupe, V.; Prudent, J.; Dassa, E.P.; Rendon, O.Z.; Shoubridge, E.A. CCDC90A (MCUR1) is a cytochrome C Oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. 2015, 21, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fieni, F.; Lee, S.B.; Jan, Y.N.; Kirichok, Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 2012, 3, 1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Nagashima, K.; Inagaki, N.; Kioka, H.; Takashima, S.; Fukuoka, H.; Noji, H.; Kakizuka, A.; Imamura, H. Glucose-stimulated single pancreatic islets sustain increased cytosolic ATP levels during initial Ca2+ influx and subsequent Ca2+ oscillations. J. Biol. Chem. 2014, 289, 2205–2216. [Google Scholar] [CrossRef] [Green Version]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R.M.; McCormack, J.G. The calcium sensitive dehydrogenases of vertebrate mitochondria. Cell Calcium 1986, 7, 377–386. [Google Scholar] [CrossRef]
- Denton, R.M.; Randle, P.J.; Martin, B.R. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem. J. 1972, 128, 161–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R.M.; Richards, D.A.; Chin, J.G. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 1978, 176, 899–906. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979, 180, 533–544. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, U.; Thevenet, J.; Hermant, A.; Dioum, E.; Wiederkehr, A. Calcium co-regulates oxidative metabolism and ATP synthase-dependent respiration in pancreatic beta cells. J. Biol. Chem. 2014, 289, 9182–9194. [Google Scholar] [CrossRef] [Green Version]
- Gilon, P.; Shepherd, R.M.; Henquin, J.C. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J. Biol. Chem. 1993, 268, 22265–22268. [Google Scholar] [CrossRef]
- Griesche, N.; Sanchez, G.; Hermans, C.; Idevall-Hagren, O. Cortical mitochondria regulate insulin secretion by local Ca(2+) buffering in rodent beta cells. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes mellitus, mitochondrial dysfunction and Ca(2+)-dependent permeability transition pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Gutierrez-Aguilar, M. The still uncertain identity of the channel-forming unit(s) of the mitochondrial permeability transition pore. Cell Calcium 2018, 73, 121–130. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef]
- Dufer, M.; Krippeit-Drews, P.; Lembert, N.; Idahl, L.A.; Drews, G. Diabetogenic effect of cyclosporin A is mediated by interference with mitochondrial function of pancreatic B-cells. Mol. Pharmacol. 2001, 60, 873–879. [Google Scholar] [PubMed]
- Fujimoto, K.; Chen, Y.; Polonsky, K.S.; Dorn, G.W. Targeting cyclophilin D and the mitochondrial permeability transition enhances beta-cell survival and prevents diabetes in Pdx1 deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 10214–10219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshkin, V.; Bikopoulos, G.; Chan, C.B.; Wheeler, M.B. The characterization of mitochondrial permeability transition in clonal pancreatic beta-cells: Multiple modes and regulation. J. Biol. Chem. 2004, 279, 41368–41376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lablanche, S.; Cottet-Rousselle, C.; Argaud, L.; Laporte, C.; Lamarche, F.; Richard, M.J.; Berney, T.; Benhamou, P.Y.; Fontaine, E. Respective effects of oxygen and energy substrate deprivation on beta cell viability. Biochim. Biophys. Acta 2015, 1847, 629–639. [Google Scholar] [CrossRef]
- Lu, A.; Chu, C.; Mulvihill, E.; Wang, R.; Liang, W. ATP-sensitive K(+) channels and mitochondrial permeability transition pore mediate effects of hydrogen sulfide on cytosolic Ca(2+) homeostasis and insulin secretion in beta-cells. Pflugers Arch. 2019, 471, 1551–1564. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef] [PubMed]
- Zoratti, M.; Szabo, I.; De Marchi, U. Mitochondrial permeability transitions: How many doors to the house? Biochim. Biophys. Acta 2005, 1706, 40–52. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. Why F-ATP synthase remains a strong candidate as the mitochondrial permeability transition pore. Front. Physiol. 2018, 9, 1543. [Google Scholar] [CrossRef] [Green Version]
- Carraro, M.; Jones, K.; Sartori, G.; Schiavone, M.; Antonucci, S.; Kucharczyk, R.; di Rago, J.P.; Franchin, C.; Arrigoni, G.; Forte, M.; et al. The unique cysteine of F-ATP synthase OSCP subunit participates in modulation of the permeability transition pore. Cell Rep. 2020, 32, 108095. [Google Scholar] [CrossRef] [PubMed]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin, D.J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, U.; Basso, E.; Szabo, I.; Zoratti, M. Electrophysiological characterization of the Cyclophilin D-deleted mitochondrial permeability transition pore. Mol. Membr. Biol. 2006, 23, 521–530. [Google Scholar] [CrossRef]
- Barbu, A.; Welsh, N.; Saldeen, J. Cytokine-induced apoptosis and necrosis are preceded by disruption of the mitochondrial membrane potential (Deltapsi(m)) in pancreatic RINm5F cells: Prevention by Bcl. Mol. Cell Endocrinol. 2002, 190, 75–82. [Google Scholar] [CrossRef]
- Contreras, J.L.; Smyth, C.A.; Bilbao, G.; Young, C.J.; Thompson, J.A.; Eckhoff, D.E. 17beta-Estradiol protects isolated human pancreatic islets against proinflammatory cytokine-induced cell death: Molecular mechanisms and islet functionality. Transplantation 2002, 74, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Lablanche, S.; Cottet-Rousselle, C.; Lamarche, F.; Benhamou, P.Y.; Halimi, S.; Leverve, X.; Fontaine, E. Protection of pancreatic INS-1 beta-cells from glucose- and fructose-induced cell death by inhibiting mitochondrial permeability transition with cyclosporin A or metformin. Cell Death Dis. 2011, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Arduino, D.M.; Wettmarshausen, J.; Vais, H.; Navas-Navarro, P.; Cheng, Y.; Leimpek, A.; Ma, Z.; Delrio-Lorenzo, A.; Giordano, A.; Garcia-Perez, C.; et al. Systematic identification of MCU modulators by orthogonal interspecies chemical screening. Mol. Cell 2017, 67, 711–723. [Google Scholar] [CrossRef]
- Woods, J.J.; Lovett, J.; Lai, B.; Harris, H.H.; Wilson, J.J. Redox stability controls the cellular uptake and activity of ruthenium-based inhibitors of the mitochondrial calcium uniporter (MCU). Angew. Chem. Int. Ed. Engl. 2020, 59, 6482–6491. [Google Scholar] [CrossRef]
- Woods, J.J.; Wilson, J.J. Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Cur. Opin. Chem. Biol. 2020, 55, 9–18. [Google Scholar] [CrossRef]
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017, 3, 17045. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Sharipov, R.R.; Boyarkin, D.P.; Belosludtseva, N.V.; Dubinin, M.V.; Krasilnikova, I.A.; Bakaeva, Z.V.; Zgodova, A.E.; Pinelis, V.G.; Surin, A.M. The effect of DS16570511, a new inhibitor of mitochondrial calcium uniporter, on calcium homeostasis, metabolism, and functional state of cultured cortical neurons and isolated brain mitochondria. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129847. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Pathak, T.; Trebak, M. Mitochondrial Ca(2+) signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assali, E.A.; Jones, A.E.; Veliova, M.; Acin-Perez, R.; Taha, M.; Miller, N.; Shum, M.; Oliveira, M.F.; Las, G.; Liesa, M.; et al. NCLX prevents cell death during adrenergic activation of the brown adipose tissue. Nat. Commun. 2020, 11, 3347. [Google Scholar] [CrossRef]
- Nita, I.I.; Hershfinkel, M.; Fishman, D.; Ozeri, E.; Rutter, G.A.; Sensi, S.L.; Khananshvili, D.; Lewis, E.C.; Sekler, I. The mitochondrial Na+/Ca2+ exchanger upregulates glucose dependent Ca2+ signalling linked to insulin secretion. PLoS ONE 2012, 7, e46649. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zhao, L.; Clish, C.B.; Clapham, D.E. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc. Natl. Acad. Sci. USA 2013, 110, E2249–E2254. [Google Scholar] [CrossRef] [Green Version]
- Dimmer, K.S.; Navoni, F.; Casarin, A.; Trevisson, E.; Endele, S.; Winterpacht, A.; Salviati, L.; Scorrano, L. LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum. Mol. Genet. 2008, 17, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Nowikovsky, K.; Froschauer, E.M.; Zsurka, G.; Samaj, J.; Reipert, S.; Kolisek, M.; Wiesenberger, G.; Schweyen, R.J. The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf-Hirschhorn syndrome. J. Biol. Chem. 2004, 279, 30307–30315. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.; Tavakoli, M.; Pfeiffer, C.; Seifert, J.; Mattarei, A.; De Stefani, D.; Zoratti, M.; Nowikovsky, K. LETM1-Mediated K(+) and Na(+) homeostasis regulates mitochondrial Ca(2+) efflux. Front. Physiol. 2017, 8, 839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimi, H.; McDonald, L.; Stribrna, E.; Lukes, J. Trypanosome Letm1 protein is essential for mitochondrial potassium homeostasis. J. Biol. Chem. 2013, 288, 26914–26925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Giorgi, C.; Bononi, A.; Marchi, S.; Patergnani, S.; Rimessi, A.; Rizzuto, R.; Pinton, P. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat. Protoc. 2013, 8, 2105–2118. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Miles, P.D.; Vargas, L.; Luan, P.; Glasco, S.; Kushnareva, Y.; Kornbrust, E.S.; Grako, K.A.; Wollheim, C.B.; Maechler, P.; et al. Inhibition of mitochondrial Na+-Ca2+ exchanger increases mitochondrial metabolism and potentiates glucose-stimulated insulin secretion in rat pancreatic islets. Diabetes 2003, 52, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Luciani, D.S.; Ao, P.; Hu, X.; Warnock, G.L.; Johnson, J.D. Voltage-gated Ca(2+) influx and insulin secretion in human and mouse beta-cells are impaired by the mitochondrial Na(+)/Ca(2+) exchange inhibitor CGP. Eur. J. Pharmacol. 2007, 576, 18–25. [Google Scholar]
- Thu, T.; Ahn, J.R.; Woo, S.H. Inhibition of L-type Ca2+ channel by mitochondrial Na+-Ca2+ exchange inhibitor CGP-37157 in rat atrial myocytes. Eur. J. Pharmacol. 2006, 552, 15–19. [Google Scholar] [CrossRef]
- Neumann, J.T.; Diaz-Sylvester, P.L.; Fleischer, S.; Copello, J.A. CGP-37157 inhibits the sarcoplasmic reticulum Ca(2)+ ATPase and activates ryanodine receptor channels in striated muscle. Mol. Pharmacol. 2011, 79, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Nita, I.I.; Hershfinkel, M.; Kantor, C.; Rutter, G.A.; Lewis, E.C.; Sekler, I. Pancreatic beta-cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria. FASEB J. 2014, 28, 3301–3312. [Google Scholar] [CrossRef] [PubMed]
- Montero, M.; Lobaton, C.D.; Hernandez-Sanmiguel, E.; Santodomingo, J.; Vay, L.; Moreno, A.; Alvarez, J. Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem. J. 2004, 384, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Heikkila, E.; Hermant, A.; Thevenet, J.; Bermont, F.; Kulkarni, S.S.; Ratajczak, J.; Santo-Domingo, J.; Dioum, E.H.; Canto, C.; Barron, D.; et al. The plant product quinic acid activates Ca(2+) -dependent mitochondrial function and promotes insulin secretion from pancreatic beta cells. Br. J. Pharmacol. 2019, 176, 3250–3263. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiser, A.; Feige, J.N.; De Marchi, U. Mitochondrial Calcium Signaling in Pancreatic β-Cell. Int. J. Mol. Sci. 2021, 22, 2515. https://doi.org/10.3390/ijms22052515
Weiser A, Feige JN, De Marchi U. Mitochondrial Calcium Signaling in Pancreatic β-Cell. International Journal of Molecular Sciences. 2021; 22(5):2515. https://doi.org/10.3390/ijms22052515
Chicago/Turabian StyleWeiser, Anna, Jerome N. Feige, and Umberto De Marchi. 2021. "Mitochondrial Calcium Signaling in Pancreatic β-Cell" International Journal of Molecular Sciences 22, no. 5: 2515. https://doi.org/10.3390/ijms22052515
APA StyleWeiser, A., Feige, J. N., & De Marchi, U. (2021). Mitochondrial Calcium Signaling in Pancreatic β-Cell. International Journal of Molecular Sciences, 22(5), 2515. https://doi.org/10.3390/ijms22052515