
PSMA-D4 Radioligand for Targeted Therapy of Prostate Cancer: Synthesis, Characteristics and Preliminary Assessment of Biological Properties

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Results
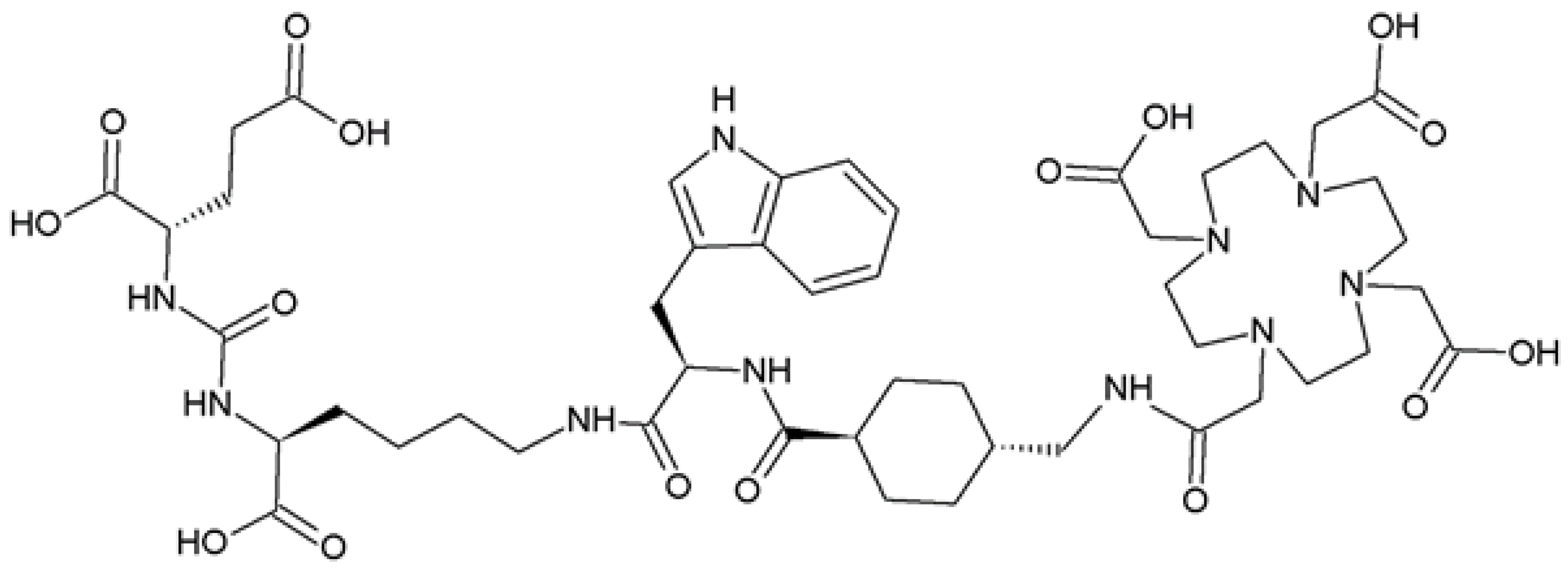
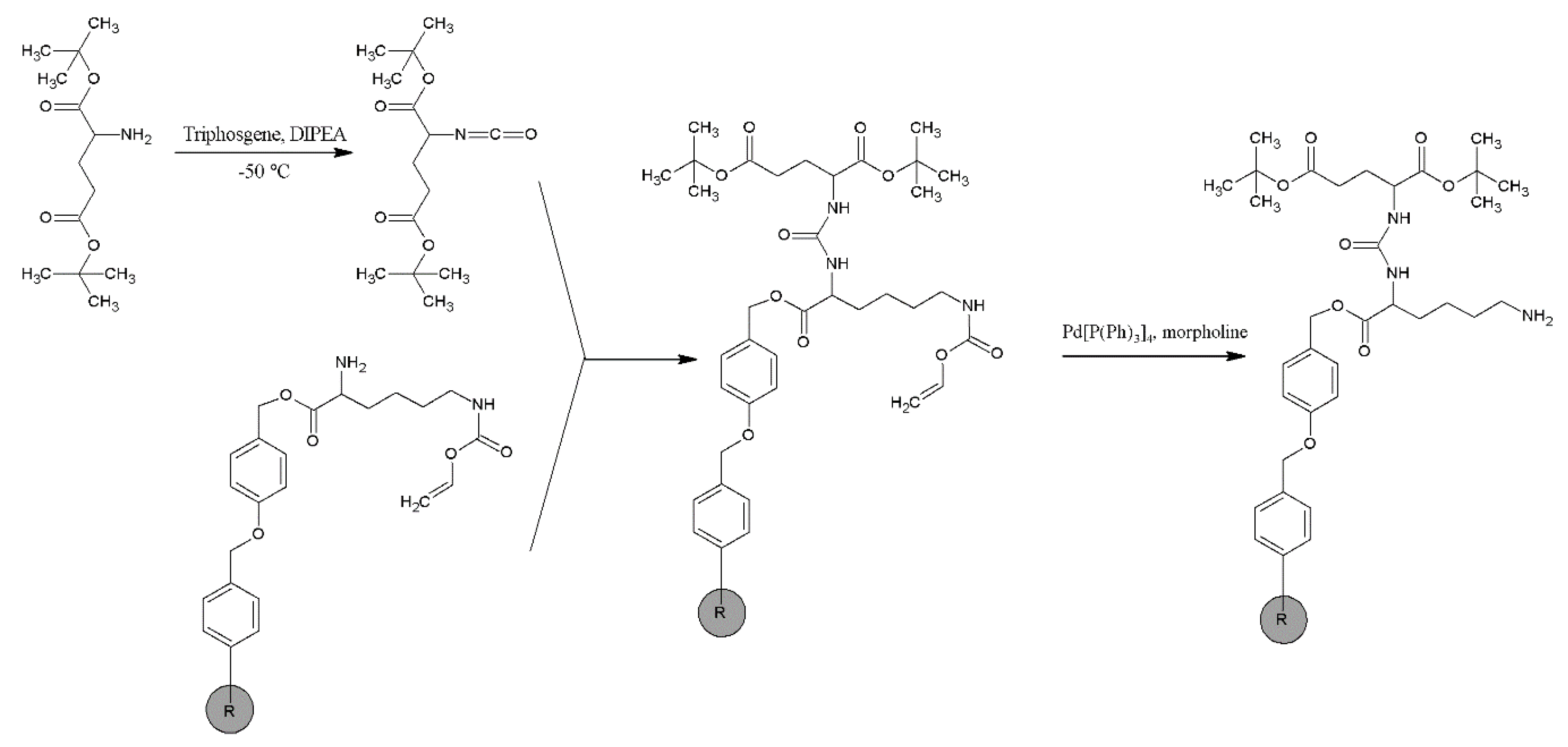
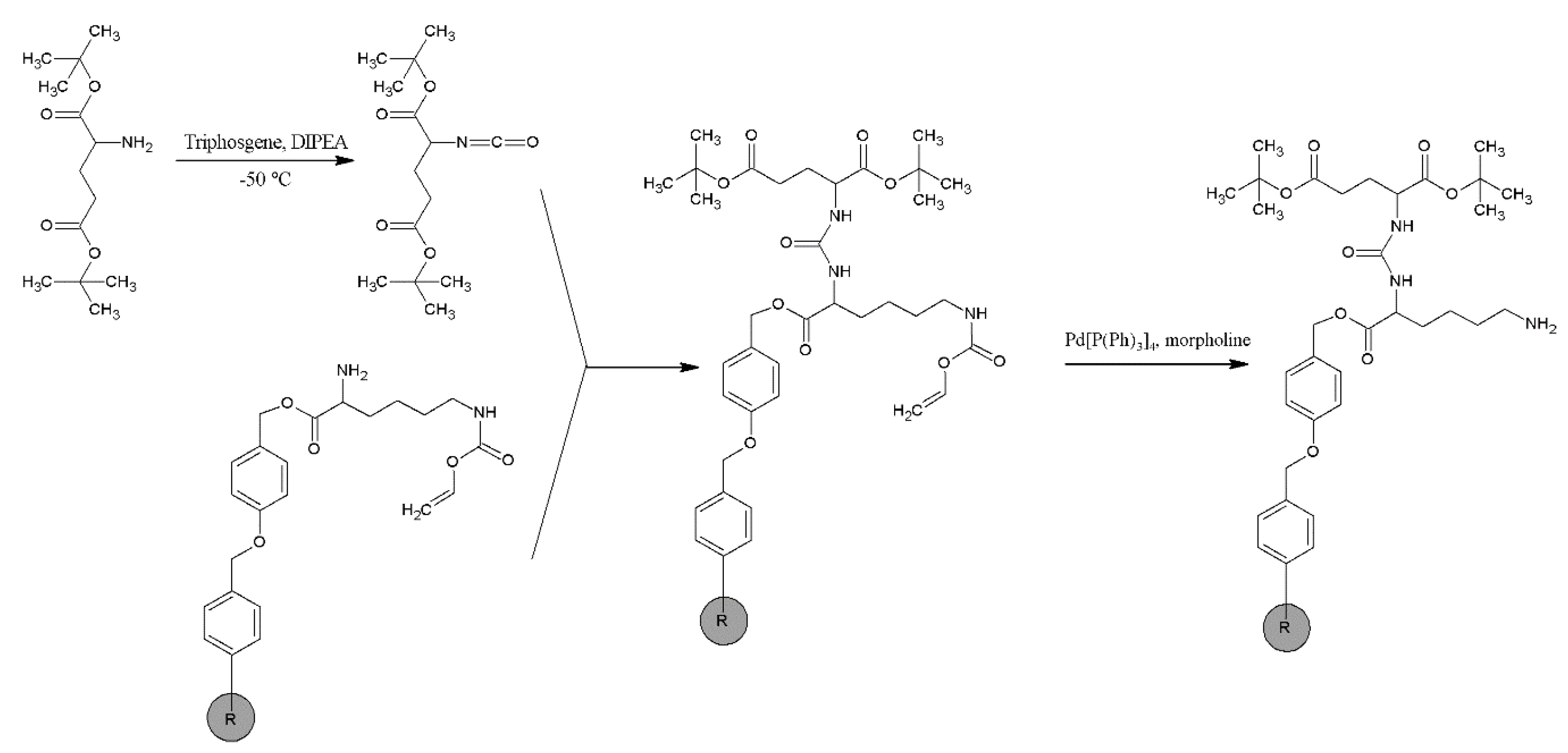
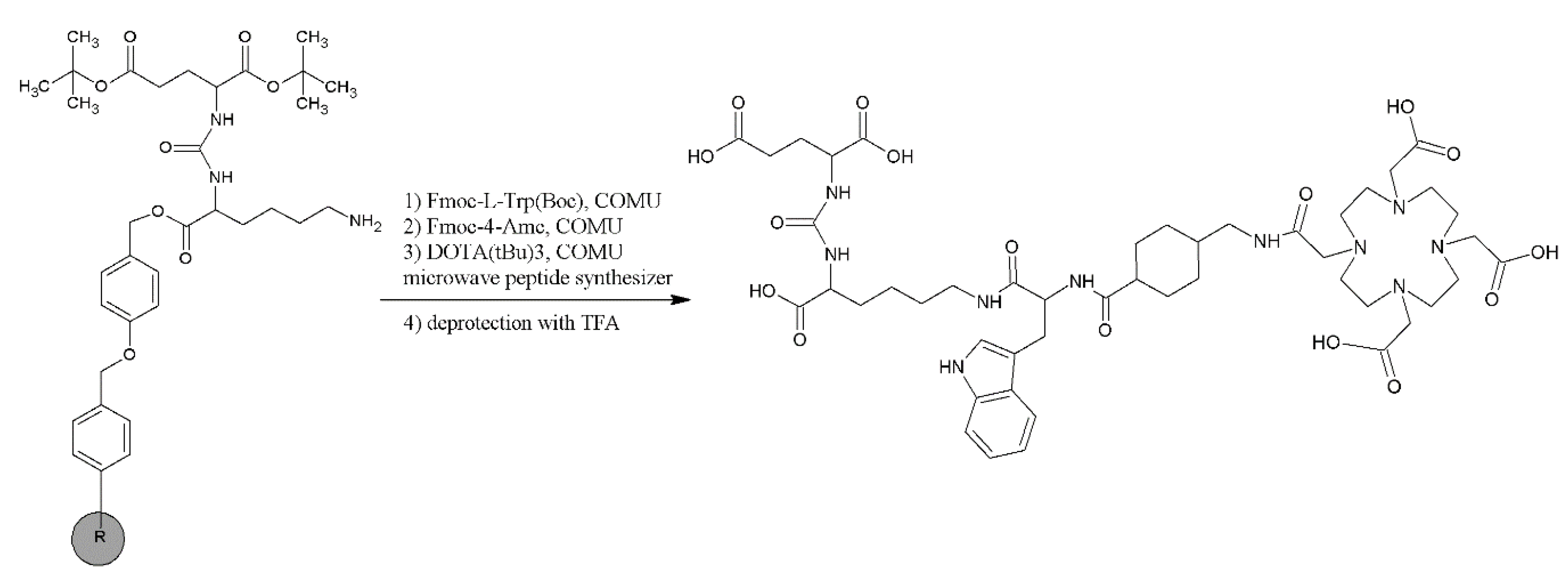
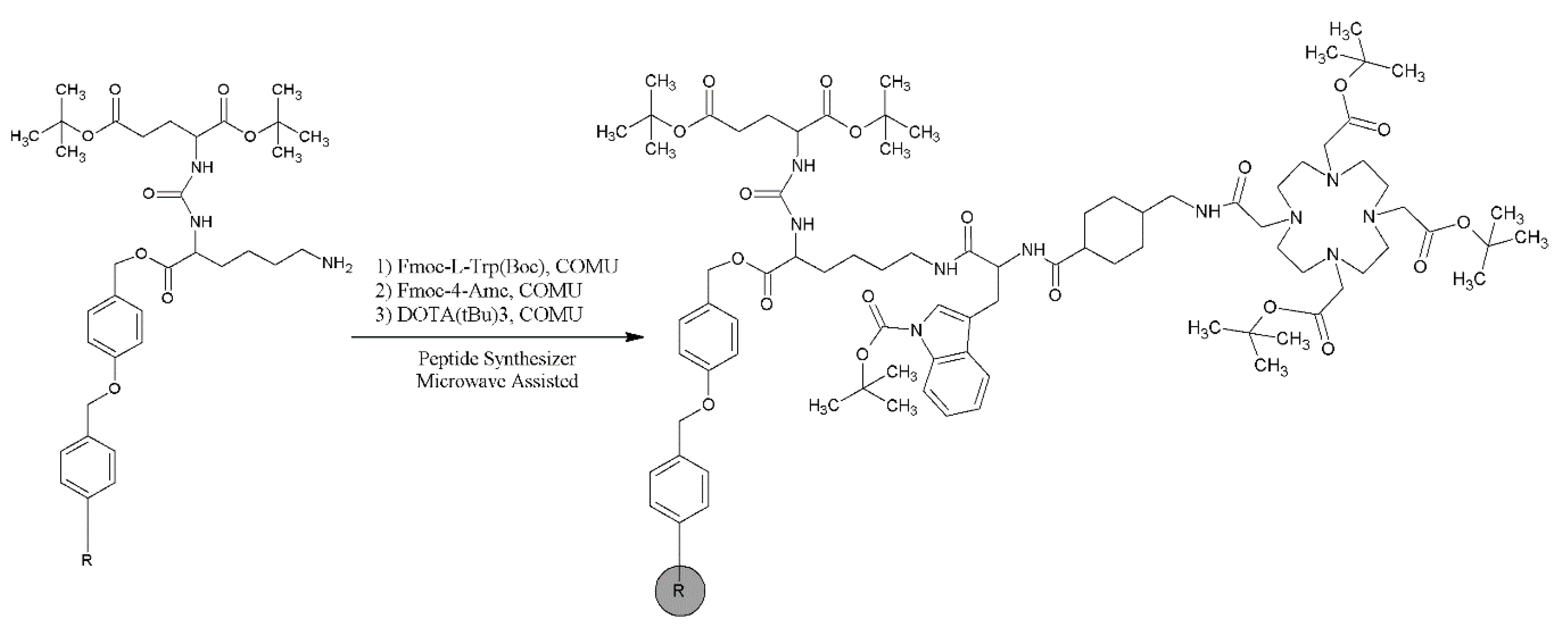
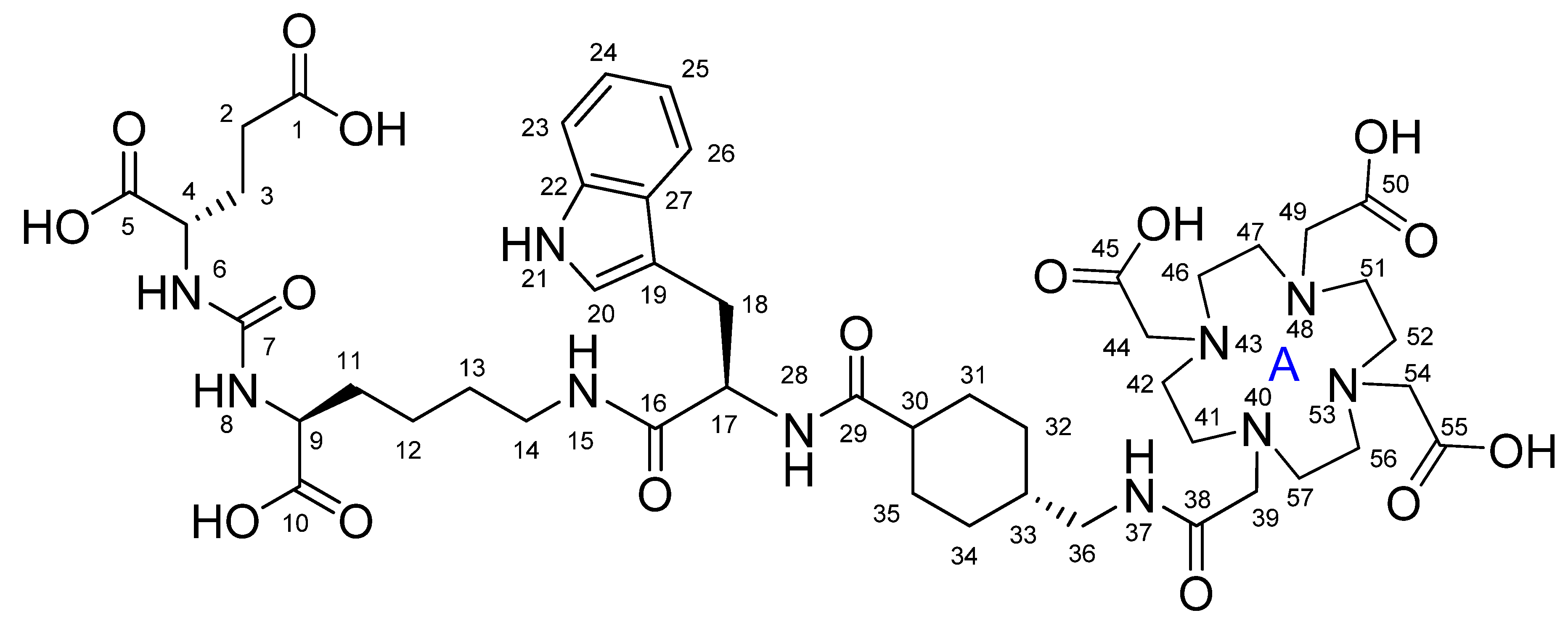
2.1. Synthesis and Physico-Chemical Characteristics of PSMA-D4
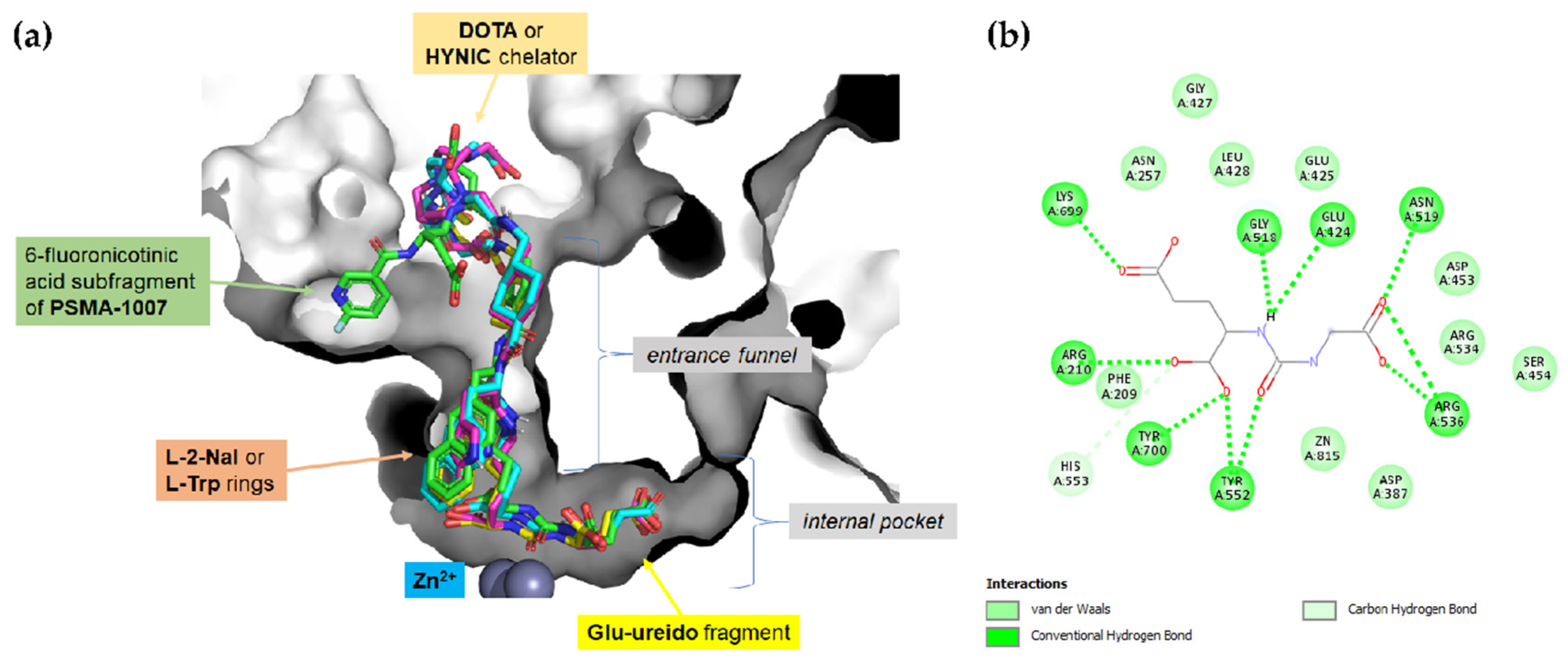
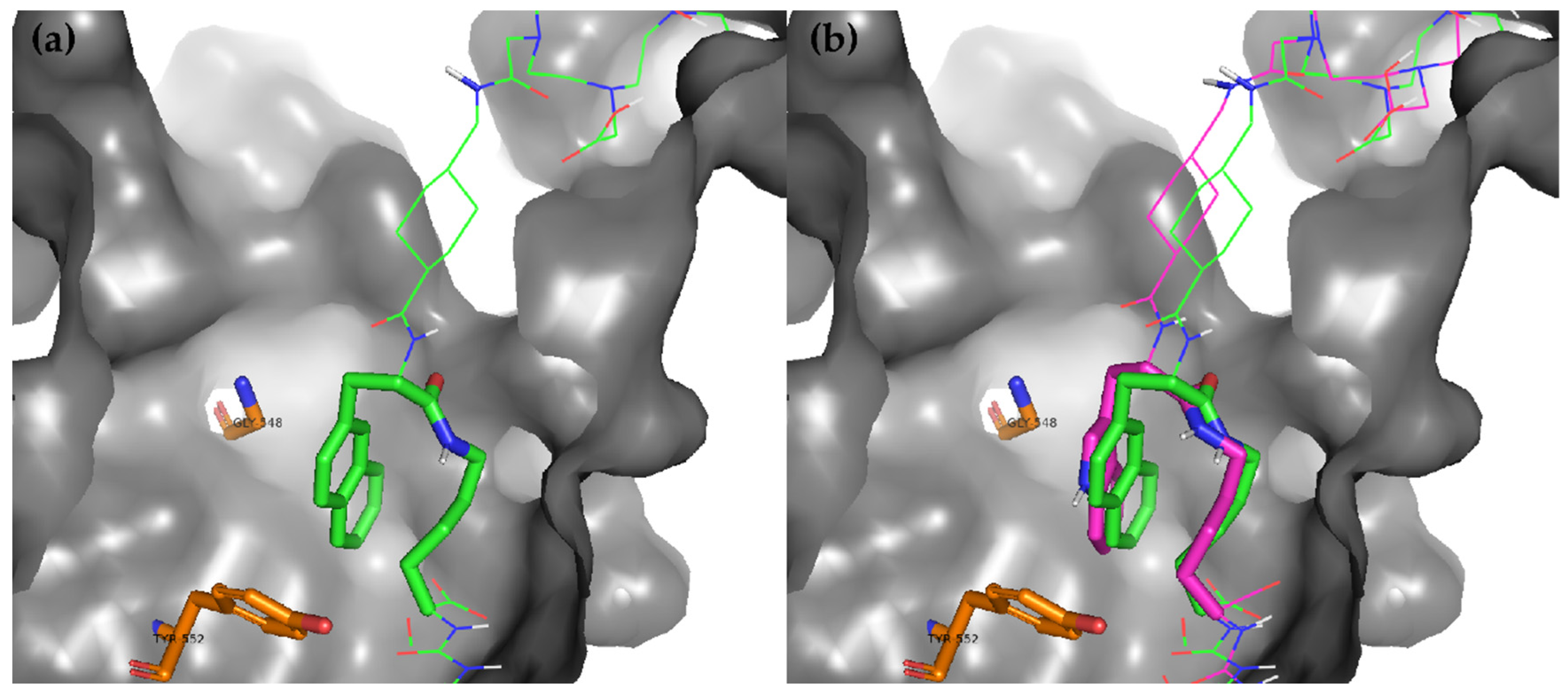
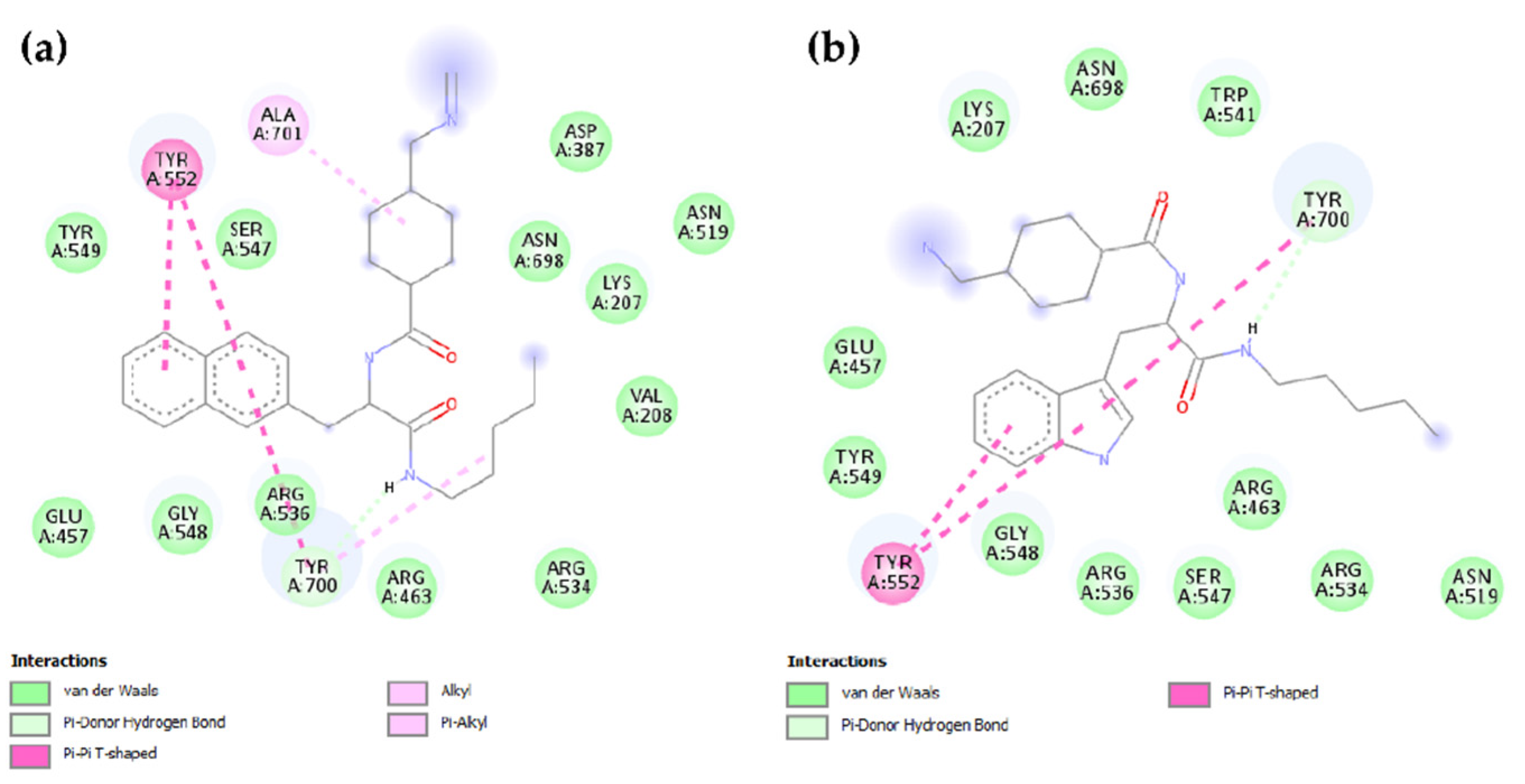
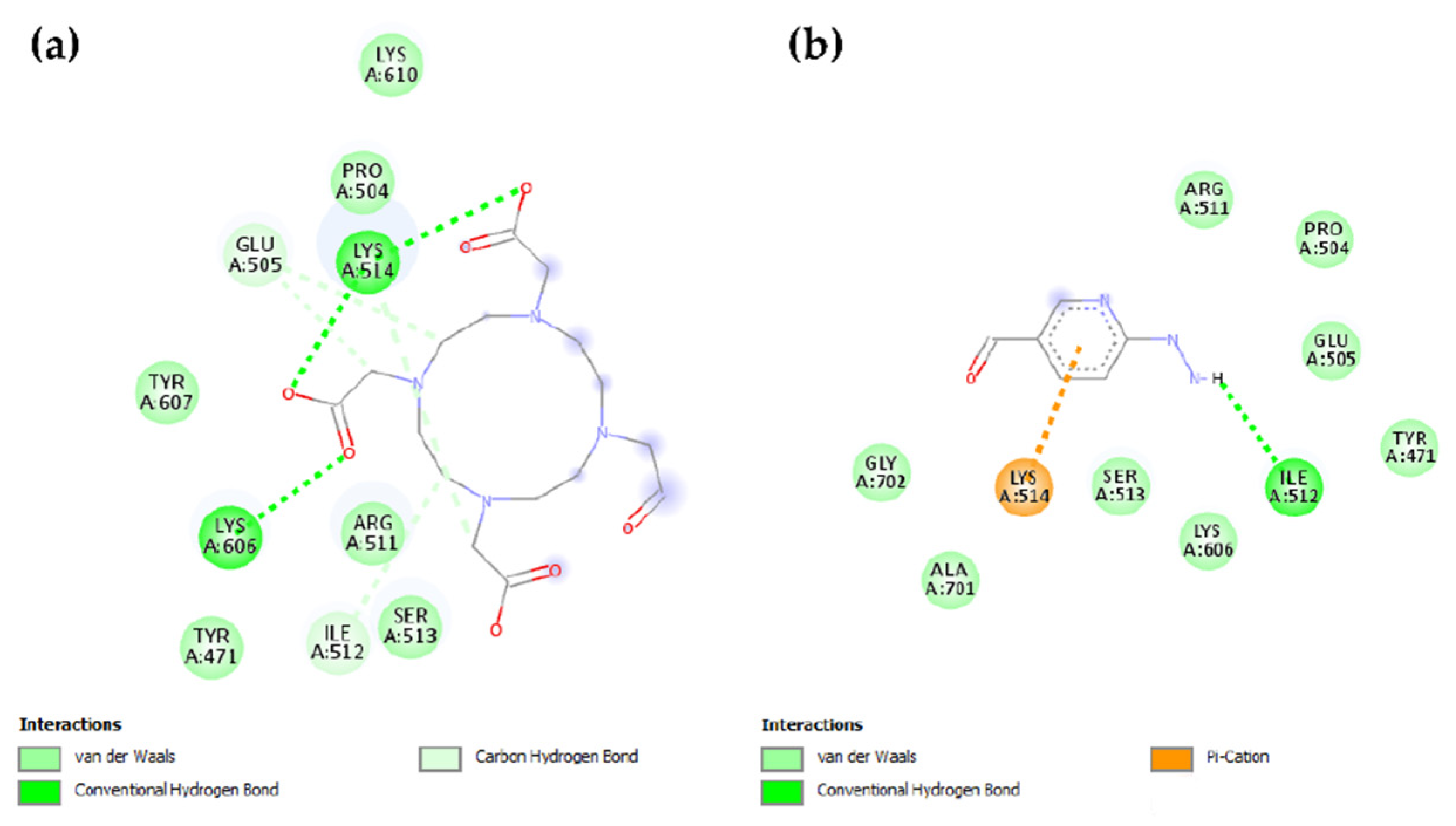
2.2. Molecular Modeling
2.3. Radiolabeling of PSMA-D4 and logD
2.4. In Vitro
2.5. Ex Vivo
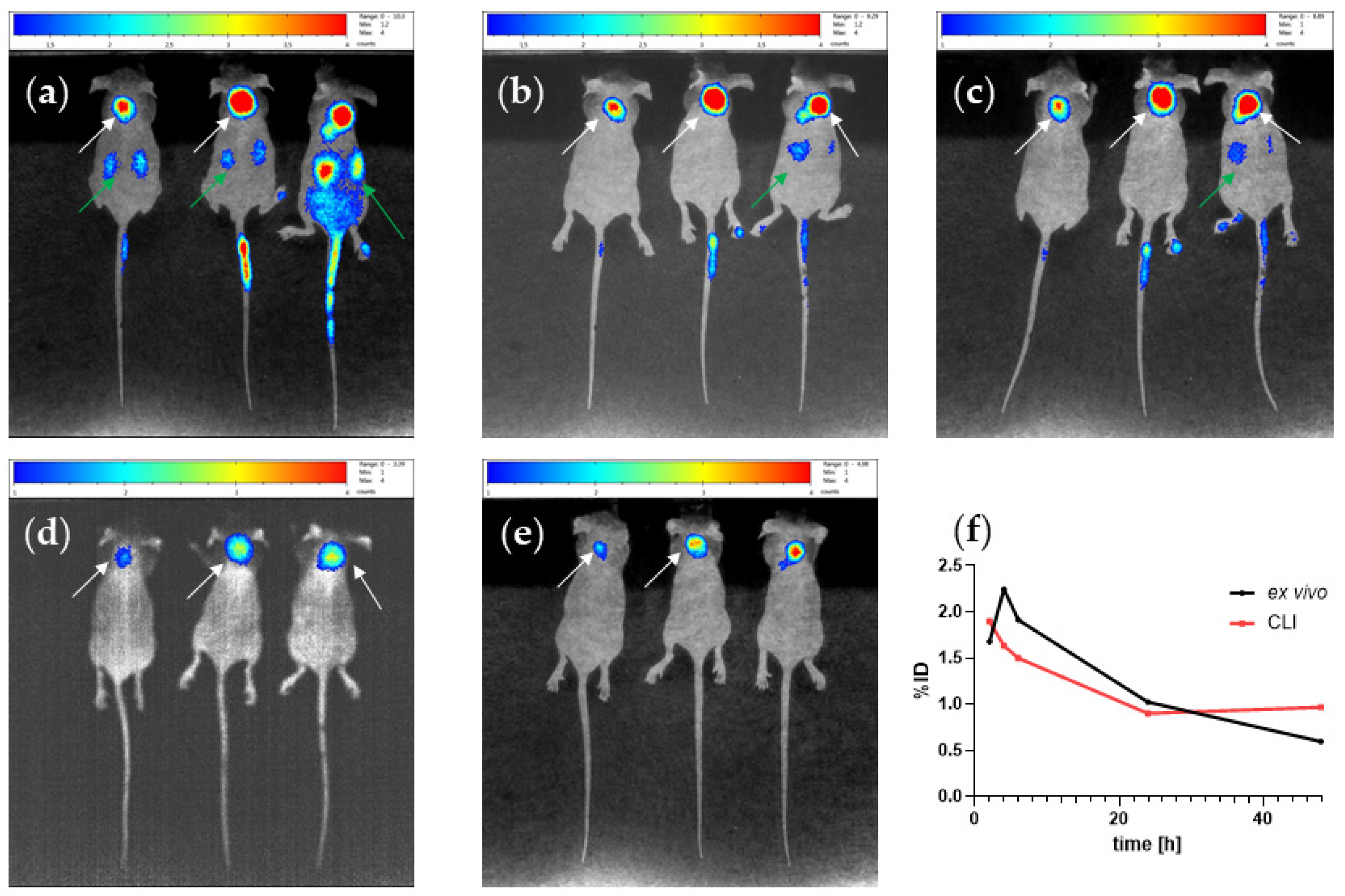
2.6. In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemicals and Radionuclides
4.2. Synthesis of PSMA-D4
4.3. Analytical Methods
4.3.1. HPLC
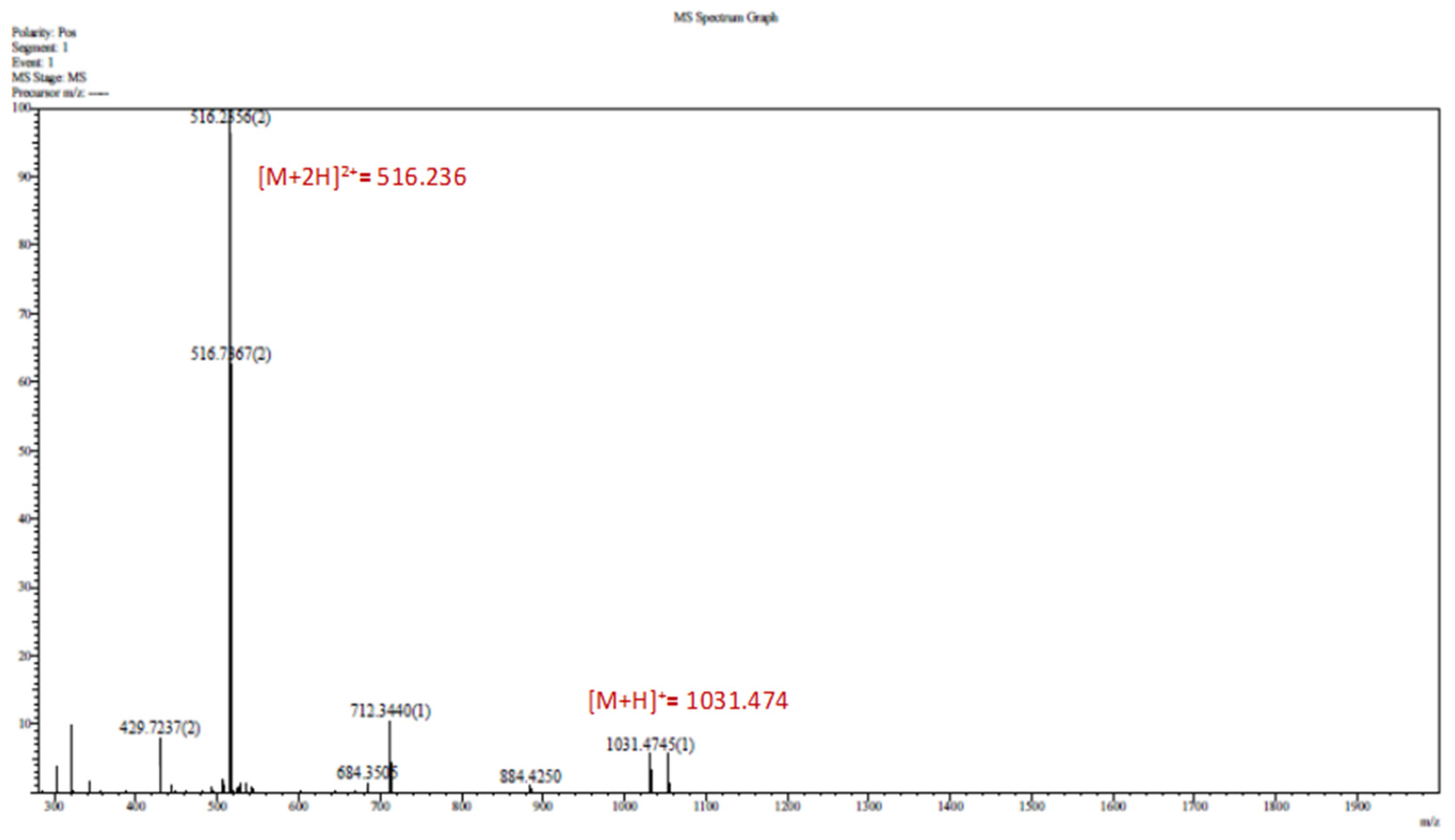
4.3.2. LC-MS
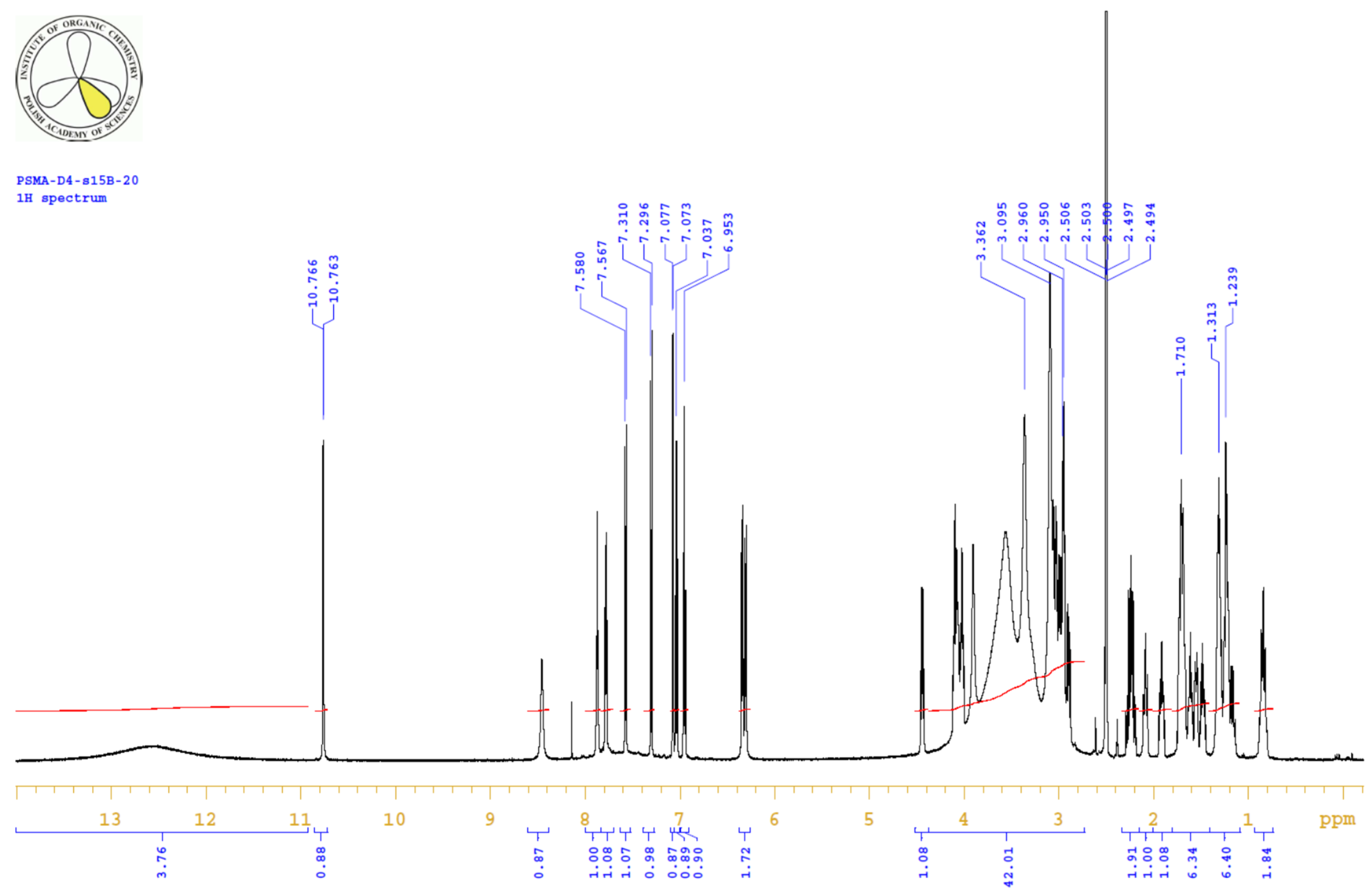
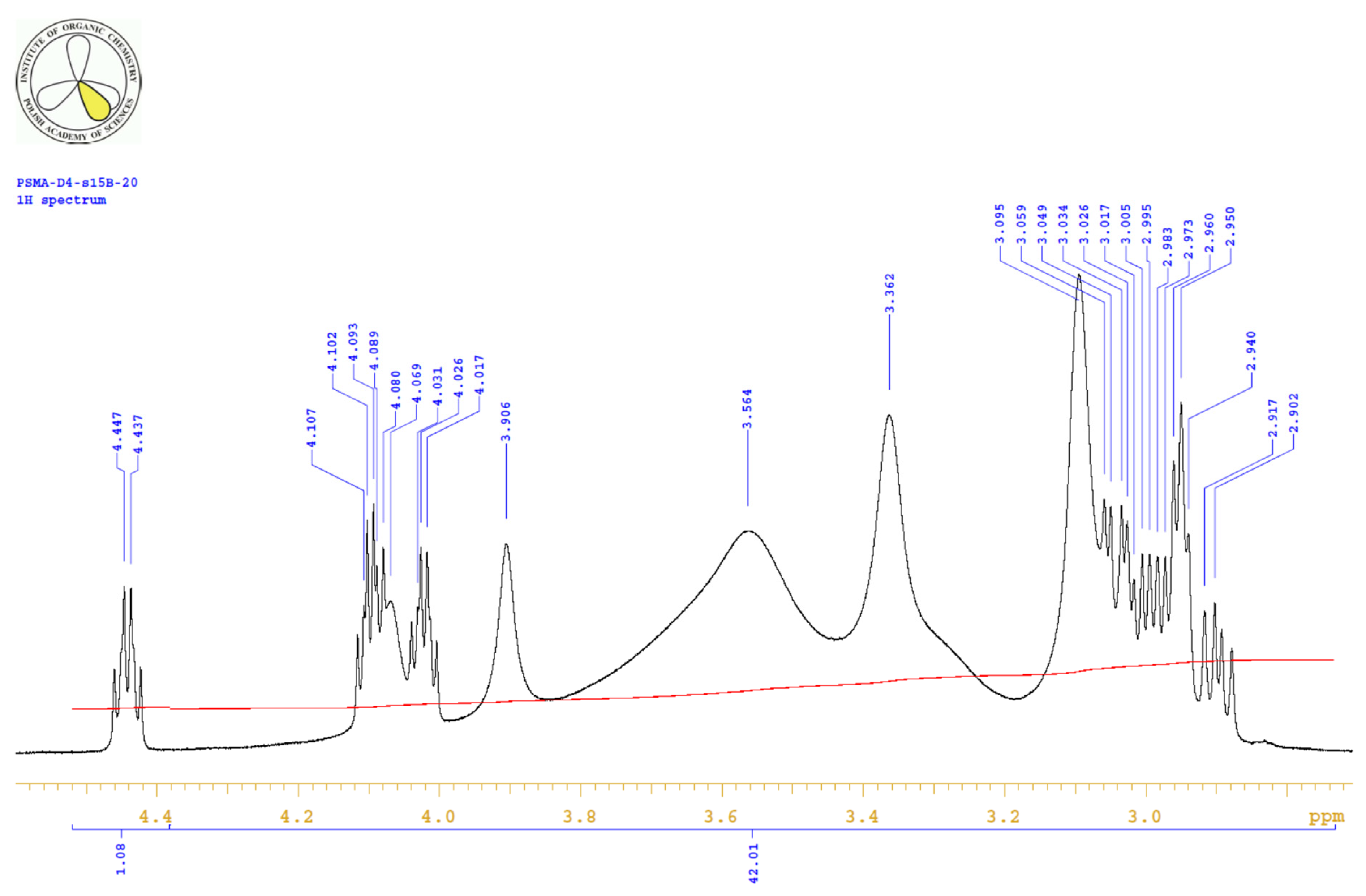
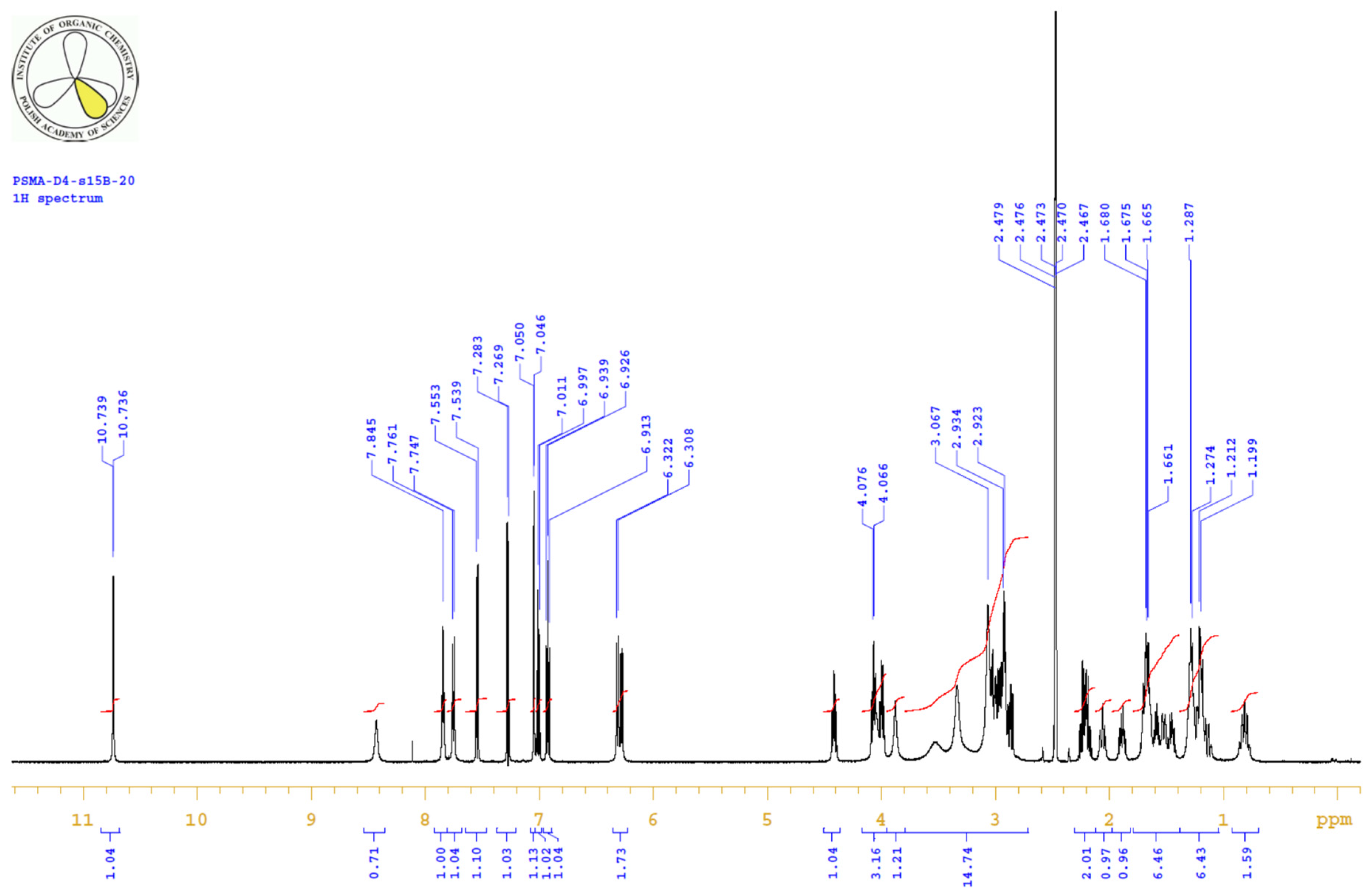
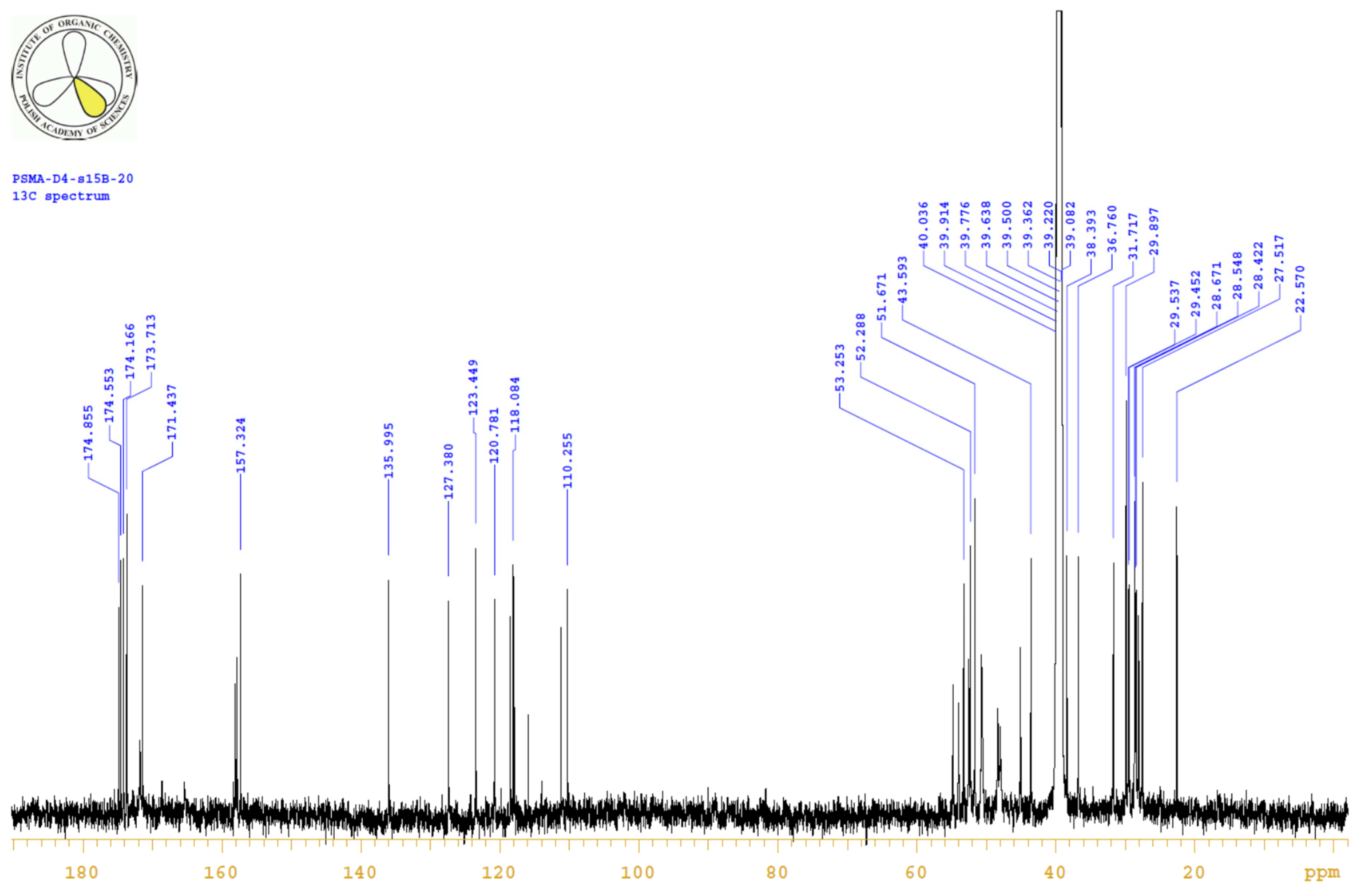
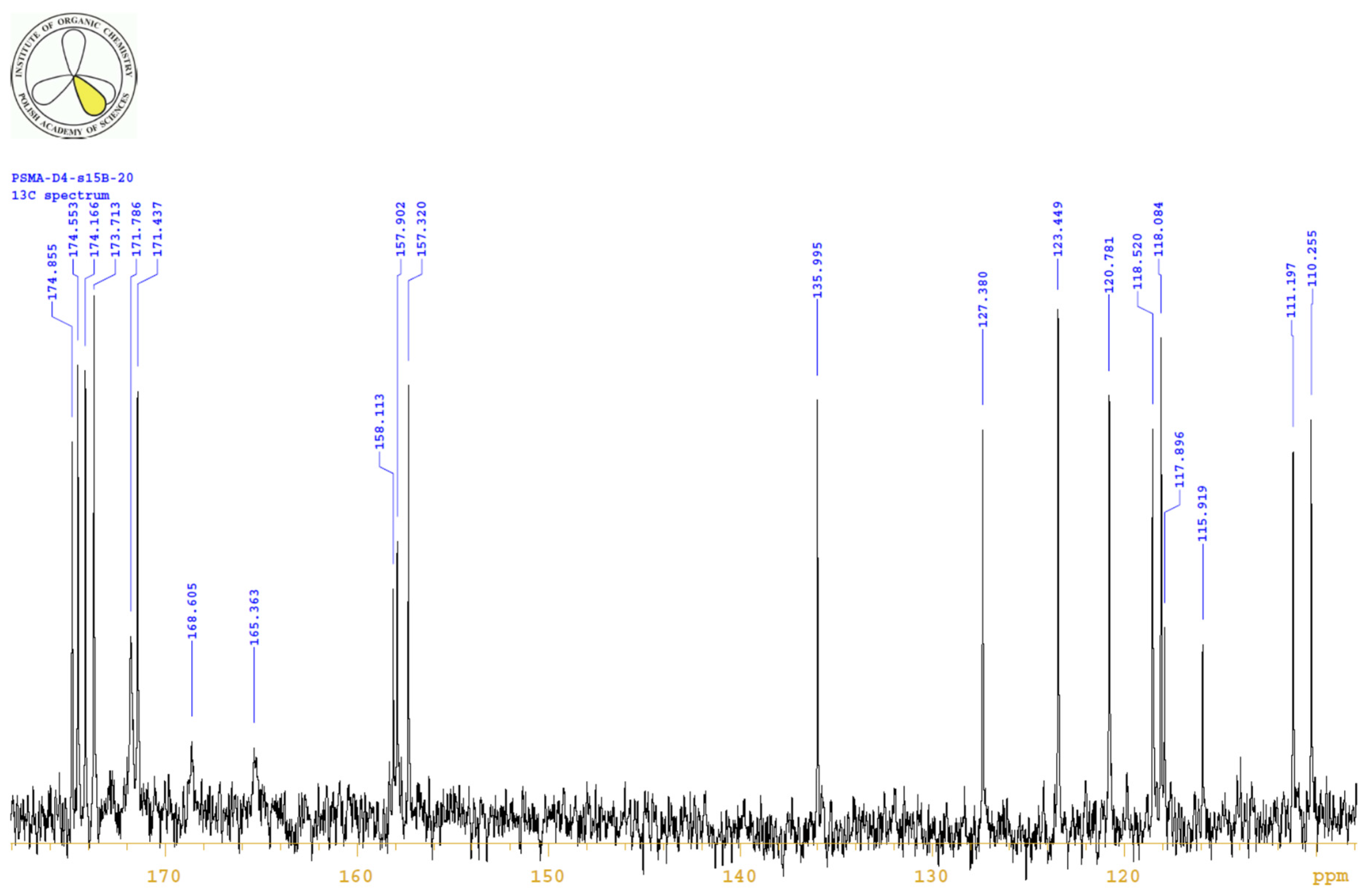
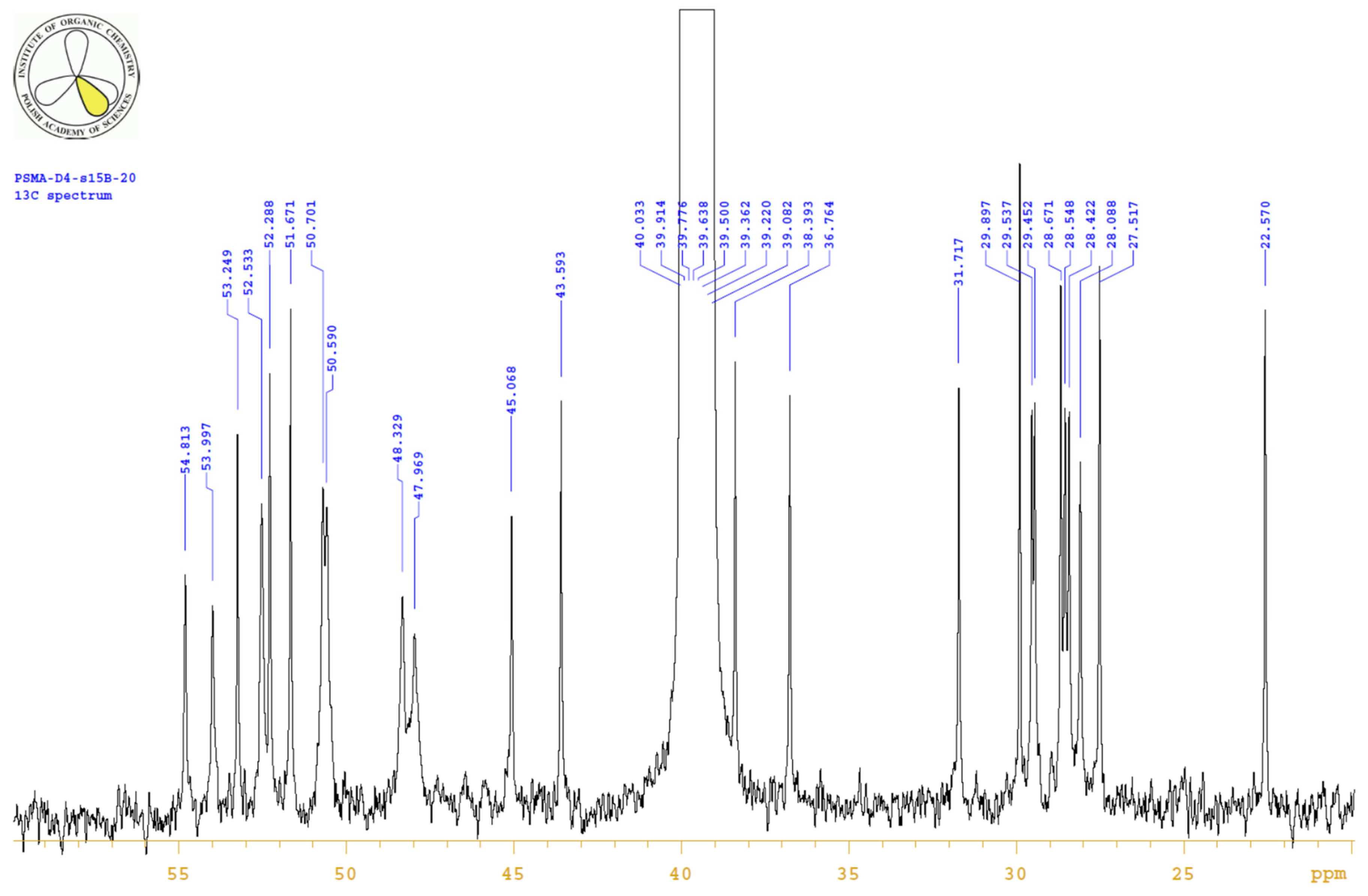
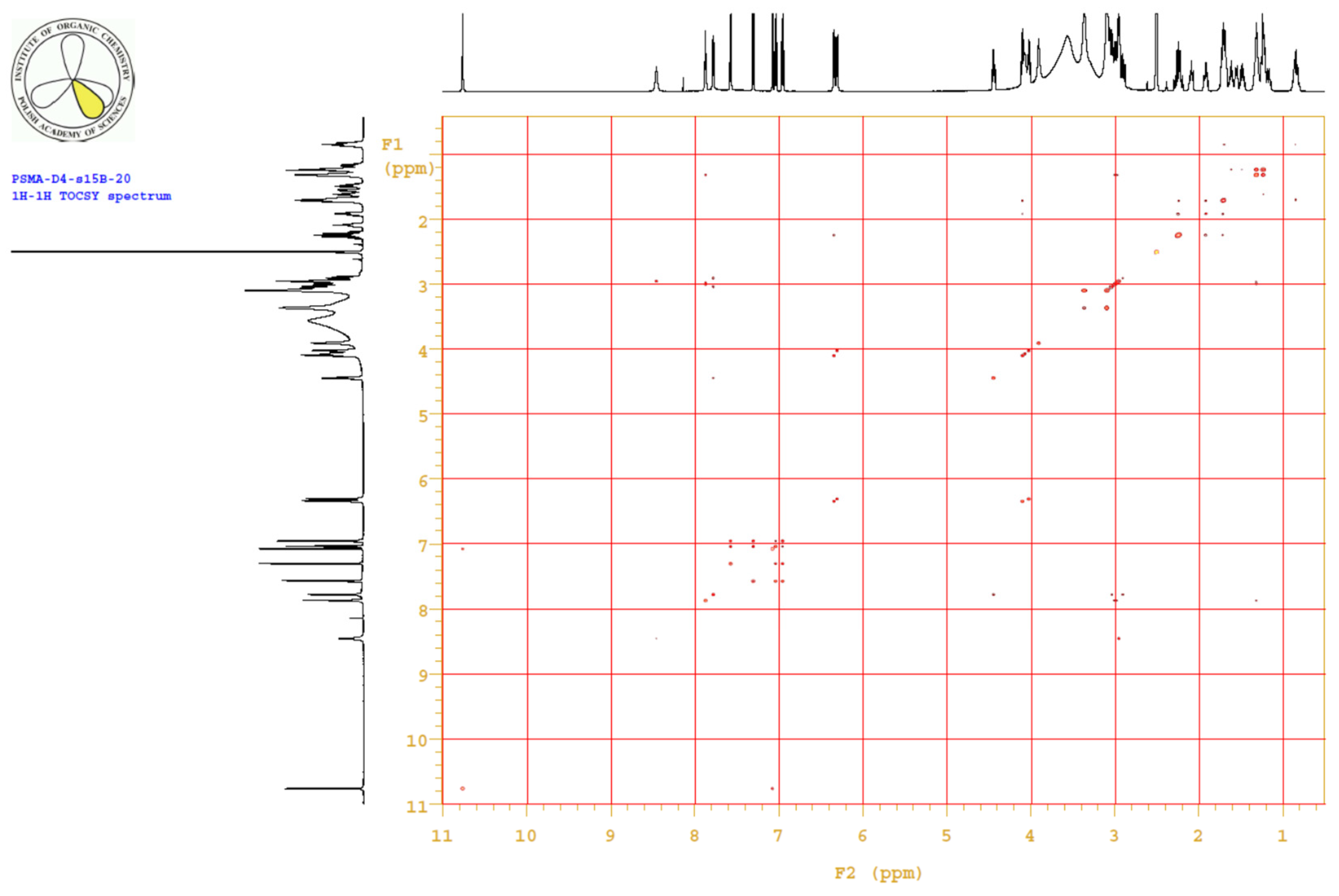
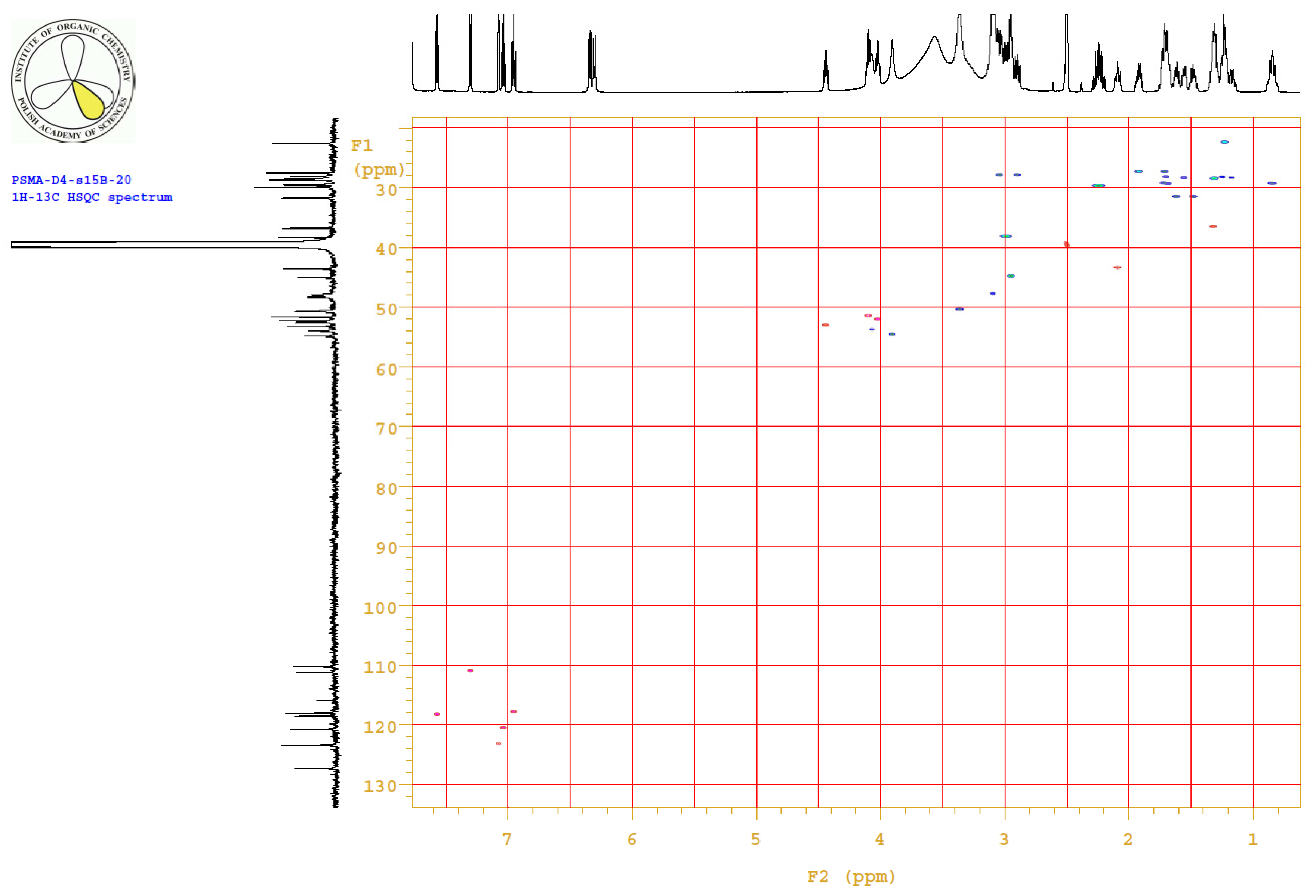
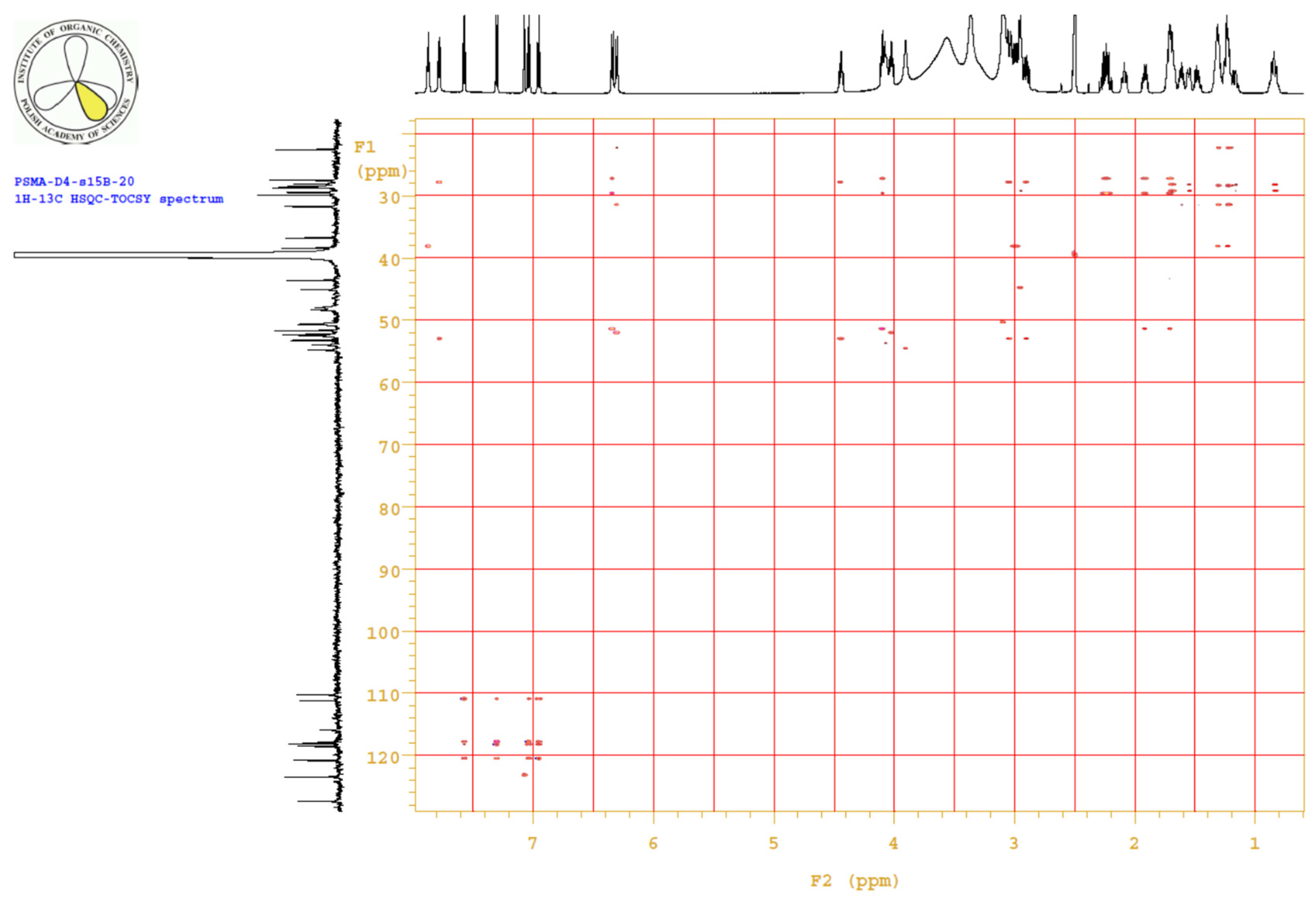
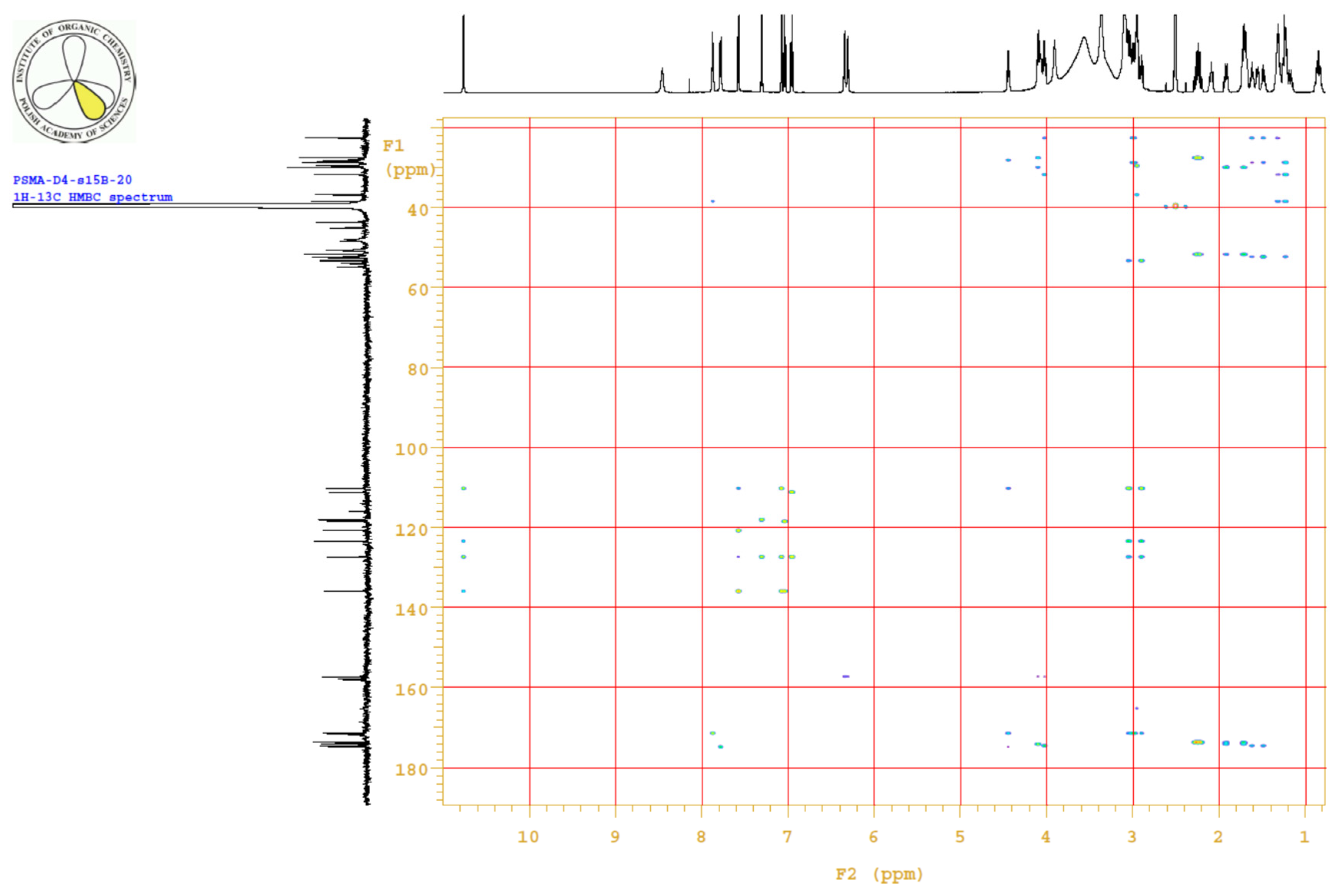
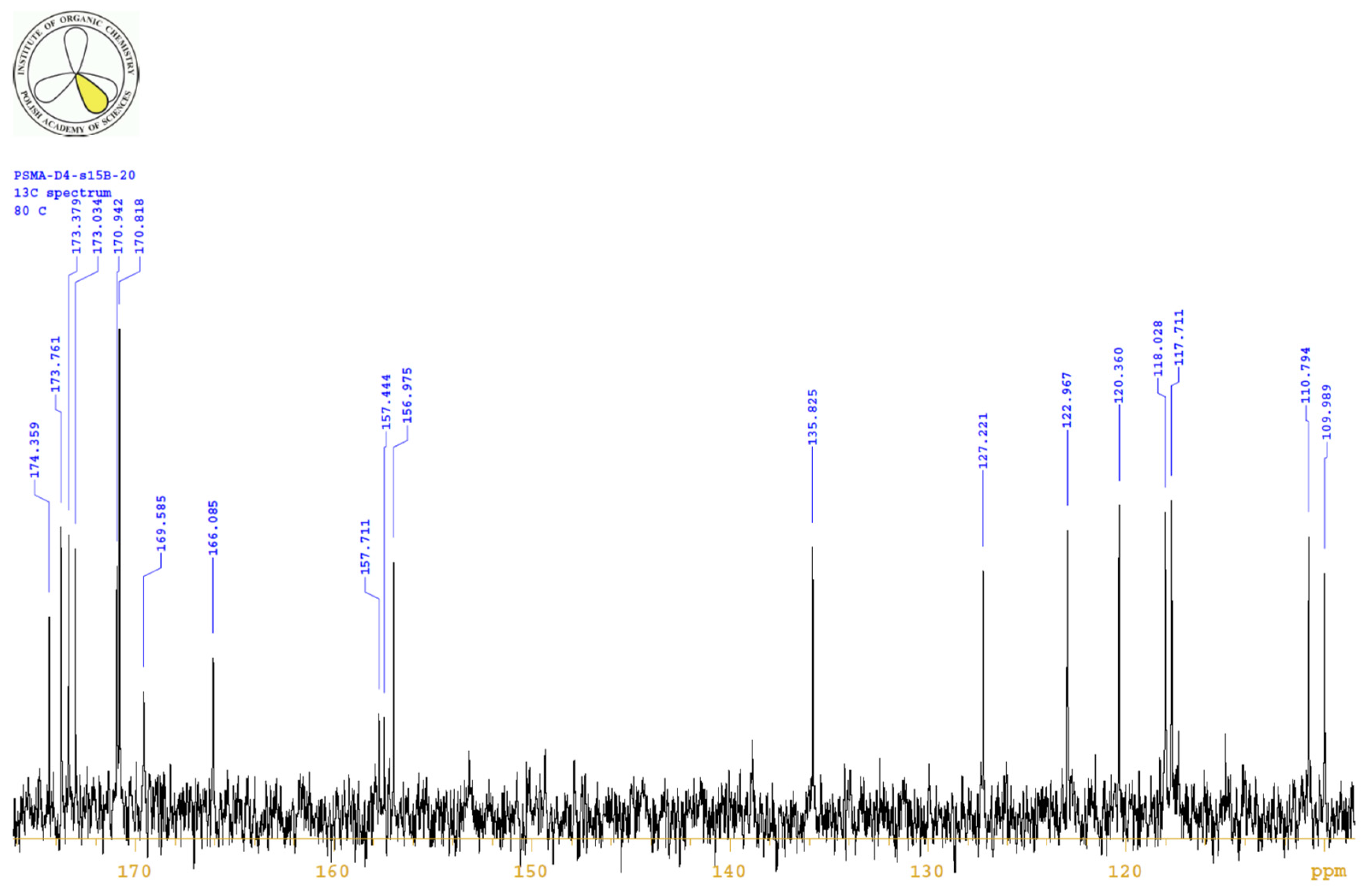
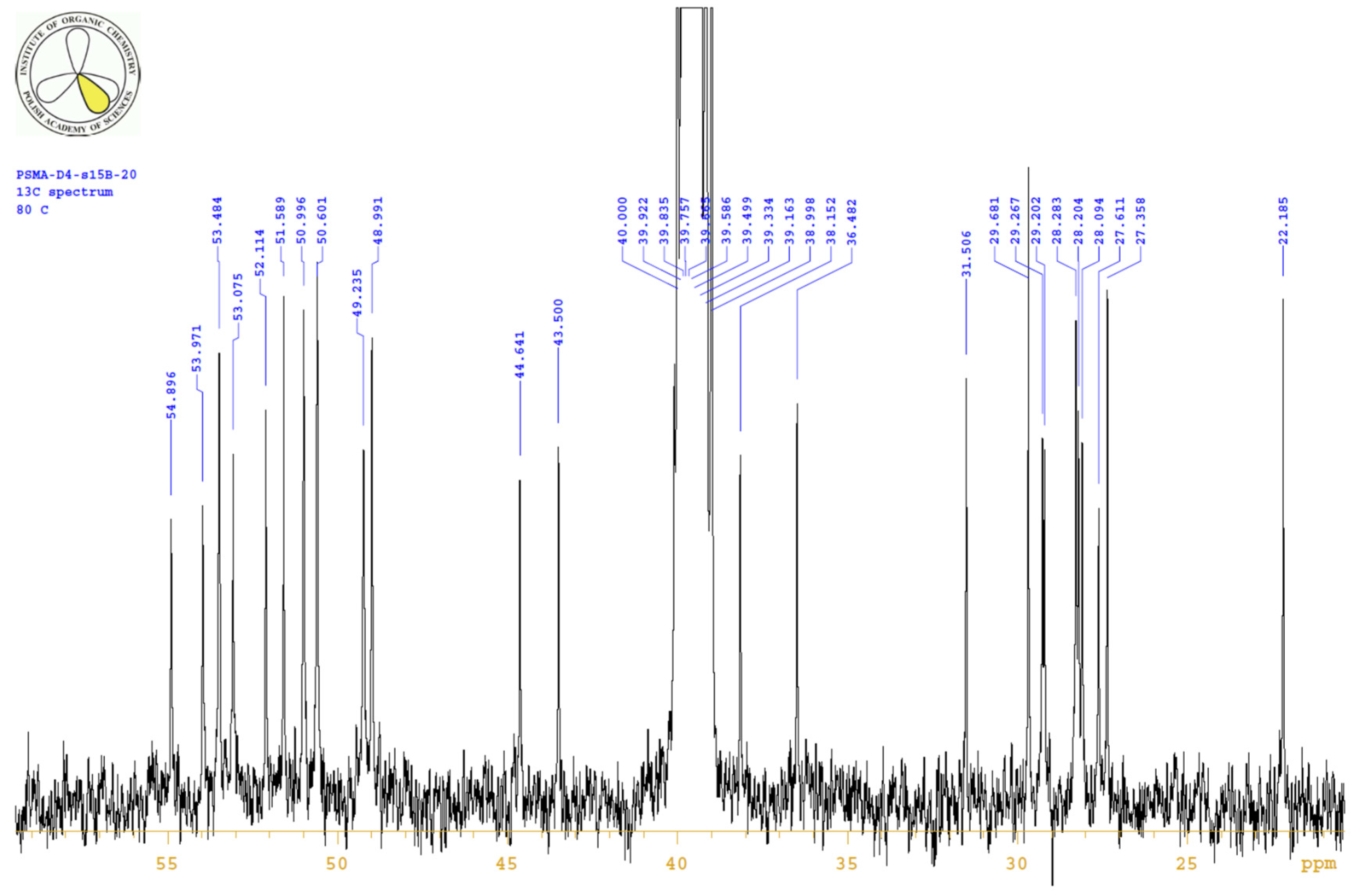
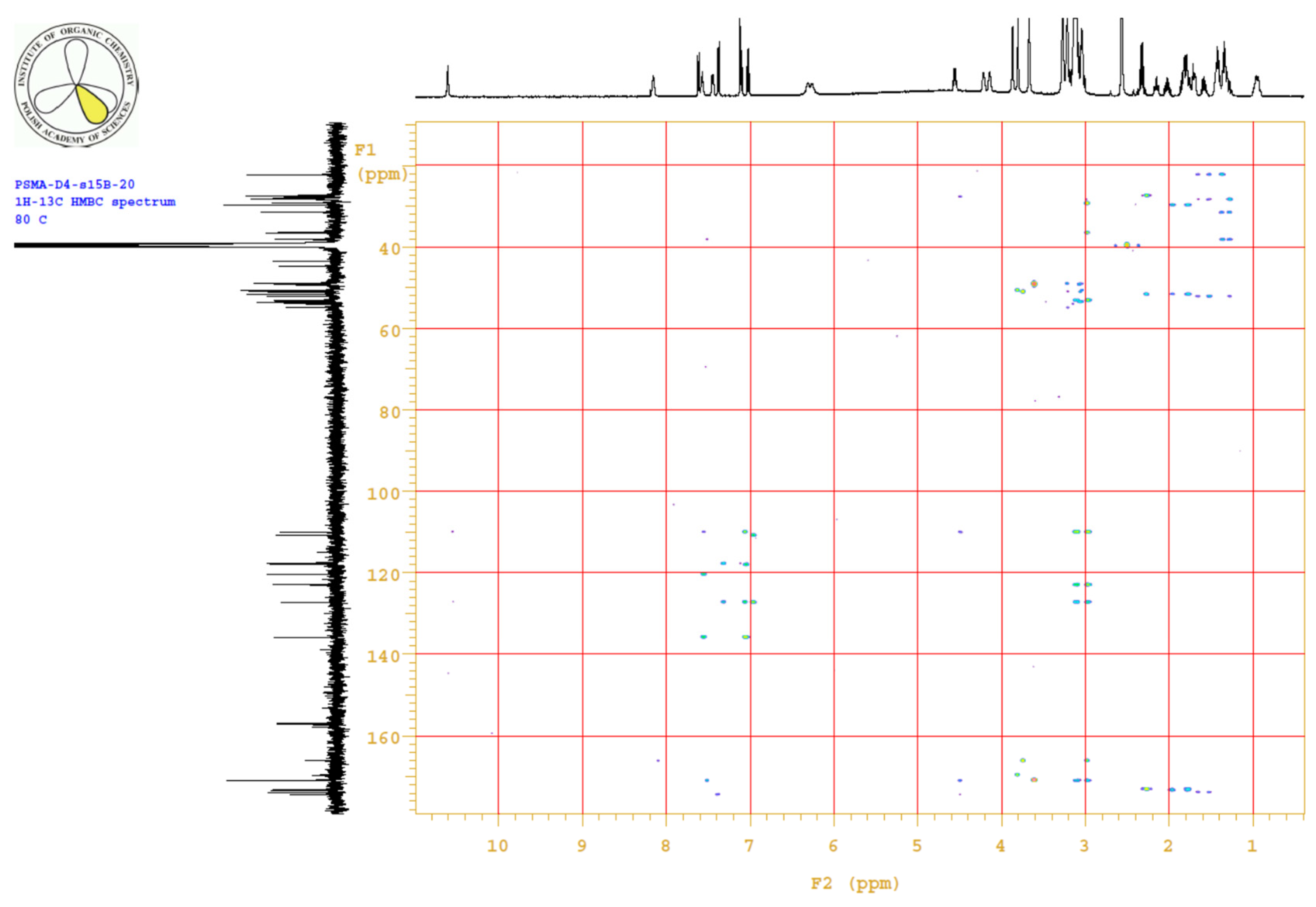
4.3.3. 1H, 13C-NMR
4.3.4. Elemental Analysis
4.3.5. Lipophilicity Determination
4.4. Docking Methodology
4.5. Radiolabeling and QC
4.6. In Vitro
4.7. Animal Study
4.8. Optical Imaging of [90Y]Y-PSMA-D4
4.9. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PCa | Prostate Cancer |
| PSMA-D4 | Prostate-Specific Membrane Antigen ligand conjugated with DOTA (Glu-CO-Lys-L-Trp-4-Amc-DOTA) |
| DOTA | 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid, tetraxetan |
| mCRPC | metastatic castration-resistant prostate cancer |
| PSA | prostate-specific antigen |
| GCPII | glutamate carboxypeptidase II |
| BFC | bifunctional chelator |
| LNCaP | Lymph Node Carcinoma of the Prostate |
| COMU | 1-[(1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)dimethylaminemorpholino)]uronium hexafluorophosphate |
| KD | binging affinity |
| Bmax | maximum receptor density |
| CLI | Cerenkov luminescence imaging |
| ROI | region of interest |
| SA | specific activity |
| RCY | radiochemical yield |
| ATCC | American Type Culture Collection |
| FBS | fetal bovine serum |
| ID | injected dose |
| SD | standard deviation |
| SE | electrospray soure |
| IT | ion trap |
| TOF | time of flight analyzer |
| TCD | Thermal conductivity detector |
Appendix A
Synthesis of PSMA-D4


Appendix B
Appendix B.1. Physico-Chemical Characteristics of PSMA-D4

Appendix B.2. Equipment and Reagents
Appendix B.2.1. Instruments
Appendix B.2.2. Reagents and Sample Preparation
Appendix B.3. Methodology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
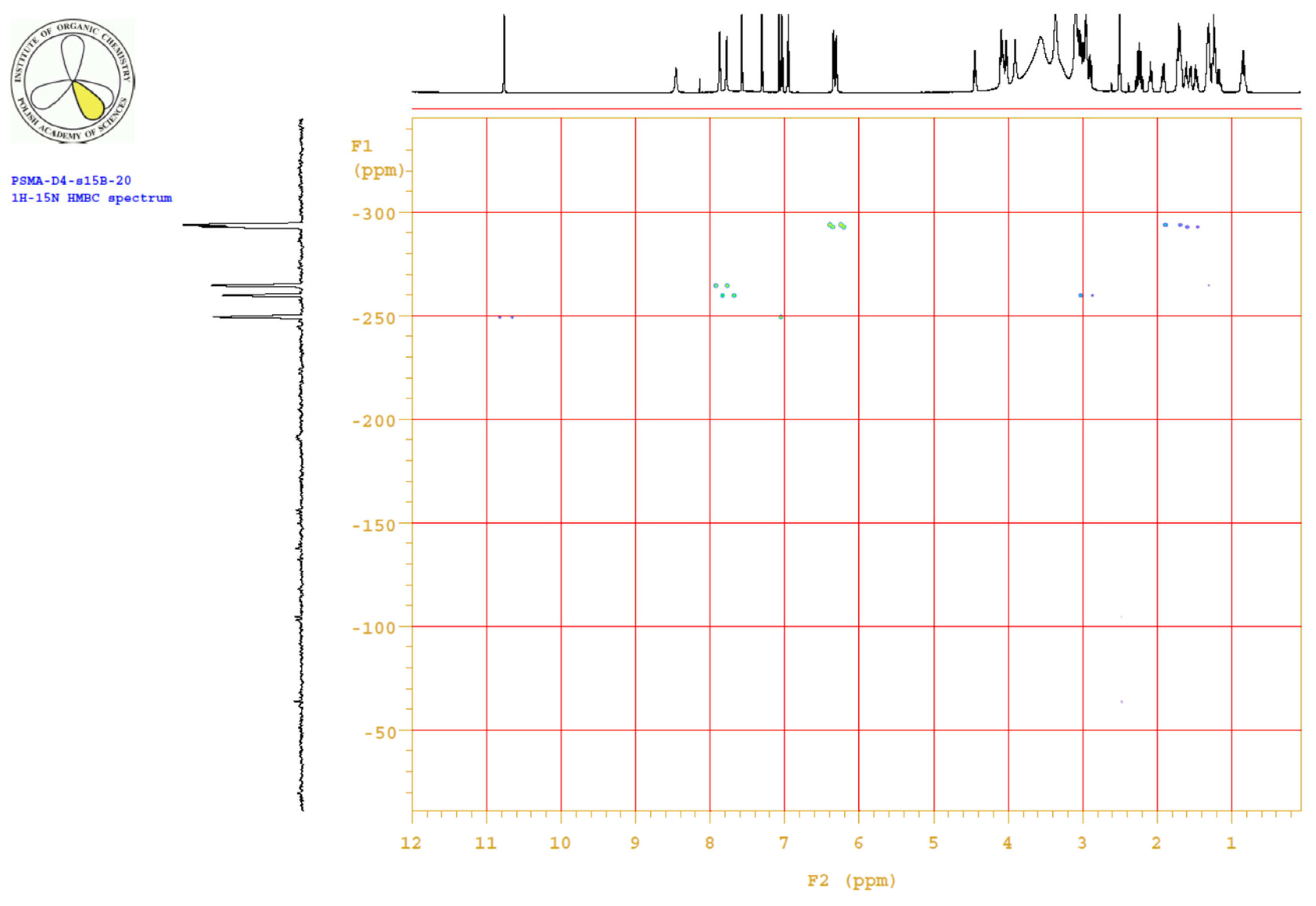
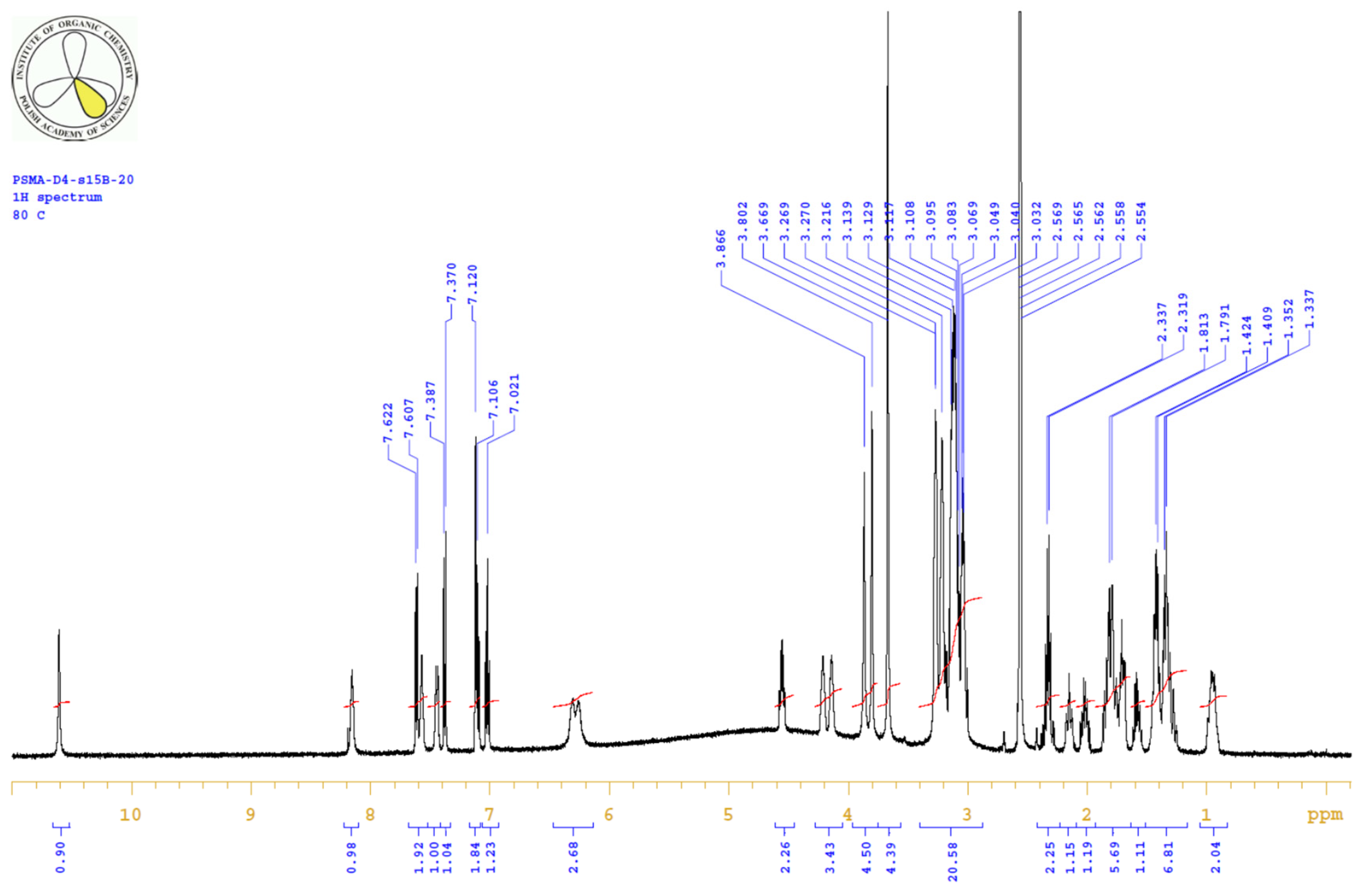
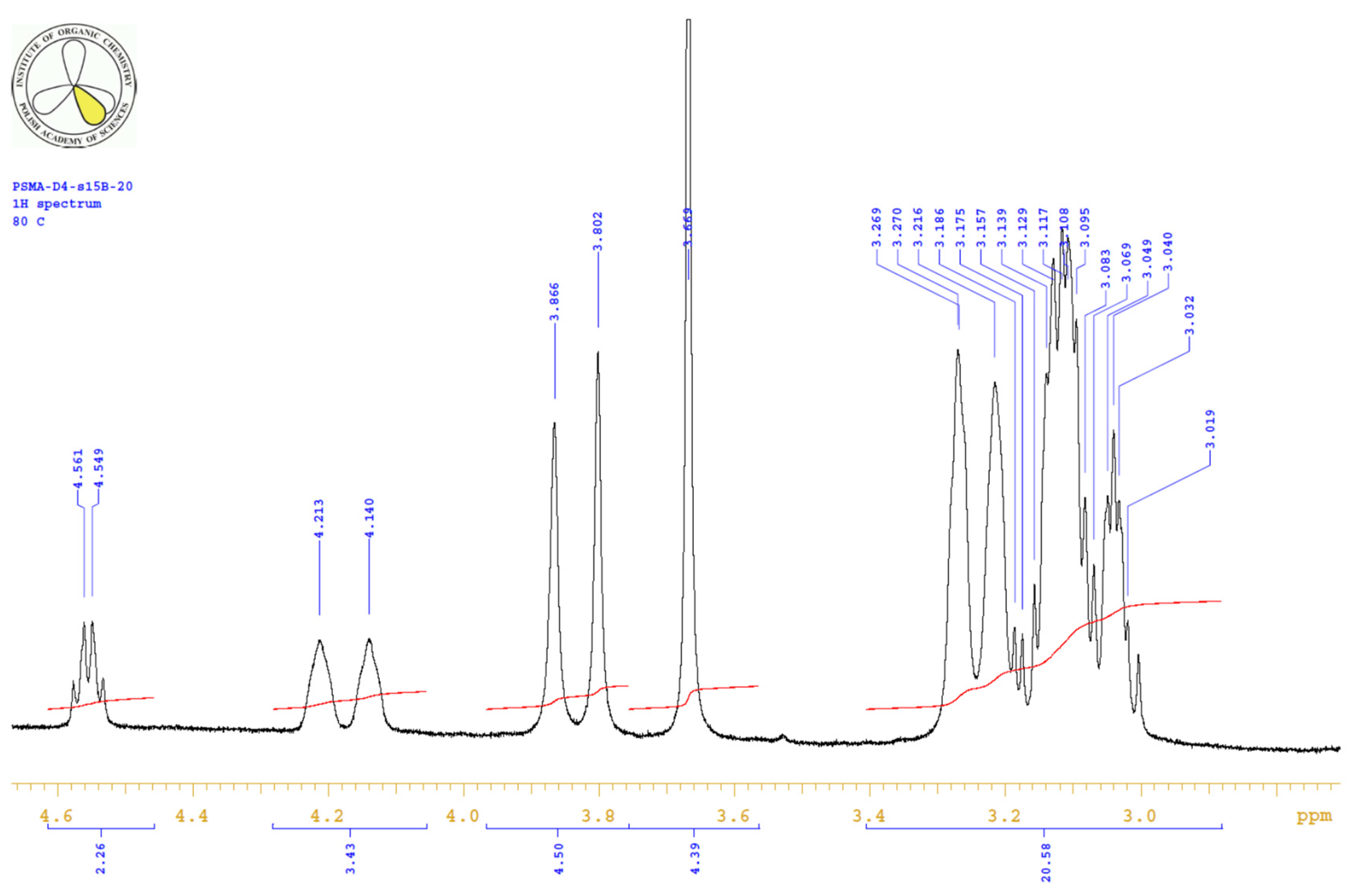
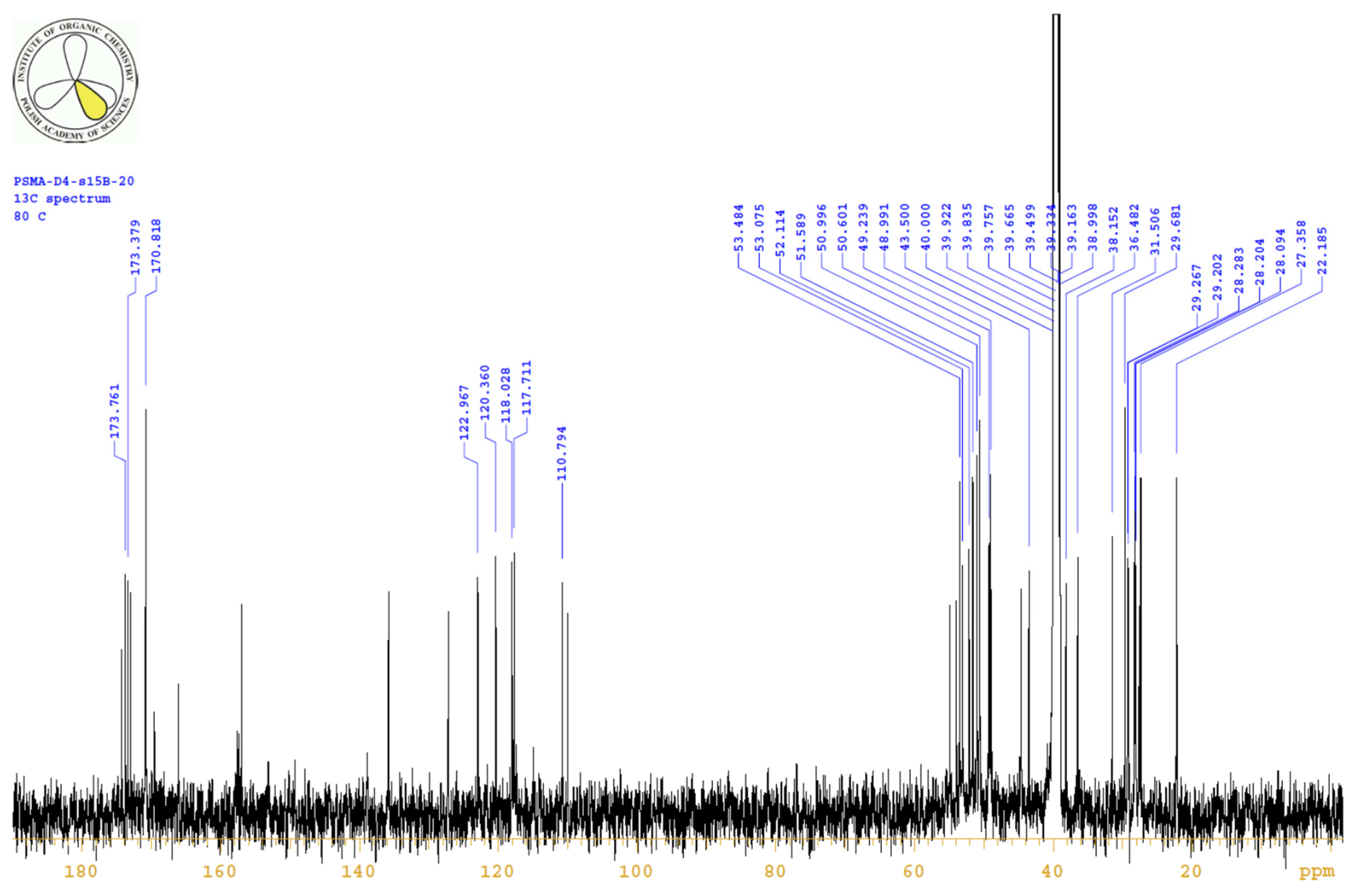
| PositionNumber | δ (15N) * [ppm] | δ (13C) ** [ppm] | δ (1H) ** [ppm] | Multiplicity (a) | (H,H) Couplings [Hz] | ||
|---|---|---|---|---|---|---|---|
| 298 K | 298 K | 353 K | 298 K | 353 K | 298 K | 298 K | |
| 1 | ----- | 173.7 | 173.0 | ----- | ----- | ----- | |
| OH (C1) | ----- | ----- | ----- | ~12.0 b | c | s (br) d | |
| 2 | ----- | 29.9 | 29.7 | 2.23 | 2.26 | m | 5.7, 9.0, 16.3 |
| 3 | ----- | 27.5 | 27.4 | 1.70/1.91 | 1.76/1.95 | m(ov)/m | |
| 4 | ----- | 51.6 | 51.6 | 4.08 | 4.14 | m | |
| 5 | ----- | 174.2 | 173.4 | ----- | ----- | ----- | |
| OH (C5) | ----- | ----- | ----- | ~12.0 b | c | s (br) d | |
| 6 | −294.0 | ----- | ----- | 6.34 | 6.23 e | d | 8.4 |
| 7 | ----- | 157.3 | 157.0 | ----- | ----- | ----- | |
| 8 | −292.9 | ----- | ----- | 6.30 | 6.20 e | d | 8.2 |
| 9 | ----- | 52.4 | 52.1 | 4.01 | 4.07 | m | |
| 10 | ----- | 174.6 | 173.8 | ----- | ----- | ----- | |
| OH (C10) | ----- | ----- | ----- | ~12.0 b | c | s (br) d | |
| 11 | ----- | 31.7 | 31.5 | 1.48/1.61 | 1.51/1.64 | m/m | |
| 12 | ----- | 22.6 | 22.2 | 1.22 | 1.27 | m(ov) | |
| 13 | ----- | 28.7 | 28.3 | 1.30 | 1.35 | m(ov) | |
| 14 | ----- | 38.4 | 38.2 | 2.98 | 3.00 | m(ov) | |
| 15 | −264.6 | ----- | ----- | 7.86 | 7.50 | t | 5.9 |
| 16 | ----- | 171.4 | 170.9 | ----- | ----- | ----- | |
| 17 | ----- | 53.3 | 53.1 | 4.44 | 4.48 | m | |
| 18 | ----- | 28.1 | 27.6 | 2.89/3.04 | 2.95/3.09 | dd/m(ov) | 8.7, 14.5/--- |
| 19 | ----- | 110.3 | 110.0 | ----- | ----- | ----- | |
| 20 | ---- | 123.5 | 123.0 | 7.07 | 7.05 | d | 2.2 |
| 21 | −249.5 | ----- | ----- | 10.75 | 10.54 | s | |
| 22 | ----- | 136.0 | 135.8 | ----- | ----- | ----- | |
| 23 | ----- | 111.2 | 110.8 | 7.29 | 7.30 | ddd | 0.9, 1.2, 8.2 |
| 24 | ----- | 120.8 | 120.4 | 7.03 | 7.03 | ddd | 1.1, 6.9, 8.2 |
| 25 | ----- | 118.1 | 117.7 | 6.94 | 6.95 | ddd | 1.0, 7.0, 7.8 |
| 26 | ----- | 118.5 | 118.0 | 7.56 | 7.54 | d | 7.8 |
| 27 | ----- | 127.4 | 127.2 | ----- | ----- | ----- | |
| 28 | −269.9 | ----- | ----- | 7.77 | 7.37 | d | 8.3 |
| 29 | ----- | 174.9 | 174.4 | ----- | ----- | ----- | |
| 30 | ----- | 43.6 | 43.5 | 2.08 | 2.08 | m | |
| 31 | ----- | 28.6 | 28.2 | 1.16/1.54 | 1.22/1.62 | m(ov)/m(ov) | |
| 32 | ----- | 29.6 | 29.2 | 0.82/1.67 | 0.90/1.73 | m(ov)/m(ov) | |
| 33 | ----- | 37.8 | 36.5 | 1.32 | 1.35 | m(ov) | |
| 34 | ----- | 29.5 | 29.3 | 0.84/1.71 | 0.88/1.71 | m(ov)/m(ov) | |
| 35 | ----- | 28.4 | 28.1 | 1.24/1.69 | 1.27/1.73 | m(ov)/m(ov) | |
| 36 | ----- | 45.1 | 44.6 | 2.94 | 2.96 | m(ov) | |
| 37 | f | ----- | ----- | 8.45 | 8.08 | s(br) | |
| 38 | ----- | 165.3 | 166.1 | ----- | ----- | ----- | |
| 39 | ----- | 54.9 | 3.72 | ----- | |||
| 40 | f | ----- | ----- | ----- | ----- | ----- | |
| 41/57 | ----- | 51.0 | 3.19 | ----- | |||
| 42/56 | ----- | 49.0 | 3.05 | ----- | |||
| 43/53 | f | ----- | ----- | ----- | ----- | ----- | |
| 44/54 | ----- | 53.5 | 3.59 | ----- | |||
| 45/55 | ----- | ----- | 170.8 | ----- | ---- | ----- | |
| 46/52 | f | ----- | 49.2 | 3.03 | ----- | ||
| 47/51 | ----- | 50.6 | 3.14 | ----- | |||
| 48 | ----- | ----- | ----- | ----- | ----- | ||
| 49 | f | ----- | 54.0 | 3.79 | ----- | ||
| 50 | ----- | 169.6 | ----- | ----- | |||
| OH g | ----- | ----- | ~12.0 b | c | ----- | ||
Appendix B.4. Results




















Appendix B.5. MS Spectrum

Appendix C


Appendix D
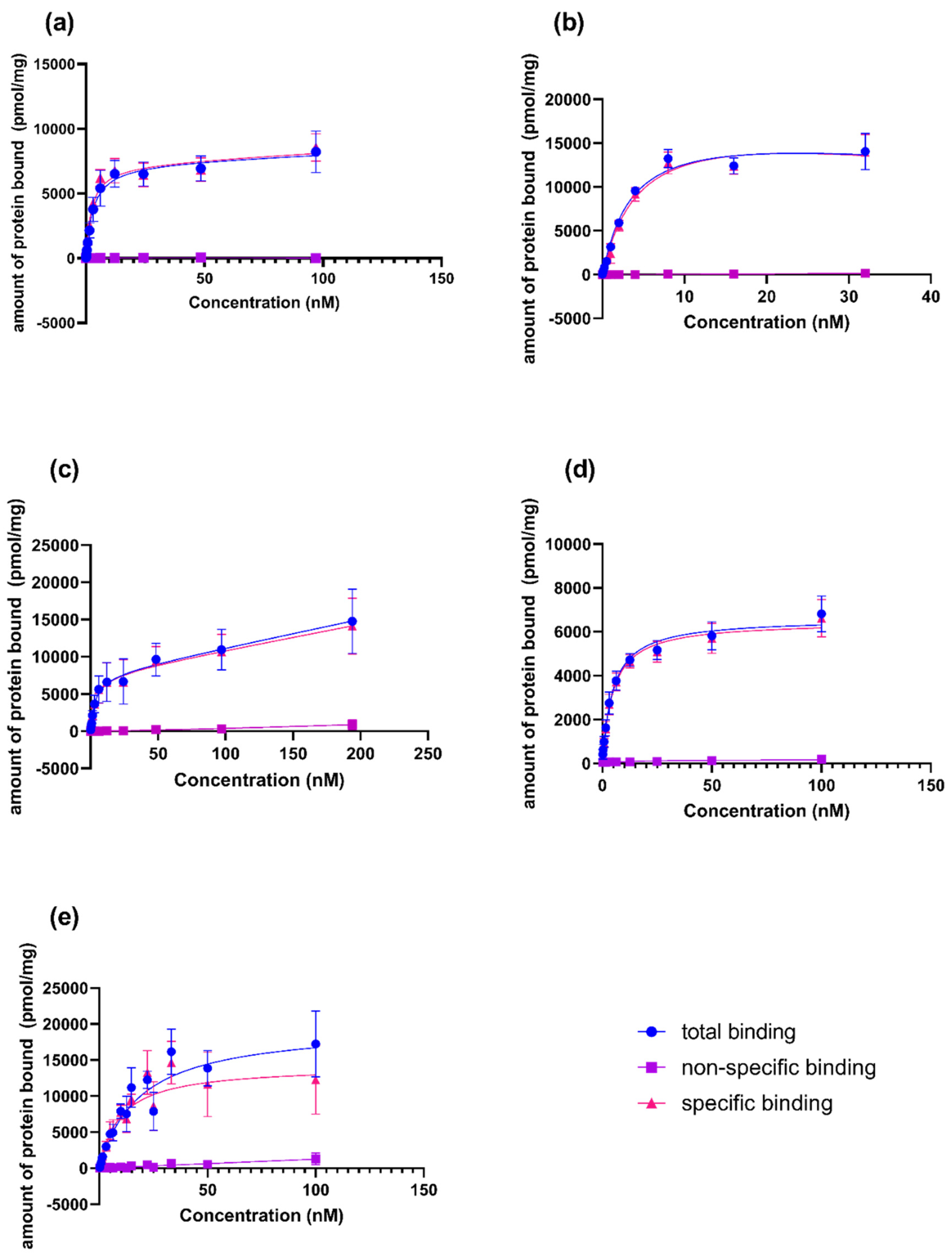
 ) and non-specific binding (
) and non-specific binding (  ) were determined by incubation PSMA ligands with LNCaP (PSMA positive) and PC3 (PSMA negative) membranes, respectively. The specific binding (
) were determined by incubation PSMA ligands with LNCaP (PSMA positive) and PC3 (PSMA negative) membranes, respectively. The specific binding (  ) was evaluated as a difference between the total and non-specific binding.
) and non-specific binding ( ) were determined by incubation PSMA ligands with LNCaP (PSMA positive) and PC3 (PSMA negative) membranes, respectively. The specific binding ( ) was evaluated as a difference between the total and non-specific binding.
) was evaluated as a difference between the total and non-specific binding.
) and non-specific binding ( ) were determined by incubation PSMA ligands with LNCaP (PSMA positive) and PC3 (PSMA negative) membranes, respectively. The specific binding ( ) was evaluated as a difference between the total and non-specific binding.
References
- The American Cancer Society. Available online: http://www.cancer.org (accessed on 1 January 2013).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Pinto, J.T.; Suffoletto, B.P.; Berzin, T.M.; Qiao, C.H.; Lin, S.; Tong, W.P.; May, F.; Mukherjee, B.; Heston, W.D. Prostate-specific membrane antigen: A novel folate hydrolase in human prostatic carcinoma cells. Clin. Cancer Res. 1996, 2, 1445–1451. [Google Scholar] [PubMed]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Perner, S.; Hofer, M.D.; Kim, R.; Shah, R.B.; Li, H.; Möller, P.; Hautmann, R.E.; Gschwend, J.E.; Kuefer, R.; Rubin, M.A. Prostate-specific membrane antigen expression as a predictor of prostate cancer progression. Hum. Pathol. 2007, 38, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.L.; Grob, B.M.; Haley, C.; Grossman, K.; Newhall, K.; Petrylak, D.; Troyer, J.; Konchuba, A.; Schellhammer, P.F.; Moriarty, R. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology 1996, 48, 326–334. [Google Scholar] [CrossRef]
- Ross, J.S.; Sheehan, C.E.; Fisher, H.A.; Kaufman, R.P., Jr.; Kaur, P.; Gray, K.; Webb, I.; Gray, G.S.; Mosher, R.; Kallakury, B.V. Correlation of primary tumor prostatę specific membrane antigen expression with disease recurrence in prostate cancer. Clin. Cancer Res. 2003, 9, 6357–6362. [Google Scholar]
- Vallabhajosula, S.; Kuji, I.; Hamacher, K.A.; Konishi, S.; Kostakoglu, L.; Kothari, P.A.; Milowski, M.I.; Nanus, D.M.; Bander, N.H.; Goldsmith, S.J. Pharmacokinetics and biodistribution of 111In- and 177Lu-labeled J591 antibody specific for pros-tate-specific membrane antigen: Prediction of 90Y-J591 radiation dosimetry based on 111In or 177Lu? J. Nucl. Med. 2005, 46, 634–641. [Google Scholar] [PubMed]
- Troyer, J.K.; Beckett, M.L.; Wright, G.L. Location of prostate-specific membrane antigen in the LNCaP prostate carcinoma cell line. Prostate 1997, 30, 232–242. [Google Scholar] [CrossRef]
- Chang, S.S. Monoclonal antibodies and prostate-specific membrane antigen. Curr. Opin. Investig. Drugs 2004, 5, 611–615. [Google Scholar]
- Haffner, M.C.; Kronberger, I.E.; Ross, J.S.; Sheehan, C.E.; Zitt, M.; Mühlmann, G.; Öfner, D.; Zelger, B.; Ensinger, C.; Yang, X.J.; et al. Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers. Hum. Pathol. 2009, 40, 1754–1761. [Google Scholar] [CrossRef]
- Hillier, S.M.; Maresca, K.P.; Femia, F.J.; Marquis, J.C.; Foss, C.A.; Nguyen, N.; Zimmerman, C.N.; Barrett, J.A.; Eckelman, W.C.; Pomper, M.G.; et al. Preclinical Evaluation of Novel Glutamate-Urea-Lysine Analogues That Target Prostate-Specific Membrane Antigen as Molecular Imaging Pharmaceuticals for Prostate Cancer. Cancer Res. 2009, 69, 6932–6940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschalis, A.; Sheehan, B.; Riisnaes, R.; Rodrigues, D.N.; Gurel, B.; Bertan, C.; Ferreira, A.; Lambros, M.B.; Seed, G.; Yuan, W.; et al. Prostate-specific Membrane Antigen Heterogeneity and DNA Repair Defects in Prostate Cancer. Eur. Urol. 2019, 76, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagawa, S.T.; Milowsky, M.I.; Morris, M.; Vallabhajosula, S.; Christos, P.; Akhtar, N.H.; Osborne, J.; Goldsmith, S.J.; Larson, S.; Taskar, N.P.; et al. Phase II Study of Lutetium-177–Labeled Anti-Prostate-Specific Membrane Antigen Monoclonal Antibody J591 for Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2013, 19, 5182–5191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Study Evaluating mCRPC Treatment Using PSMA [Lu-177]-PNT2002 Therapy after Second-line Hormonal Treatment. Available online: ClinicalTrials.gov (accessed on 31 December 2020).
- Phase II Trial Shows Novel, Radiolabeled PSMA-Targeted Treatment Provides High Response Rates in Men with Meta-static Prostate Cancer|ASCO. Available online: https://www.asco.org (accessed on 31 December 2020).
- 177Lu-PSMA-617 Therapy and Olaparib in Patients with Metastatic Castration Resistant Prostate Cancer (LuPARP). NCT03874884. Available online: ClinicalTrials.gov (accessed on 19 February 2021).
- Kratochwil, C.; Haberkorn, U.; Giesel, F.L. 225Ac-PSMA-617 for Therapy of Prostate Cancer. Semin. Nucl. Med. 2020, 50, 133–140. [Google Scholar] [CrossRef]
- De Vincentis, G.; Gerritsen, W.; Gschwend, J.; Hacker, M.; Lewington, V.; O’Sullivan, J.; Oya, M.; Pacilio, M.; Parker, C.; Shore, N.; et al. Advances in targeted alpha therapy for prostate cancer. Ann. Oncol. 2019, 30, 1728–1739. [Google Scholar] [CrossRef]
- Czerwińska, M.; Bilewicz, A.; Kruszewski, M.; Wegierek-Ciuk, A.; Lankoff, A. Targeted Radionuclide Therapy of Prostate Cancer—From Basic Research to Clinical Perspectives. Molecules 2020, 25, 1743. [Google Scholar] [CrossRef] [Green Version]
- Eder, M.; Neels, O.; Müller, M.; Bauder-Wüst, U.; Remde, Y.; A Schafer, M.; Hennrich, U.; Eisenhut, M.; Afshar-Oromieh, A.; Haberkorn, U.; et al. Novel Preclinical and Radiopharmaceutical Aspects of [68Ga]Ga-PSMA-HBED-CC: A New PET Tracer for Imaging of Prostate Cancer. Pharmaceuticals 2014, 7, 779–796. [Google Scholar] [CrossRef]
- Eder, M.; Schäfer, M.; Bauder-Wüst, U.; Hull, W.-E.; Wängler, C.; Mier, W.; Haberkorn, U.; Eisenhut, M. 68Ga-Complex Lipophilicity and the Targeting Property of a Urea-Based PSMA Inhibitor for PET Imaging. Bioconj. Chem. 2012, 23, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Kopka, K.; Benešová, M.; Bařinka, C.; Haberkorn, U.; Babich, J. Glu-Ureido–Based Inhibitors of Prostate-Specific Membrane Antigen: Lessons Learned During the Development of a Novel Class of Low-Molecular-Weight Theranostic Radiotracers. J. Nucl. Med. 2017, 58, 17S–26S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Eiber, M.; Schwaiger, M.; Wester, H. Development and first in human evalu-ation of PSMA I&T—A ligand for diagnostic imaging and endoradiotherapy of prostate cancer. JNM 2014, 55 (Suppl. 1), 1083. [Google Scholar]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Baum, R.P.; Yildiz, A.; Beykan, S.; Kulkarni, H.R.; Lassmann, M.; Klette, I.; Eiber, M.; et al. 68Ga- and 177Lu-Labeled PSMA I&T: Optimization of a PSMA-Targeted Theranostic Concept and First Proof-of-Concept Human Studies. J. Nucl. Med. 2015, 56, 1169–1176. [Google Scholar] [CrossRef] [Green Version]
- Benešová, M.; Bauder-Wüst, U.; Schäfer, M.; Klika, K.D.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Linker Modification Strategies To Control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic Properties of DOTA-Conjugated PSMA Inhibitors. J. Med. Chem. 2016, 59, 1761–1775. [Google Scholar] [CrossRef]
- PSMA Inhibitor Derivatives for Labelling with 99mTc via HYNIC, a Radiopharmaceutical kit, Radiopharmaceutical Prepa-rations and Their Use in Prostate Cancer Diagnostics. EP3721907A1. Available online: https://worldwide.espacenet.com (accessed on 31 December 2020).
- Sergieva, S.; Mangaldjiev, R.; Dimcheva, M.; Bozhil, R. SPECT-Computed Tomography with New 99mTc Pros-tate-Specific Membrane Antigen-T4 Tracer in Patients with Recurrent Prostate Cancer. 14th International Conference on Radiopharmaceutical Therapy (ICRT 2019) & World Association of Radiopharmaceutical and molecular therapy (WARMTH), Nanjing, China, 22–25 August, 2019. World J. Nucl. Med. 2019, 18, 317–323. [Google Scholar]
- Sergieva, S.; Robev, B.; Dimcheva, M. Clinical Application of SPECT-CT Imaging with 99mTc-PSMA-T4 in patients with Reccurrent Prostate Cancer. J. Nuclear Med. 2020, 61 (Suppl. 1), 473. [Google Scholar]
- Cardinale, J.; Roscher, M.; Schaefer, M.; Geerlings, M.; Benešová, M.; Bauder-Wüst, U.; Remde, Y.; Eder, M.; Nováková, Z.; Motlová, L.; et al. Development of PSMA-1007-Related Series of 18F-Labeled Glu-Ureido-Type PSMA Inhibitors. J. Med. Chem. 2020, 63, 10897–10907. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and Auto-DockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlicek, J.; Ptacek, J.; Barinka, C. Glutamate Carboxypeptidase II: An Overview of Structural Studies and Their Im-portance for Structure-Based Drug Design and Deciphering the Reaction Mechanism of the Enzyme. Curr. Med. Chem. 2012, 19, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Tönnesmann, R.; Meyer, P.T.; Eder, M.; Baranski, A.-C. [177Lu]Lu-PSMA-617 Salivary Gland Uptake Characterized by Quantitative In Vitro Autoradiography. Pharmaceuticals 2019, 12, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical Trial of Ac225-PSMA Radioligand Therapy of Metastatic Castration-Resistant Prostate Cancer. NCT04225910. Available online: ClinicalTrials.gov (accessed on 19 February 2021).
- Benešová, M.; Schäfer, M.; Bauder-Wüst, U.; Afshar-Oromieh, A.; Kratochwil, C.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. J. Nucl. Med. 2015, 56, 914–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schottelius, M.; Wurzer, A.; Wissmiller, K.; Beck, R.; Koch, M.; Gkorpas, D.; Notni, J.; Buckle, T.; Van Oosterom, M.N.; Steiger, K.; et al. Synthesis and Preclinical Characterization of the PSMA-Targeted Hybrid Tracer PSMA-I&F for Nuclear and Fluorescence Imaging of Prostate Cancer. J. Nucl. Med. 2019, 60, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schottelius, M.; Wirtz, M.; Eiber, M.; Maurer, T.; Wester, H.-J. [111In]PSMA-I&T: Expanding the spectrum of PSMA-I&T applications towards SPECT and radioguided surgery. EJNMMI Res. 2015, 5, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shao, G.; Wu, J.; Cui, C.; Zang, S.; Qiu, F.; Jia, R.; Wang, Z.; Wang, F. Preparation of 68Ga-PSMA-11 with a Synthesis Module for Micro PET-CT Imaging of PSMA Expression during Prostate Cancer Progression. Contrast Media Mol. Imaging 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbricht, C.A.; Benešová, M.; Schmid, R.M.; Türler, A.; Schibli, R.; Van Der Meulen, N.P.; Müller, C. 44Sc-PSMA-617 for radiotheragnostics in tandem with 177Lu-PSMA-617—preclinical investigations in comparison with 68Ga-PSMA-11 and 68Ga-PSMA-617. EJNMMI Res. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, D.; Wojdowska, W.; Parus, L.J.; Cieszykowska, I.; Zoltowska, M.; Garnuszek, P.; Mikolajczak, R. Comparison of separation methods for 47Ca/47Sc radionuclide generator. Appl. Radiat. Isot. 2019, 151, 140–144. [Google Scholar] [CrossRef]
- Abu-Baker, S.; Garber, P.; Hina, B.; Reed, T.; Shahrokh, G.; Al-Saghir, M.; Lorigan, G. Microwave Assisted Peptide Synthesis as a New Gold Standard in Solid Phase Peptide Synthesis: Phospholamban as an Example. Open J. Synth. Theory Appl. 2014, 3, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Collins, J.M.; Porter, K.A.; Singh, S.K.; Vanier, G.S. High-Efficiency Solid Phase Peptide Synthesis (HE-SPPS). Org. Lett. 2014, 16, 940–943. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. COMU: A third generation of uronium-type coupling reagents. J. Pept. Sci. 2009, 16, 6–9. [Google Scholar] [CrossRef]
- Wängler, B.; Beck, C.; Wagner-Utermann, U.; Schirrmacher, E.; Bauer, C.; Rösch, F.; Schirrmacher, R.; Eisenhut, M. Application of tris-allyl-DOTA in the preparation of DOTA–peptide conjugates. Tetrahedron Lett. 2006, 47, 5985–5988. [Google Scholar] [CrossRef]
- Waterhouse, R.N. Determination of lipophilicity and its use as a predictor of blood–brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2003, 5, 376–389. [Google Scholar] [CrossRef]
- A Wilson, A.; Jin, L.; Garcia, A.; DaSilva, J.N.; Houle, S. An admonition when measuring the lipophilicity of radiotracers using counting techniques. Appl. Radiat. Isot. 2001, 54, 203–208. [Google Scholar] [CrossRef]
- Aime, S.; Barge, A.; Botta, M.; Fasano, M.; Ayala, J.D.; Bombieri, G. Crystal structure and solution dynamics of the lutetium(III) chelate of DOTA. Inorganica Chim. Acta 1996, 246, 423–429. [Google Scholar] [CrossRef]
- BIOVIA, Dassault Systèmes. Discovery Studio Visualizer, v 17.2. 0; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. Available online: https://github.com/schrodinger/pymol-open-source (accessed on 31 December 2020).
- Fichna, J.; Krajewska, U.; Rozalski, M.; Mirowski, M.; Janecka, A. Characterization of the [125I]endomorphin-2 binding sites in the MCF7 breast cancer cell line. Peptides 2005, 26, 295–299. [Google Scholar] [CrossRef]
- Hames, E.D. Fmoc Solid Phase Peptide Synthesis. A Practical Approach; Chan, W.C., White, P.D., Eds.; Oxford University Press Inc.: New York, NY, USA, 2004; pp. 47–48. [Google Scholar]
- Eissler, S.; Kley, M.; Bächle, D.; Loidl, G.; Meier, T.; Samson, D. Substitution determination of Fmoc-substituted resins at different wavelengths. J. Pept. Sci. 2017, 23, 757–762. [Google Scholar] [CrossRef]
- Subirós-Funosas, R.; Nieto-Rodriguez, L.; Jensen, K.J.; Albericio, F. COMU: Scope and limitations of the latest innovation in peptide acyl transfer reagents. J. Pept. Sci. 2013, 19, 408–414. [Google Scholar] [CrossRef] [PubMed]







| Compound | RCY [%] | SA [GBq mcmol−1] | pH |
|---|---|---|---|
| [177Lu]Lu-PSMA-D4 | 99.6 ± 0.1 | 17.2 | 4.5–5.0 |
| [90Y]Y-PSMA-D4 | 99.7 ± 0.1 | 17.2 | 4.5–5.0 |
| [47Sc]Sc-PSMA-D4 | 99.6 ± 0.1 | 4.57 | 4.0–4.5 |
| [225Ac]Ac-PSMA-D4 | 99.3 ± 0.2 | 0.116 | 8.5–9.0 |
| Compound | logD |
|---|---|
| [177Lu]Lu-PSMA-D4 | −5.16 ± 0.07 |
| [90Y]Y-PSMA-D4 | −5.18 ± 0.04 |
| [177Lu]Lu-PSMA-617 | −5.12 ± 0.07 |
| [90Y]Y-PSMA-617 | −5.08 ± 0.13 |
| [68Ga]Ga-PSMA-11 | −3.46 ± 0.06 |
| [177Lu]Lu-PSMA-I&T | −4.47 ± 0.14 |
| Compound | IC50 [nM] | p |
|---|---|---|
| PSMA-D4 | 28.7 ± 5.2 | - |
| PSMAI&T | 61.1 ± 7.8 | 0.0059 |
| PSMA-11 | 84.5 ± 26.5 | <0.0001 |
| PSMA-617 | 2.7 ± 0.1 [34] * | 0.0259 |
| Compound | Total Binding (%) | Internalization (%) | KD (nM) | Specific Binding (%) |
|---|---|---|---|---|
| [177Lu]Lu-PSMA-D4 | 10.2 | 37.0 ± 3.3 | 2.4 ± 0.3 | 99.1 ± 0.3 |
| [90Y]Y-PSMA-D4 | 16.8 | 35.4 | 4.5 ± 2.1 | 99.9 ± 0.1 |
| [47Sc]Sc-PSMA-D4 | n.d. | n.d. | 3.4 ± 2.0 | 98.8 ± 0.9 |
| [177Lu]Lu-PSMA I&T | n.d. | n.d. | 5.1 ± 2.6 | 97.1 ± 2.9 |
| [68Ga]Ga-PSMA-11 | 13.1 | 2.6 | 11.4 ± 7.1 | 96.8 ± 2.7 |
| [177Lu]Lu-PSMA 617 | 12.8 ± 1.0 | 38.9 ± 1.7 | 2.0 ± 0.3 [34] * | n.d. |
| 2 h | 4 h | 6 h | 24 h | 48 h | ||
|---|---|---|---|---|---|---|
| [90Y]Y-PSMA-D4 | blood | 0.06 ± 0.01 | 0.05 ± 0.03 | 0.06 ± 0.01 | 0.03 ± 0.03 | 0.04 ± 0.02 |
| liver | 0.08 ± 0.00 | 0.08 ± 0.01 | 0.31 ± 0.07 | 0.04 ± 0.01 | 0.17 ± 0.07 | |
| spleen | 0.19 ± 0.06 | 0.17 ± 0.06 | 1.05 ± 0.29 | 0.12 ± 0.06 | 0.97 ± 0.62 | |
| kidneys | 2.70 ± 0.20 | 1.70 ± 0.13 | 0.98 ± 0.38 | 0.30 ± 0.12 | 0.12 ± 0.03 | |
| small intestine | 0.32 ± 0.13 | 0.30 ± 0.14 | 0.14 ± 0.11 | 0.20 ± 0.20 | 0.14 ± 0.07 | |
| large intestine | 0.12 ± 0.05 | 0.32 ± 0.16 | 0.20 ± 0.02 | 0.41 ± 0.15 | 0.84 ± 0.56 | |
| stomach | 0.10 ± 0.04 | 0.11 ± 0.05 | 0.04 ± 0.03 | 0.06 ± 0.05 | 0.03 ± 0.03 | |
| tumor | 20.24 ± 5.52 | 16.40 ± 6.88 | 8.28 ± 1.97 | 8.33 ± 3.09 | 8.03 ± 7.31 | |
| muscle | 0.21 ± 0.13 | 0.05 ± 0.02 | 0.01 ± 0.01 | 0.02 ± 0.02 | 0.02 ± 0.01 | |
| urine [%ID] | 96.08 ± 0.53 | 95.47 ± 3.07 | 96.57 ± 2.44 | 94.79 ± 7.51 | 97.75 ± 0.34 | |
| T/B | 323.7 | 312.9 | 131.2 | 257.6 | 193.0 | |
| T/M | 97.0 | 324.4 | 983.7 | 387.4 | 351.4 | |
| T/K | 7.6 | 9.7 | 8.4 | 29.0 | 68.4 | |
| 1 h | 4 h | 6 h | 24 h | 48 h | ||
| [47Sc]Sc-PSMA-D4 | blood | 0.29 ± 0.08 | 0.08 ± 0.04 | 0.04 ± 0.01 | 0.01 ± 0.01 | 0.05 ± 0.03 |
| liver | 0.13 ± 0.02 | 0.08 ± 0.04 | 0.06 ± 0.01 | 0.03 ± 0.01 | 0.04 ± 0.01 | |
| spleen | 0.52 ± 0.19 | 0.57 ± 0.31 | 0.08 ± 0.04 | 0.04 ± 0.01 | 0.10 ± 0.09 | |
| kidneys | 9.46 ± 1.44 | 2.16 ± 0.86 | 1.42 ± 0.28 | 0.29 ± 0.09 | 0.26 ± 0.14 | |
| small intestine | 0.35 ± 0.19 | 0.41 ± 0.30 | 0.11 ± 0.07 | 0.34 ± 0.18 | 0.45 ± 0.13 | |
| large intestine | 0.09 ± 0.03 | 0.70 ± 0.48 | 0.69 ± 0.17 | 0.12 ± 0.12 | 1.63 ± 0.31 | |
| stomach | 0.23 ± 0.09 | 0.39 ± 0.17 | 0.08 ± 0.09 | 0.05 ± 0.06 | 0.03 ± 0.04 | |
| tumor | 9.24 ± 5.13 | 9.22 ± 2.21 | 8.65 ± 3.19 | 5.28 ± 3.01 | 4.11 ± 1.08 | |
| muscle | 0.10 ± 0.06 | 0.39 ± 0.17 | 0.03 ± 0.02 | 0.02 ± 0.02 | 0.06 ± 0.06 | |
| urine [%ID] | 89.23 ± 2.63 | 93.42 ± 2.07 | 94.89 ± 1.34 | 97.89 ± 1.22 | 95.37 ± 1.89 | |
| T/B | 31.6 | 120.9 | 207.6 | 467.3 | 78.8 | |
| T/M | 135.2 | 24.3 | 251.0 | 229.0 | 66.7 | |
| T/K | 1.0 | 4.3 | 6.1 | 18.4 | 16.0 |
| a | %ID g−1 | Time p.i.v. | Dose | Mice | Refs. | ||
|---|---|---|---|---|---|---|---|
| Spleen | Kidneys | Tumor | |||||
| [177Lu]Lu-PSMA I&T | 5.85 ± 2.26 | 107.24 ± 15.61 | 7.96 ± 1.76 | 1 h | 0.2 nmol | CD-1 nu/nu | [14] |
| [177Lu]Lu-PSMA-I&T | 7.9 ± 2.6 | 105.3 ± 15.8 | 8.0 ± 1.7 | 1 h | 0.2 nmol | CD-1 nu/nu | [37] |
| [90Y]Y-PSMA-D4 | 0.19 ± 0.06 | 2.70 ± 0.20 | 20.24 ± 5.52 | 2 h | 0.1 nmol | BALC/c Nude | - |
| [47Sc]Sc-PSMA-D4 | 0.52 ± 0.19 | 9.46 ± 1.44 | 9.24 ± 5,13 | 1 h | 0.2 nmol | BALC/c Nude | - |
| [111In]In-PSMA-I&T | 47.4 ± 13.2 | 207.9 ± 23.9 | 8.1 ± 1.1 | 1 h | 0.2 nmol | CB17 SCID | [38] |
| [68Ga]Ga-PSMA I&T | 3.88 ± 1.46 | 53.26 ± 9.02 | 4.95 ± 1.57 | 1 h | 0.2 nmol | CD-1 nu/nu | [14] |
| [68Ga]Ga-PSMA-I&F | 12.8 ± 6.5 | 105.8 ± 22.7 | 4.5 ± 1.8 | 1 h | n.d. | SHO | [37] |
| [68Ga]Ga-PSMA-I&T | 5.3 ± 1.3 | 52.6 ± 13.2 | 5.0 ± 1.6 | 1 h | 0.2 nmol | CB17 SCID | [38] |
| [68Ga]Ga-PSMA-I&T | 3.9 ± 1.5 | 53.3 ± 9.0 | 4.9 ± 1.6 | 1 h | n.d. | SHO | [37] |
| [68Ga]Ga-PSMA-11 | n.d. | n.d. | 7.28 ± 0.82 | 1 h | n.d. | Nude | [39] |
| b | %ID g−1 | Time p.i.v. | Ddose | Mice | Refs. | ||
| Spleen | Kidneys | Tumor | |||||
| [47Sc]Sc-PSMA-617 | 0.81 ± 0.34 | 5.97 ± 0.90 | 46.7 ± 4.36 | 2 h | 1 nmol | BALB/c Nude | [40] |
| [177Lu]Lu-PSMA-617 | 0.21 ± 0.03 | 3.97 ± 0.56 | 45.8 ± 4.02 | 2 h | 1 nmol | BALB/c Nude | [40] |
| [68Ga]Ga-PSMA-617 | 0.91 ± 0.14 | 3.48 ± 0.18 | 55.8 ± 14.2 | 2 h | 1 nmol | BALB/c Nude | [40] |
| [68Ga]Ga-PSMA-11 | 2.18 ± 0.38 | 58.8 ± 7.62 | 40.0 ± 2.55 | 2 h | 1 nmol | BALB/c Nude | [40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garnuszek, P.; Karczmarczyk, U.; Maurin, M.; Sikora, A.; Zaborniak, J.; Pijarowska-Kruszyna, J.; Jaroń, A.; Wyczółkowska, M.; Wojdowska, W.; Pawlak, D.; et al. PSMA-D4 Radioligand for Targeted Therapy of Prostate Cancer: Synthesis, Characteristics and Preliminary Assessment of Biological Properties. Int. J. Mol. Sci. 2021, 22, 2731. https://doi.org/10.3390/ijms22052731
Garnuszek P, Karczmarczyk U, Maurin M, Sikora A, Zaborniak J, Pijarowska-Kruszyna J, Jaroń A, Wyczółkowska M, Wojdowska W, Pawlak D, et al. PSMA-D4 Radioligand for Targeted Therapy of Prostate Cancer: Synthesis, Characteristics and Preliminary Assessment of Biological Properties. International Journal of Molecular Sciences. 2021; 22(5):2731. https://doi.org/10.3390/ijms22052731
Chicago/Turabian StyleGarnuszek, Piotr, Urszula Karczmarczyk, Michał Maurin, Arkadiusz Sikora, Jolanta Zaborniak, Justyna Pijarowska-Kruszyna, Antoni Jaroń, Monika Wyczółkowska, Wioletta Wojdowska, Dariusz Pawlak, and et al. 2021. "PSMA-D4 Radioligand for Targeted Therapy of Prostate Cancer: Synthesis, Characteristics and Preliminary Assessment of Biological Properties" International Journal of Molecular Sciences 22, no. 5: 2731. https://doi.org/10.3390/ijms22052731
APA StyleGarnuszek, P., Karczmarczyk, U., Maurin, M., Sikora, A., Zaborniak, J., Pijarowska-Kruszyna, J., Jaroń, A., Wyczółkowska, M., Wojdowska, W., Pawlak, D., Lipiński, P. F. J., & Mikołajczak, R. (2021). PSMA-D4 Radioligand for Targeted Therapy of Prostate Cancer: Synthesis, Characteristics and Preliminary Assessment of Biological Properties. International Journal of Molecular Sciences, 22(5), 2731. https://doi.org/10.3390/ijms22052731