
A Review of the Pharmacological Activities and Recent Synthetic Advances of γ-Butyrolactones


Abstract
:1. Introduction
2. Pharmacological Activities of γ-Butyrolactones
2.1. Approved Drugs
2.2. Biologically Active γ-Butyrolactones
2.2.1. Anti-Inflammation
2.2.2. Anticancer
2.2.3. Antibiotic
2.2.4. Antifungal
2.2.5. Immunosuppressive
2.2.6. Neuroprotective
2.2.7. Antioxidant
2.2.8. Hypoglycemic
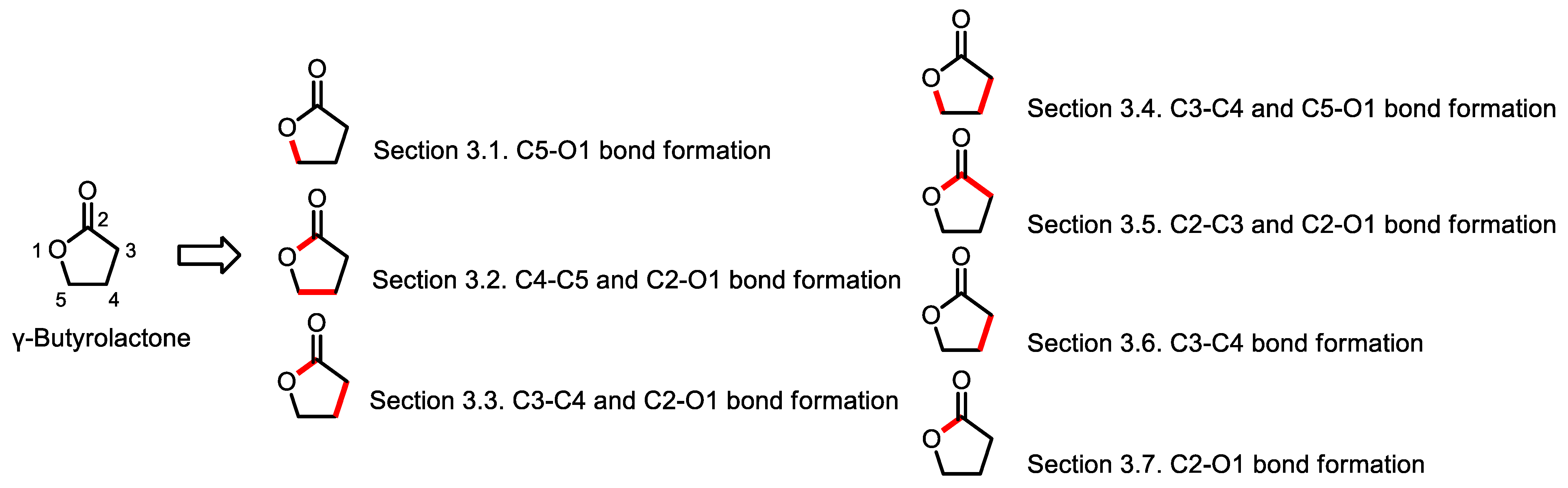
3. Synthesis of γ-Butyrolactones
3.1. Synthesis of γ-Butyrolactone via C5-O1 Bond Formation
3.1.1. Oxidative Lactonization of Pentenoic Acid
3.1.2. Halolactonization of Pentenoic Acid
3.1.3. Acid-Promoted Cyclopropane Opening
3.1.4. Au-Catalyzed Oxaallylation
3.1.5. Photoredox-Catalyzed Lactonization
3.2. Synthesis of γ-Butyrolactone via C4-C5 and C2-O1 Bonds Formation
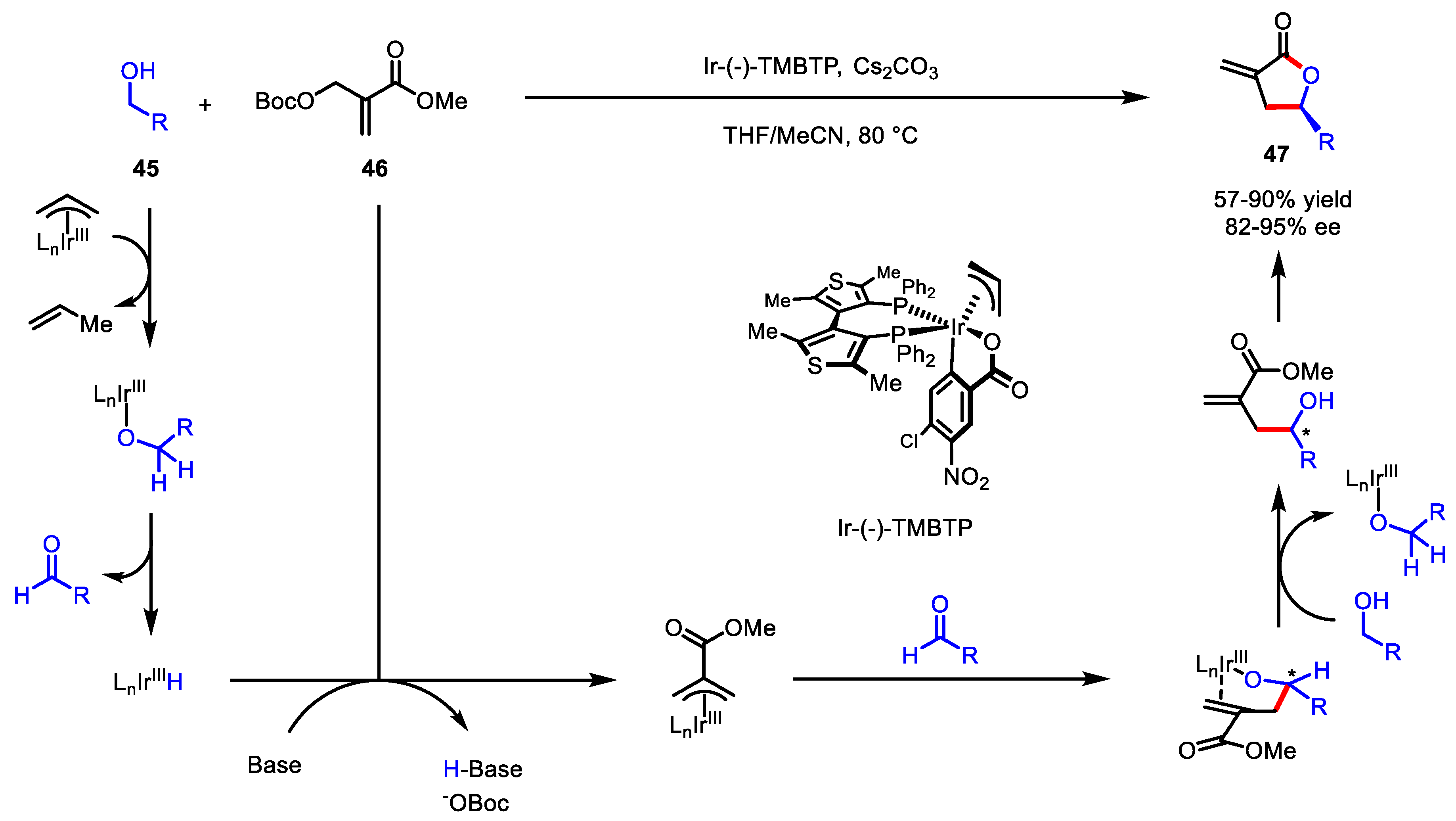
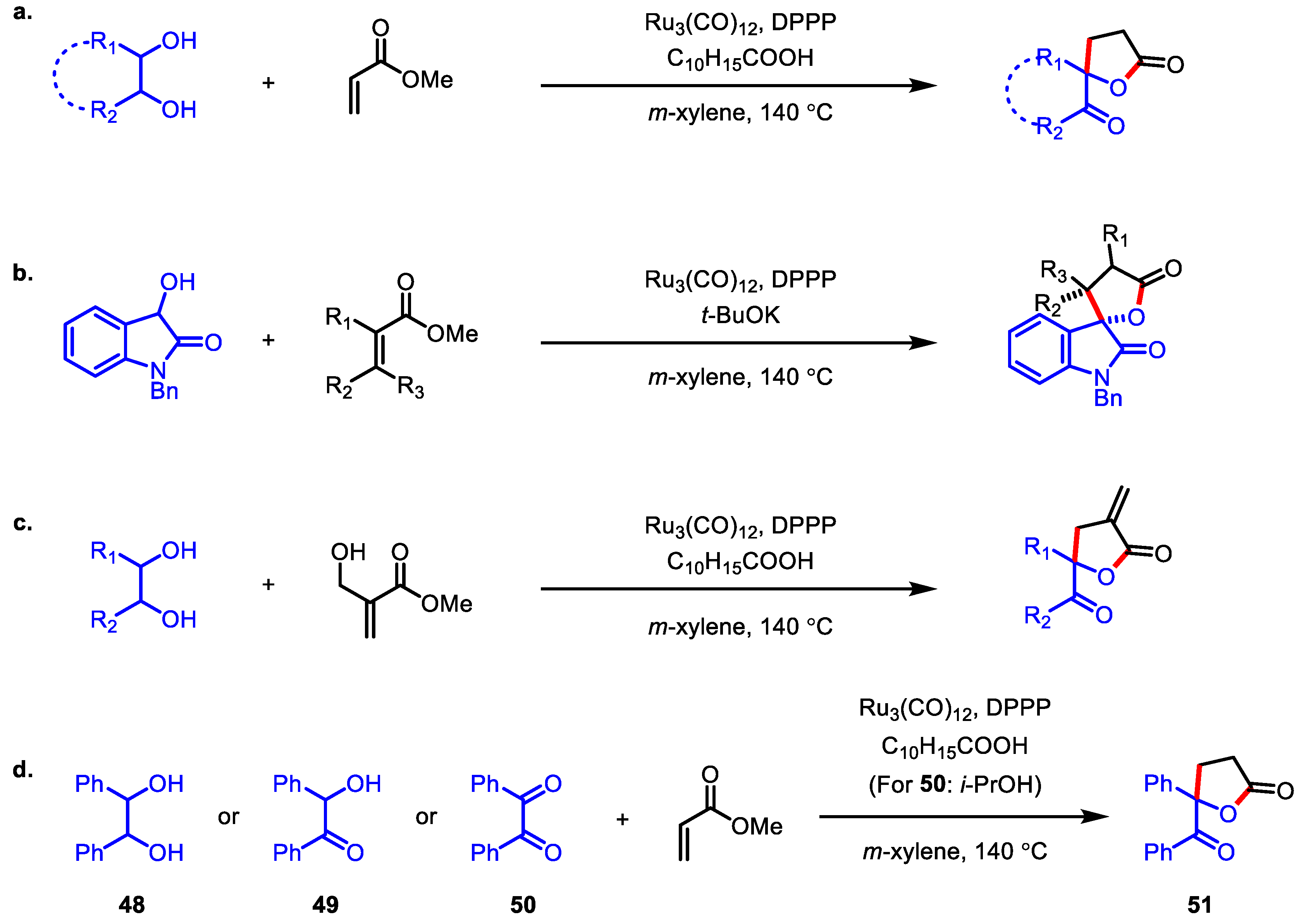
3.2.1. Transition-Metal Catalyzed C-C Bond Coupling
3.2.2. NHC-Catalyzed C-C Bond Coupling
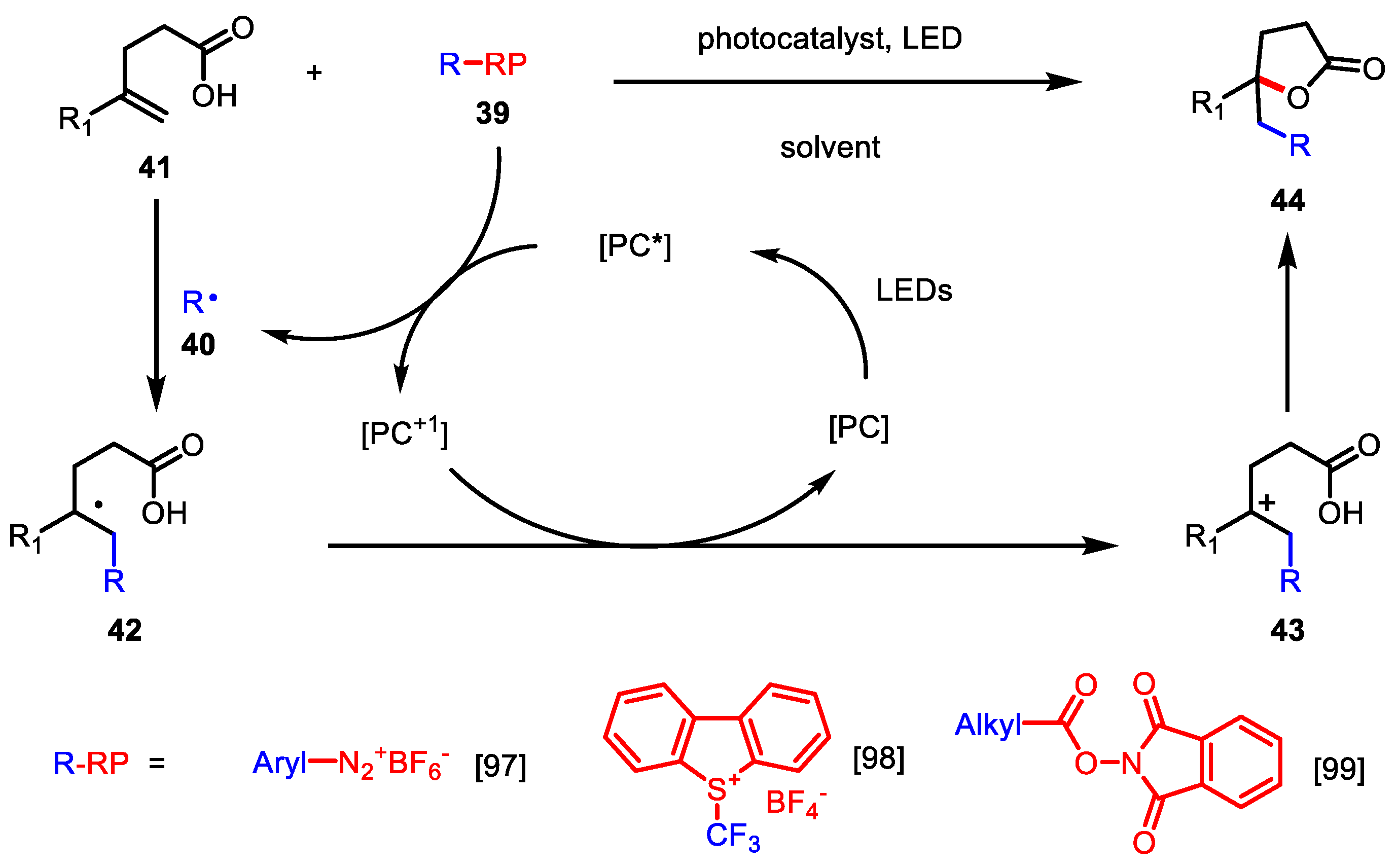
3.2.3. Photoredox-Catalyzed C-C Bond Coupling
3.2.4. Miscellsious γ-Butyrolactone Formation
3.3. Synthesis of γ-Butyrolactones via C3-C4 and C2-O1 Bond Formation
3.4. Synthesis of Butyrolactone via C3-C4 and C5-O1 Bonds Formation
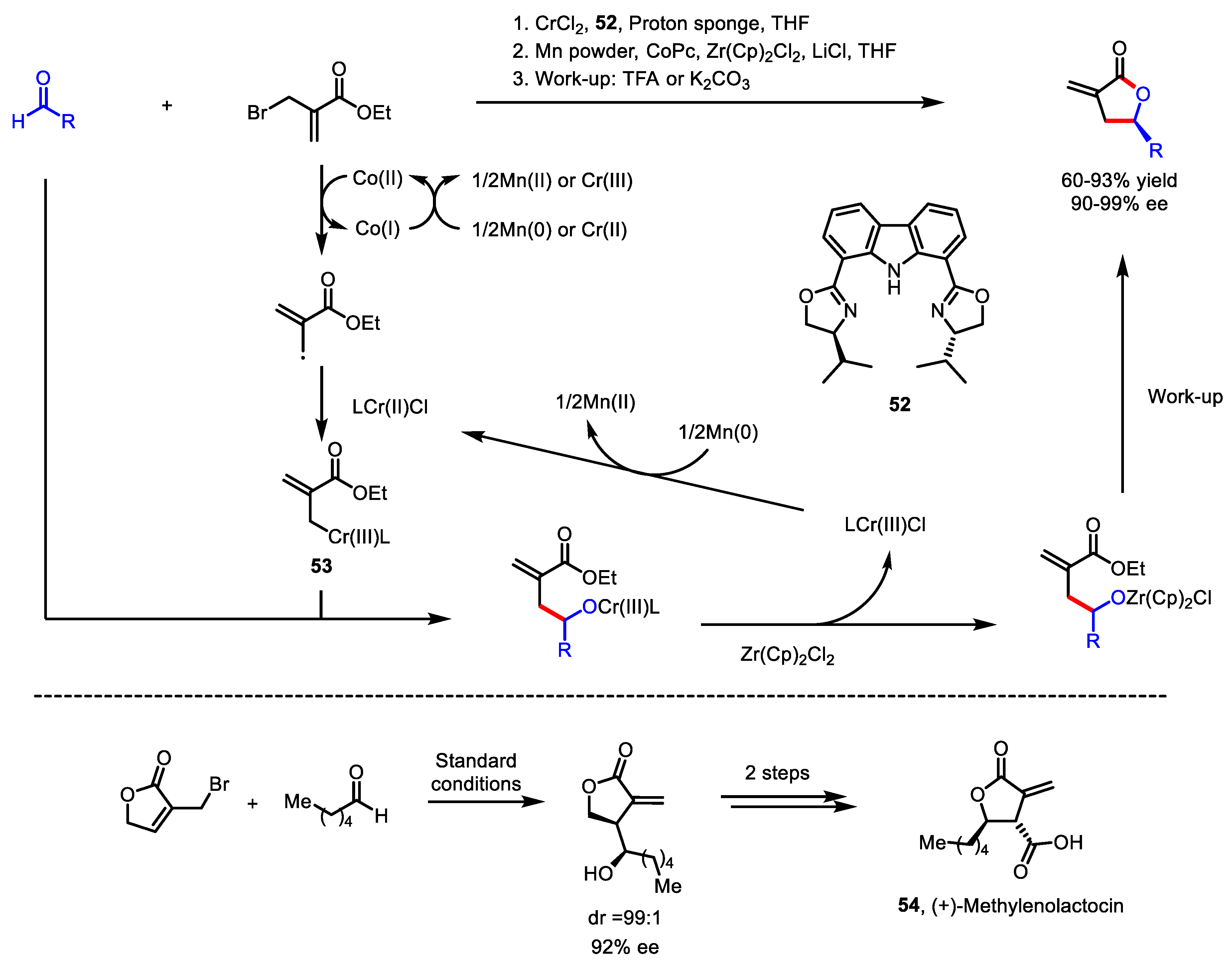
3.4.1. Polar Radical Crossover Cycloaddition (PRCC)
3.4.2. Atom-Transfer Radical Addition (ATRA)
3.4.3. Mn(OAc)3-Mediated Radical Lactonization
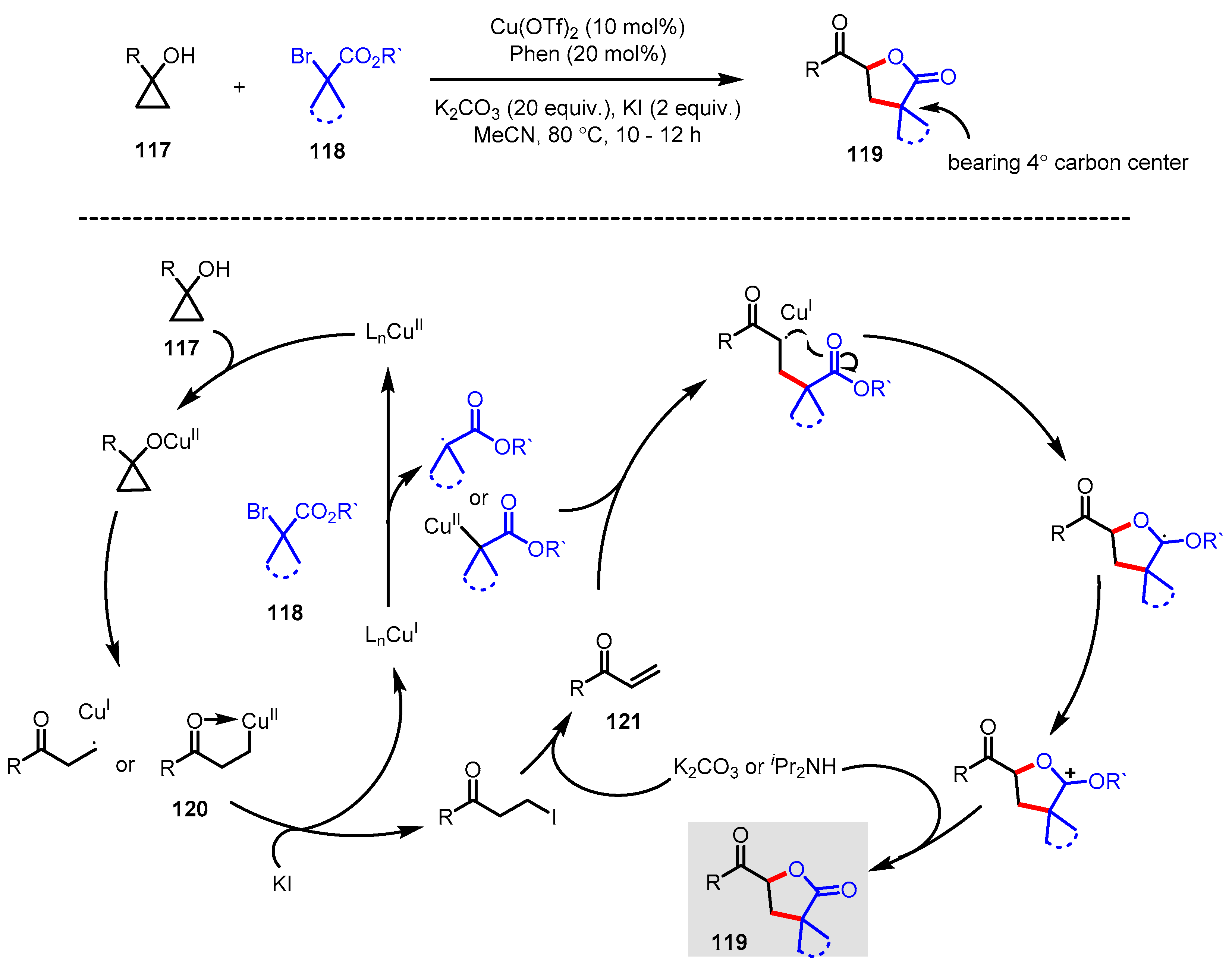
3.4.4. Copper-Catalyzed Cyclopropanol Ring-Opening Cross-Coupling Reaction
3.5. Synthesis of γ-Butyrolactones via C2-C3 and C2-O1 Bonds Formation
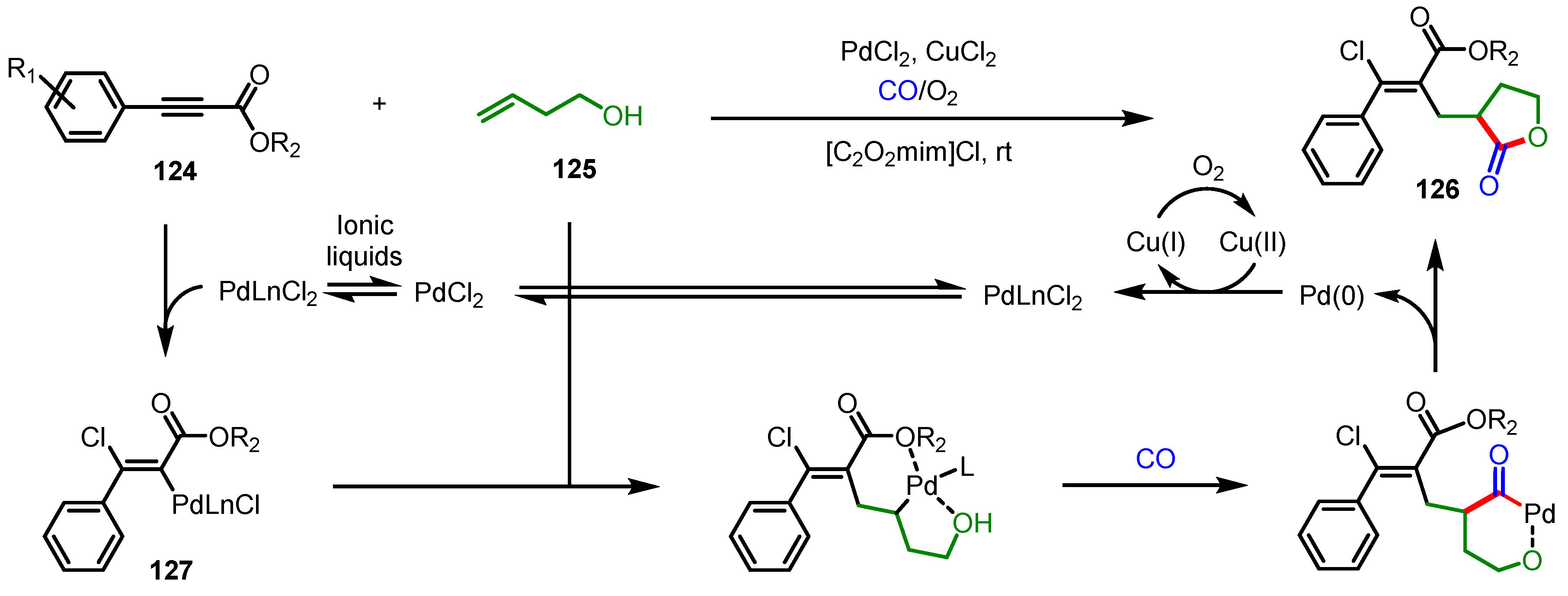
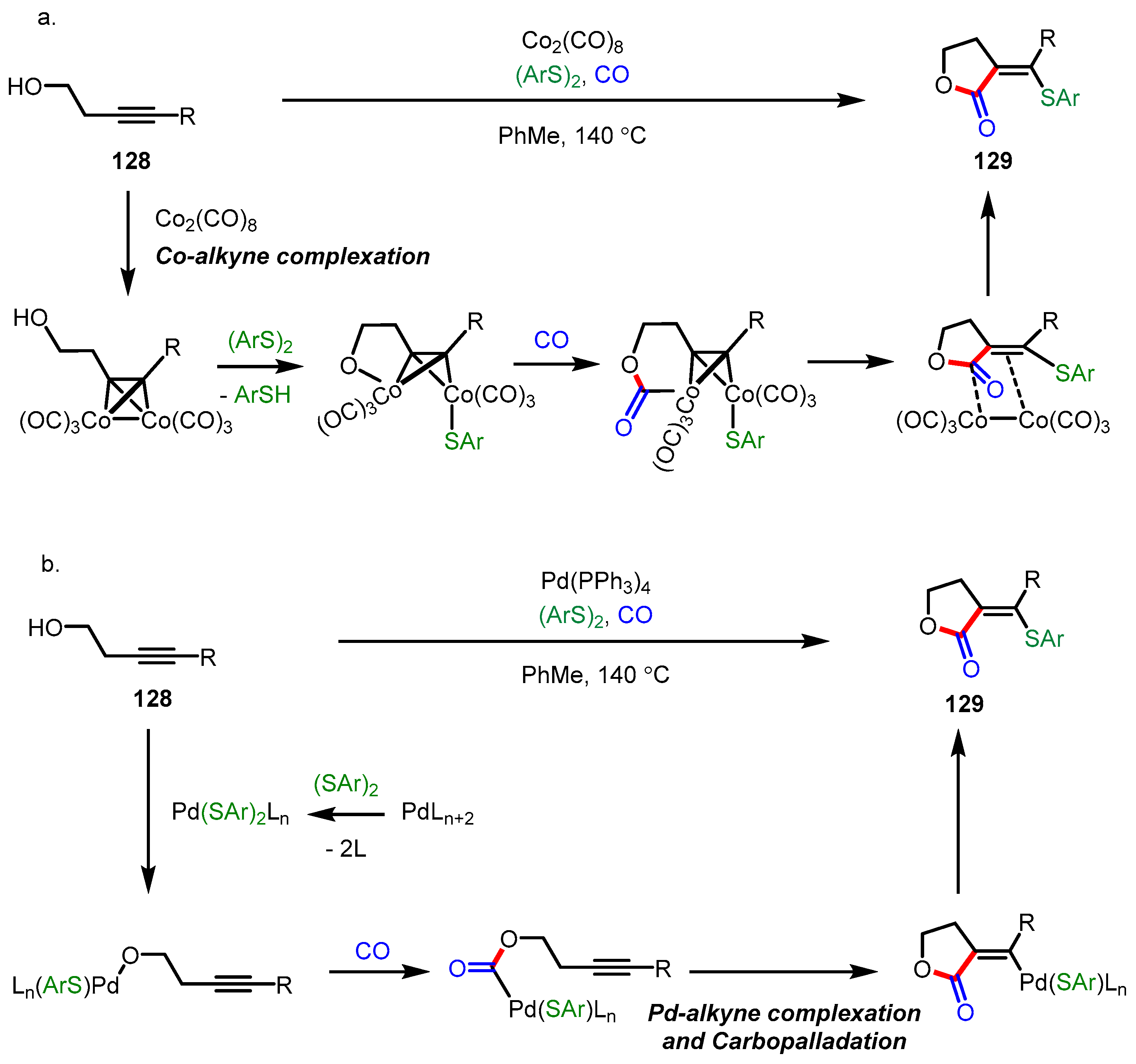
3.5.1. Carbonylative Lactonization
3.5.2. Hydroformylation-Oxidation
3.5.3. Carboxylation-Lactonization
3.6. Synthesis of γ-Butyrolactones via C3-C4 Bond Formation
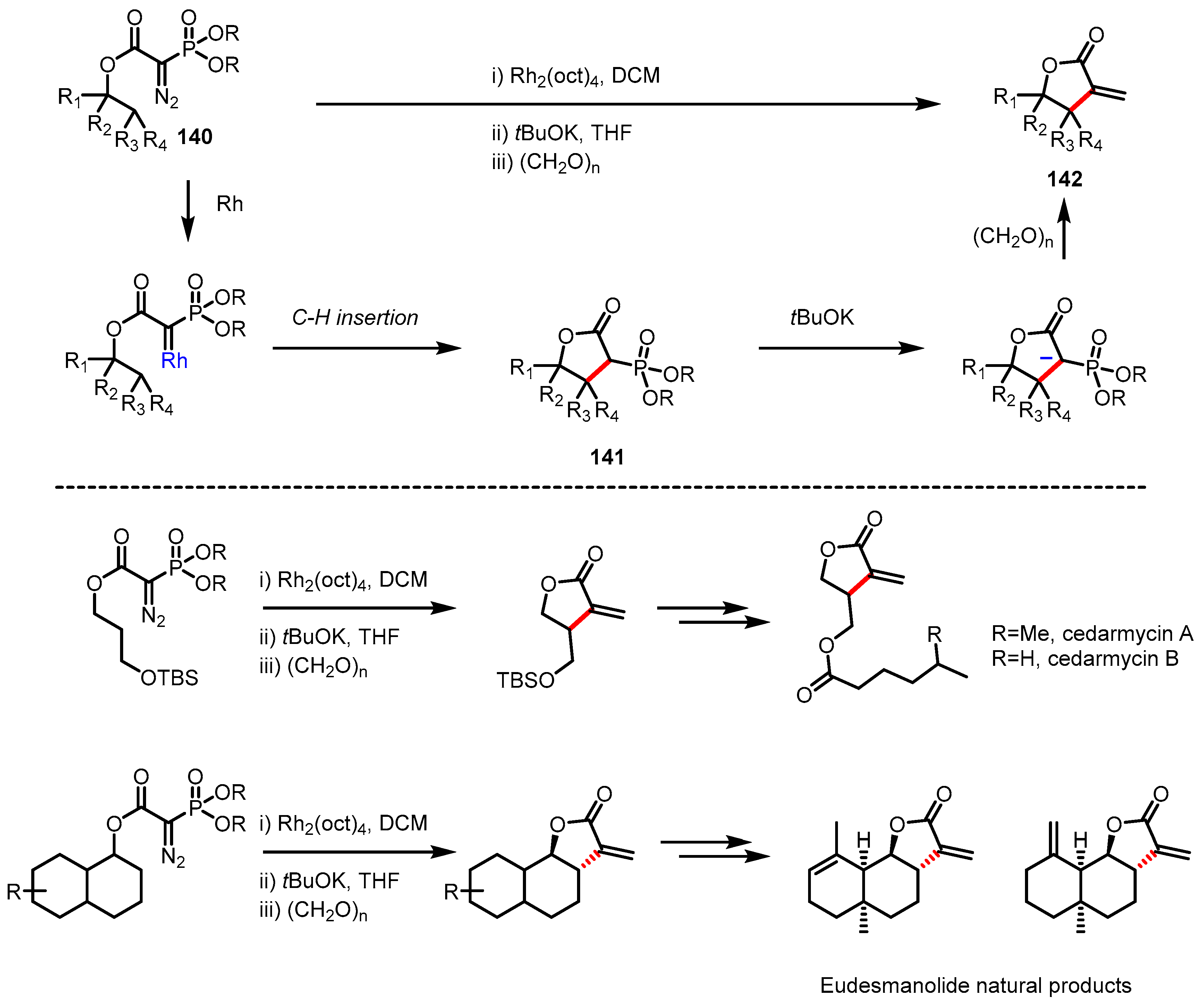
C-H Insertion
3.7. Synthesis of γ-Butyrolactones via Oxidative C2-O1 Bond Formation
 .
.4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| [C2O2 mim]Cl | 1-carboxymethyl-3-methylimidazolium chloride |
| Ac | Acetyl |
| acac | Acetylacetone |
| Acr | Acridinium |
| Ar | Aryl |
| ATRA | Atom-transfer radical addition |
| Bn | Benzyl |
| Boc | tert-Butyloxycarbonyl |
| bpy | 2,2′- bipyridine |
| Bu | Butyl |
| CDI | Carbonyldiimidazole |
| cod | 1,5-Cyclooctadiene |
| Cp | Cyclopentadienyl |
| DBU | 1,8-Diazabicyclo(5.4.0)undec-7-ene |
| DCE | 1,2-Dichloroethane |
| dF(CF3)ppy | 2-(2,4-Difluorophenyl)-5-(trifluoromethyl)pyridine |
| DFT | Density functional theory |
| DKR | Dynamic kinetic resolution |
| Dmim | 1,3-Dimethylimidazolium |
| DMSO | Dimethyl sulfoxide |
| DPPP | 1,3-Bis(diphenylphosphino)propane |
| dtbbpy | 4,4′-Di-tert-butyl-2,2′-bipyridine |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| Et | Ethyl |
| FMO | Flavin-containing monooxygenase |
| HAT | Hydrogen atom transfer |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium3-oxide hexafluorophosphate |
| Hbim | 1-Dutylimidazolium |
| HLADH | Horse liver alcohol dehydrogenase |
| HOBt | 1-Hydroxybenzotriazole |
| LED | Light-emitting diode |
| Me | Methyl |
| Mes | Mesitylene |
| MS | Molecular sieve |
| NBS | N-Bromosuccinimide |
| neoc | Neocuproine |
| NHC | N-heterocyclic carbene |
| Pc | Phthalocyanine |
| PCC | Pyridinium chlorochromate |
| PCR | Peptide coupling reagent |
| PET | Photoinduced electron transfer |
| Ph | Phenyl |
| Phen | Phenanthroline |
| Pr | Propyl |
| PRCC | Polar radical crossover cycloaddition |
| PTSA | p-Toluenesulfonic acid |
| SET | single-electron transfer |
| TBAF | Tetra-n-butylammonium fluoride |
| TBAP | Tetra-n-butylammonium phosphate |
| TBS | tert-Butyldimethylsilyl |
| TEA | Triethylamine |
| TEMPO | 2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| Tf | Trifluoromethanesulfonyl |
| TFA | trifluoroacetic acid |
| THF | tetrahydrofuran |
| TMBTP | (-)-2,2′,5,5′-tetramethyl-3,3′-bis(diphenylphosphine)-4,4′-bithiophene |
| TMS | Trimethylsilyl |
| Ts | p-Toluenesulfonyl |
References
- Omura, S.; Tanaka, H.; Okada, Y.; Marumo, H. Isolation and structure of nanaomycin D, an enantiomer of the antibiotic kalafungin. J. Chem. Soc. Chem. Commun. 1976, 213, 320–321. [Google Scholar] [CrossRef]
- Yamawaki, M.; Nishi, K.; Nishimoto, S.; Yamauchi, S.; Akiyama, K.; Kishida, T.; Maruyama, M.; Nishiwaki, H.; Sugahara, T. Immunomodulatory effect of (-)-matairesinol in vivo and ex vivo. Biosci. Biotechnol. Biochem. 2011, 75, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Zhangabylov, N.S.; Dederer, L.Y.; Gorbacheva, L.B.; Vasil’eva, S.V.; Terekhov, A.S.; Adekenov, S.M. Sesquiterpene lactone arglabin influences DNA synthesis in P388 leukemia cells in vivo. Pharm. Chem. J. 2004, 38, 651–653. [Google Scholar] [CrossRef]
- Rudolphi, K.; Gerwin, N.; Verzijl, N.; van der Kraan, P.; van den Berg, W. Pralnacasan, an inhibitor of interleukin-1β converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthr. Cart. 2003, 11, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Ghosh, S.K. Organo-catalyzed enantioselective synthesis of some β-silyl γ-alkyl γ-butyrolactones as intermediates for natural products. Tetrahedron Asymmetry 2011, 22, 1895–1900. [Google Scholar] [CrossRef]
- Ok, T.; Jeon, A.; Lee, J.; Jung, H.L.; Chang, S.H.; Lee, H.S. Enantiomerically pure synthesis of β-substituted γ-butyrolactones: A key intermediate to concise synthesis of pregabalin. J. Org. Chem. 2007, 72, 7390–7393. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.; Daugan, A. An easy preparation of (-) and (+)-β-piperonyl-γ-butyrolactones, key-intermediates for the synthesis of optically active lignans. Tetrahedron Lett. 1985, 26, 3997–3998. [Google Scholar] [CrossRef]
- Bielitza, M.; Pietruszka, J. An enantioselective Mukaiyama aldol reaction as the key step towards the tetrahydropyran core of psymberin via a γ-butyrolactone intermediate. Synlett 2012, 23, 1625–1628. [Google Scholar]
- Givens, R.S.; Oettle, W.F. Photorearrangement of a γ-butyrolactone: Generation of intermediates in photochemical reactions. Anal. Proc. 1969, 1164–1165. [Google Scholar] [CrossRef]
- Vinet, L.; Zhedanov, A. A “missing” family of classical orthogonal polynomials. J. Phys. A Math. Theor. 2011, 44, 51. [Google Scholar] [CrossRef]
- Murauski, K.J.R.; Jaworski, A.A.; Scheidt, K.A. A continuing challenge: N-heterocyclic carbene-catalyzed syntheses of γ-butyrolactones. Chem. Soc. Rev. 2018, 47, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Kitson, R.R.A.; Millemaggi, A.; Taylor, R.J.K. The renaissance of α-methylene-γ-butyrolactones: New synthetic approaches. Angew. Chem. Int. Ed. 2009, 48, 9426–9451. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Fañanás-Mastral, M.; Feringa, B.L. Catalytic asymmetric synthesis of butenolides and butyrolactones. Chem. Rev. 2017, 117, 10502–10566. [Google Scholar] [CrossRef]
- Seitz, M.; Reiser, O. Synthetic approaches towards structurally diverse γ-butyrolactone natural-product-like compounds. Curr. Opin. Chem. Biol. 2005, 9, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Vivino, F.; Al-Hashimi, I.; Khan, Z. Pilocarpine Tablets for the Treatment of Dry Mouth and Dry Eye Symptoms in Patients With Sjogren Syndrome. Arch. Intern. Med. 1999, 159, 174–181. [Google Scholar] [CrossRef]
- Kagawa, C.M.; Cella, J.A.; Van Arman, C.G. Action of New Steroids in Blocking Effects of Aldosterone and Deoxycorticosterone on Salt. Science 1957, 126, 1015–1016. [Google Scholar] [CrossRef]
- Struthers, A.; Krum, H.; Williams, G.H. A comparison of the aldosterone-blocking agents eplerenone and spironolactone. Clin. Cardiol. 2008, 31, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Krattenmacher, R. Drospirenone: Pharmacology and pharmacokinetics of a unique progestogen. Contraception 2000, 62, 29–38. [Google Scholar] [CrossRef]
- Xu, H.; Lv, M.; Tian, X. A Review on Hemisynthesis, Biosynthesis, Biological Activities, Mode of Action, and Structure-Activity Relationship of Podophyllotoxins: 2003–2007. Curr. Med. Chem. 2009, 16, 327–349. [Google Scholar] [CrossRef]
- Yang, J.; Bogni, A.; Schuetz, E.G.; Ratain, M.; Eileen Dolan, M.; McLeod, H.; Gong, L.; Thorn, C.; Relling, M.V.; Klein, T.E.; et al. Etoposide pathway. Pharm. Genom. 2009, 19, 552–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, P.I.; Slevin, M.L. The Clinical Pharmacology of Etoposide and Teniposide. Clin. Pharm. 1987, 12, 223–252. [Google Scholar] [CrossRef] [PubMed]
- Tantry, U.S.; Liu, F.; Chen, G.; Gurbel, P.A. Vorapaxar in the secondary prevention of atherothrombosis. Expert Rev. Cardiovasc. Ther. 2015, 13, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, G.; Gao, S.; Guo, R.; Zhao, Z.; Yuan, H.; Liu, S.; Wu, J.; Lu, X.; Yuan, X.; et al. Discovery of Potent Small-Molecule Inhibitors of Ubiquitin-Conjugating Enzyme UbcH5c from α-Santonin Derivatives. J. Med. Chem. 2017, 60, 6828–6852. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, W. Tetrahydronaphtho[1,2-b]furan-2(3H)-One Derivatives and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Rheumatoid Arthritis. WO2019011285, 17 January 2019. [Google Scholar]
- Chen, L.Z.; Wu, J.; Li, K.; Wu, Q.Q.; Chen, R.; Liu, X.H.; Ruan, B.F. Novel phthalide derivatives: Synthesis and anti-inflammatory activity in vitro and in vivo. Eur. J. Med. Chem. 2020, 206, 112722. [Google Scholar] [CrossRef] [PubMed]
- Ruan, B.; Li, Y. Resveratrol-Phthalide Hybrid Compound for Anti-Inflammatory Research and Its Preparation Method. CN110105316, 9 August 2019. [Google Scholar]
- Tran, Q.T.N.; Wong, W.S.F.; Chai, C.L.L. The identification of naturally occurring labdane diterpenoid calcaratarin D as a potential anti-inflammatory agent. Eur. J. Med. Chem. 2019, 174, 33–44. [Google Scholar] [CrossRef]
- Siedle, B.; García-Piñeres, A.J.; Murillo, R.; Schulte-Mönting, J.; Castro, V.; Rüngeler, P.; Klaas, C.A.; Da Costa, F.B.; Kisiel, W.; Merfort, I. Quantitative structure-activity relationship of sesquiterpene lactones as inhibitors of the transcription factor NF-κB. J. Med. Chem. 2004, 47, 6042–6054. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Sanchini, S.; Sarlah, D.; Lu, G.; Wu, T.R.; Nomura, D.K.; Cravatt, B.F.; Cubitt, B.; De La Torre, J.C.; Hessell, A.J.; et al. Design, synthesis, and biological evaluation of a biyouyanagin compound library. Proc. Natl. Acad. Sci. USA 2011, 108, 6715–6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.N.; Huang, X.Y.; Feng, Z.M.; Jiang, J.S.; Zhang, P.C. New Butyrolactone Type Lignans from Arctii Fructus and Their Anti-inflammatory Activities. J. Agric. Food Chem. 2015, 63, 7958–7966. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Mittal, A.; Bhardwaj, A.; Kaur, S.; Kumar, S. 1-Toluene-sulfonyl-3-[(3′-hydroxy-5′-substituted)-γ-butyrolactone]-indoles: Synthesis, COX-2 inhibition and anti-cancer activities. Bioorganic Med. Chem. Lett. 2008, 18, 85–89. [Google Scholar] [CrossRef]
- Brethon, A.; Chantalat, L.; Christin, O.; Clary, L.; Fournier, J.F.; Gastreich, M.; Harris, C.S.; Isabet, T.; Pascau, J.; Thoreau, E.; et al. New Caspase-1 inhibitor by scaffold hopping into bio-inspired 3D-fragment space. Bioorganic Med. Chem. Lett. 2017, 27, 5373–5377. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Itazaki, H.; Yoshida, T. Cinatrins, a novel family of phospholipase a2 inhibitors: II. Biological activities. J. Antibiot. 1992, 45, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Brisdelli, F.; Perilli, M.; Sellitri, D.; Piovano, M.; Garbarino, J.A.; Nicoletti, M.; Bozzi, A.; Amicosante, G.; Celenza, G. Cytotoxic activity and antioxidant capacity of purified lichen metabolites: An in vitro study. Phyther. Res. 2013, 27, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.K.; Roy, S.; Ueda, K. New cytotoxic cembranolides from an Okinawan soft coral, Lobophytum sp. Fitoterapia 2019, 136, 104162. [Google Scholar] [CrossRef] [PubMed]
- Salaski, E.J.; Krishnamurthy, G.; Ding, W.D.; Yu, K.; Insaf, S.S.; Eid, C.; Shim, J.; Levin, J.I.; Tabei, K.; Toral-Barza, L.; et al. Pyranonaphthoquinone lactones: A new class of AKT selective kinase inhibitors alkylate a regulatory loop cysteine. J. Med. Chem. 2009, 52, 2181–2184. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Hasuda, K.; Ito, A.; Koide, Y.; Ishii, F.; Haneda, I.; Chihara, S.; Koyama, Y. A new antibiotic, medermycin. J. Antibiot. 1976, 29, 765–768. [Google Scholar] [CrossRef] [Green Version]
- Bergy, M.E. Kalafungin, a new broade spectrum antibiotic. J. Antibiot. 1968, 21, 454–457. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Kōra, A.; Takahashi, Y.; Hayashi, T.; Awaya, J.; Masuma, R.; Ōiwa, R.; Ōmura, S. Production of deoxyfrenolicin and a new antibiotic, frenolicin B by streptomyces roseofulvus strain AM-3867. J. Antibiot. 1978, 31, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-H.; Rice, G.K.; Hall, I.H.; Amarnath, V. Antitumor agents. 86. Synthesis and cytotoxicity of .alpha.-methylene-.gamma.-lactone-bearing purines. J. Med. Chem. 1987, 30, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Huth, J.R.; Park, C.; Petros, A.M.; Kunzer, A.R.; Wendt, M.D.; Wang, X.; Lynch, C.L.; Mack, J.C.; Swift, K.M.; Judge, R.A.; et al. Discovery and Design of Novel HSP90 Inhibitors Using Multiple Fragment-based Design Strategies. Chem. Biol. Drug Des. 2007, 70, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Katayama, N.; Harada, S.; Ono, H.; Okazaki, H. Lactivicin, a naturally occurring non-β-lactam antibiotic having β-lactam-like action: Biological activities and mode of action. J. Antibiot. 1989, 42, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozaki, Y.; Katayama, N.; Ono, H.; Tsubotani, S.; Harada, S.; Okazaki, H.; Nakao, Y. Binding of a non-β-lactam antibiotic to penicillin-binding proteins. Nature 1987, 325, 179–180. [Google Scholar] [CrossRef] [PubMed]
- Gal, Z.; Koncz, A.; Szabo, I.; Deak, E.; Benko, I.; Barabas, G.; Hernandi, F.; Kovacs, P. A synthetic γ-lactone group with β-lactamase inhibitory and sporulation initiation effects. J. Chemother. 2000, 12, 274–279. [Google Scholar] [CrossRef]
- Pavlović, D.; Mutak, S.; Andreotti, D.; Biondi, S.; Cardullo, F.; Paio, A.; Piga, E.; Donati, D.; Lociuro, S. Synthesis and structure-activity relationships of α-amino-γ-lactone ketolides: A novel class of macrolide antibiotics. ACS Med. Chem. Lett. 2014, 5, 1133–1137. [Google Scholar] [CrossRef] [Green Version]
- Kochikyan, T.V.; Arutyunyan, E.V.; Samvelyan, M.A.; Arutyunyan, V.S.; Avetisyan, A.A.; Paronikyan, R.V.; Stepanyan, G.M. Synthesis and antibacterial properties of hydrazonothiazolyl derivatives of saturated 2,4,4-substituted butanolides. Pharm. Chem. J. 2009, 43, 144–147. [Google Scholar] [CrossRef]
- Mazur, M.; Gładkowski, W.; Podkowik, M.; Bania, J.; Nawrot, J.; Białońska, A.; Wawrzeńczyk, C. Lactones 43. New biologically active lactones: β-cyclocitral derivatives. Pest Manag. Sci. 2014, 70, 286–294. [Google Scholar] [CrossRef]
- Hamann, H.J.; Abutaleb, N.S.; Pal, R.; Seleem, M.N.; Ramachandran, P.V. β,γ-Diaryl α-methylene-γ-butyrolactones as potent antibacterials against methicillin-resistant Staphylococcus aureus. Bioorg. Chem. 2020, 104, 104183. [Google Scholar] [CrossRef] [PubMed]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Białońska, A.; Poradowski, D.; Drynda, A.; Urbaniak, M. Synthesis and anticancer activity of novel halolactones with β-aryl substituents from simple aromatic aldehydes. Tetrahedron 2013, 69, 10414–10423. [Google Scholar] [CrossRef]
- Włoch, A.; Stygar, D.; Bahri, F.; Bażanów, B.; Kuropka, P.; Chełmecka, E.; Pruchnik, H.; Gładkowski, W. Antiproliferative, antimicrobial and antiviral activity of β-aryl-δ-iodo-γ-lactones, their effect on cellular oxidative stress markers and biological membranes. Biomolecules 2020, 10, 1594. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.T.; Ma, Z.Q.; Li, J.H.; He, J.; Xu, H.; Zhang, X. Synthesis and antifungal activity of carabrone derivatives. Molecules 2010, 15, 6485–6492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun-Tao, F.; De-Long, W.; Yong-Ling, W.; He, Y.; Xing, Z. New antifungal scaffold derived from a natural pharmacophore: Synthesis of α-methylene-γ-butyrolactone derivatives and their antifungal activity against Colletotrichum lagenarium. Bioorganic Med. Chem. Lett. 2013, 23, 4393–4397. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, D.; Gao, Y.; Feng, J.; Zhang, X. New α-methylene-γ-butyrolactone derivatives as potential fungicidal agents: Design, synthesis and antifungal activities. Molecules 2016, 21, 130. [Google Scholar] [CrossRef] [Green Version]
- Björn Bode, H.; Irschik, H.; Wenzel, S.C.; Reichenbach, H.; Müller, R.; Höfle, G. The leupyrrins: A structurally unique family of secondary metabolites from the myxobacterium Sorangium cellulosum. J. Nat. Prod. 2003, 66, 1203–1206. [Google Scholar] [CrossRef]
- Herkommer, D.; Thiede, S.; Wosniok, P.R.; Dreisigacker, S.; Tian, M.; Debnar, T.; Irschik, H.; Menche, D. Stereochemical determination of the leupyrrins and total synthesis of leupyrrin A1. J. Am. Chem. Soc. 2015, 137, 4086–4089. [Google Scholar] [CrossRef] [PubMed]
- Wosniok, P.R.; Knopf, C.; Dreisigacker, S.; Orozco-Rodriguez, J.M.; Hinkelmann, B.; Mueller, P.P.; Brönstrup, M.; Menche, D. SAR Studies of the Leupyrrins: Design and Total Synthesis of Highly Potent Simplified Leupylogs. Chem. A Eur. J. 2020, 26, 15074–15078. [Google Scholar] [CrossRef]
- Yang, N.; Wang, Q.-H.; Wang, W.-Q.; Wang, J.; Li, F.; Tan, S.-P.; Cheng, M.-S. The design, synthesis and in vitro immunosuppressive evaluation of novel isobenzofuran derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 53–56. [Google Scholar] [CrossRef]
- Reinhardt, J.K.; Klemd, A.M.; Danton, O.; De Mieri, M.; Smieško, M.; Huber, R.; Bürgi, T.; Gründemann, C.; Hamburger, M. Sesquiterpene Lactones from Artemisia argyi: Absolute Configuration and Immunosuppressant Activity. J. Nat. Prod. 2019, 82, 1424–1433. [Google Scholar] [CrossRef] [PubMed]
- Chinthakindi, P.K.; Singh, J.; Gupta, S.; Nargotra, A.; Mahajan, P.; Kaul, A.; Ahmed, Z.; Koul, S.; Sangwan, P.L. Synthesis of α-santonin derivatives for diminutive effect on T and B-cell proliferation and their structure activity relationships. Eur. J. Med. Chem. 2017, 127, 1047–1058. [Google Scholar] [CrossRef]
- Xiang, M.; Liu, T.; Tan, W.; Ren, H.; Li, H.; Liu, J.; Cao, H.; Cheng, Q.; Liu, X.; Zhu, H.; et al. Effects of kinsenoside, a potential immunosuppressive drug for autoimmune hepatitis, on dendritic cells/CD8 + T cells communication in mice. Hepatology 2016, 64, 2135–2150. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiang, M. Kinsenoside Having Good Therapeutic Effect on Autoimmune Hepatitis (AIH), and Its Application and Preparation Method. CN106317142, 11 January 2017. [Google Scholar]
- Liu, X.; Fu, J.; Yao, X.J.; Yang, J.; Liu, L.; Xie, T.G.; Jiang, P.C.; Jiang, Z.H.; Zhu, G.Y. Phenolic Constituents Isolated from the Twigs of Cinnamomum cassia and Their Potential Neuroprotective Effects. J. Nat. Prod. 2018, 81, 1333–1342. [Google Scholar] [CrossRef]
- Wang, S.; Jin, D.Q.; Xie, C.; Wang, H.; Wang, M.; Xu, J.; Guo, Y. Isolation, characterization, and neuroprotective activities of sesquiterpenes from Petasites japonicus. Food Chem. 2013, 141, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, S.; Peng, N.; Xie, Z.; Wang, P.; Zhao, C.; Wei, L.; Yang, H.; Zhao, B.; Miao, J.; et al. Butyrolactone derivative 3-benzyl-5-((2-nitrophenoxy) methyl)-dihydrofuran- 2(3H)-one protects against amyloid-β peptides-induced cytotoxicity in PC12 cells. J. Alzheimer’s Dis. 2012, 28, 345–356. [Google Scholar] [CrossRef]
- Wei, L.; Yang, H.; Xie, Z.; Yang, S.; Yang, H.; Zhao, C.; Wang, P.; Xu, S.; Miao, J.; Zhao, B.; et al. A Butyrolactone Derivative 3BDO Alleviates Memory Deficits and Reduces Amyloid-β Deposition in an AβPP/PS1 Transgenic Mouse Model. J. Alzheimer’s Dis. 2012, 30, 531–543. [Google Scholar] [CrossRef]
- Min, B.S.; Na, M.K.; Oh, S.R.; Ahn, K.S.; Jeong, G.S.; Li, G.; Lee, S.K.; Joung, H.; Lee, H.K. New furofuran and butyrolactone lignans with antioxidant activity from the stem bark of Styrax japonica. J. Nat. Prod. 2004, 67, 1980–1984. [Google Scholar] [CrossRef]
- Lohezic-Le Devehat, F.; Tomasi, S.; Elix, J.A.; Bernard, A.; Rouaud, I.; Uriac, P.; Boustie, J. Stictic acid derivatives from the lichen Usnea articulata and their antioxidant activities. J. Nat. Prod. 2007, 70, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Liu, L.; Zhu, H.; Sun, Y.; Wu, Y.; Liao, H.; Gui, Y.; Li, L.; Liu, L.; Sun, F.; et al. Butyrolactone-I, an efficient α-glucosidase inhibitor, improves type 2 diabetes with potent TNF-α–lowering properties through modulating gut microbiota in db/db mice. FASEB J. 2019, 33, 12616–12629. [Google Scholar] [CrossRef] [Green Version]
- Trécant, C.; Dlubala, A.; George, P.; Pichat, P.; Ripoche, I.; Troin, Y. Synthesis and biological evaluation of analogues of M6G. Eur. J. Med. Chem. 2011, 46, 4035–4041. [Google Scholar] [CrossRef] [PubMed]
- Prévost, C. Iodo-Silver Benzoate and Its Use in the Oxidation of Ethylene Derivatives into α-Glycols. Compt. Rend 1933, 196, 1129–1131. [Google Scholar]
- Woodward, R.B.; Brutcher, F.V., Jr. cis-Hydroxylation of a synthetic steroid intermediate with iodine, silver acetate and wet acetic acid. J. Am. Chem. Soc. 1958, 80, 209–211. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Marko, I.; Mungall, W.S.; Schroeder, G.; Sharpless, K.B. Asymmetric dihydroxylation via ligand-accelerated catalysis. J. Am. Chem. Soc. 1988, 110, 1968–1970. [Google Scholar] [CrossRef]
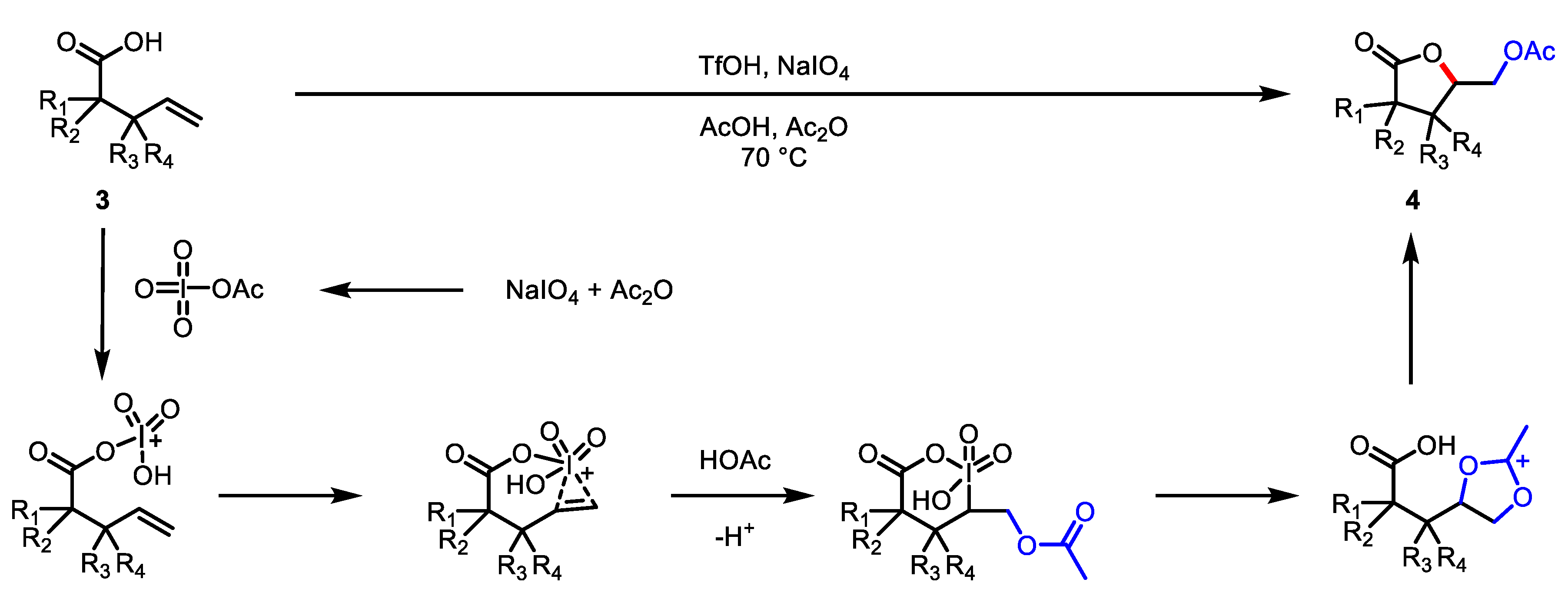
- Kang, Y.-B.; Gade, L.H. Triflic Acid Catalyzed Oxidative Lactonization and Diacetoxylation of Alkenes Using Peroxyacids as Oxidants. J. Org. Chem. 2012, 77, 1610–1615. [Google Scholar] [CrossRef]
- Rosatella, A.A.; Afonso, C.A.M. Brønsted Acid-Catalyzed Dihydroxylation of Olefins in Aqueous Medium. Adv. Synth. Catal. 2011, 353, 2920–2926. [Google Scholar] [CrossRef]
- Kang, Y.-B.; Chen, X.-M.; Yao, C.-Z.; Ning, X.-S. Direct oxidative lactonization of alkenoic acids mediated solely by NaIO4: Beyond a simple oxidant. Chem. Commun. 2016, 52, 6193–6196. [Google Scholar] [CrossRef]
- Triandafillidi, I.; Raftopoulou, M.; Savvidou, A.; Kokotos, C.G. Organocatalytic Synthesis of Lactones by the Oxidation of Alkenoic Acids. ChemCatChem 2017, 9, 4120–4124. [Google Scholar] [CrossRef]
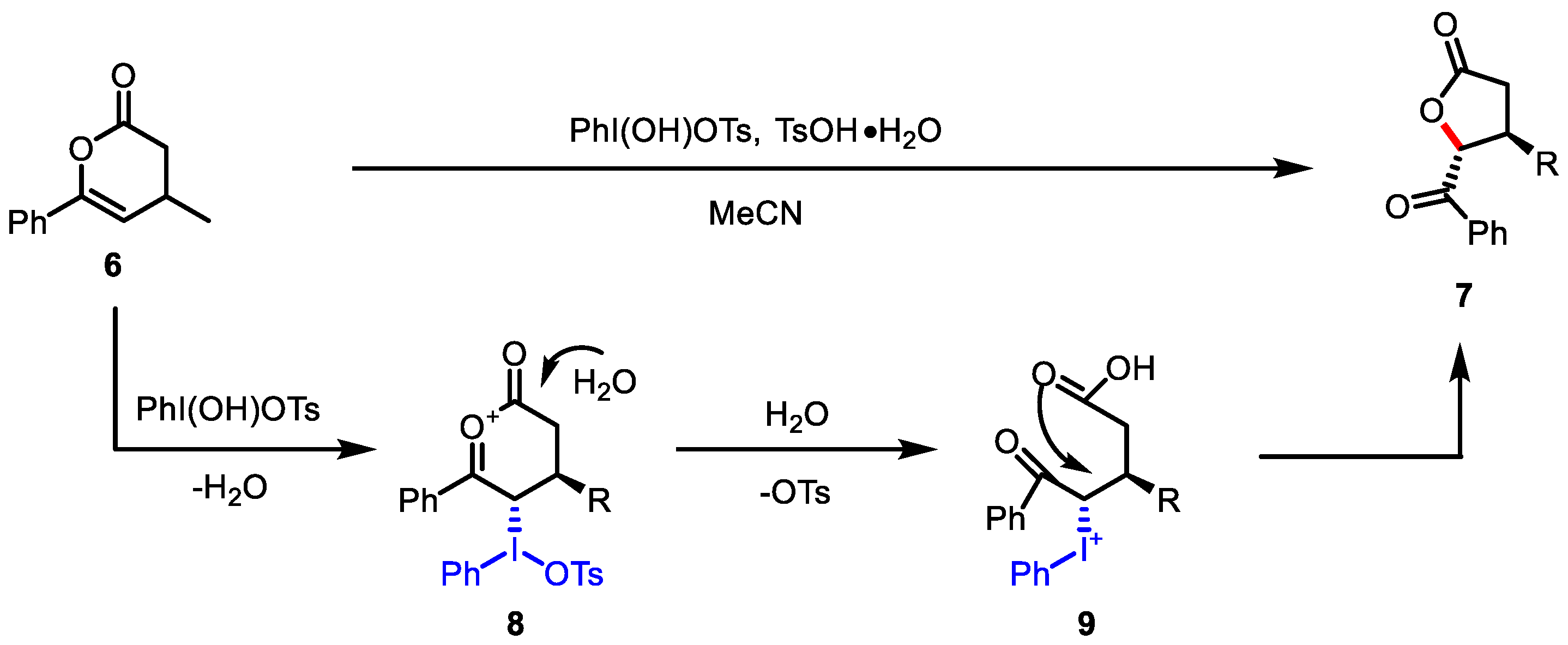
- Dagenais, R.; Lussier, T.; Legault, C.Y. Iodine(III)-Mediated Contraction of 3,4-Dihydropyranones: Access to Polysubstituted γ-Butyrolactones. Org. Lett. 2019, 21, 5290–5294. [Google Scholar] [CrossRef]
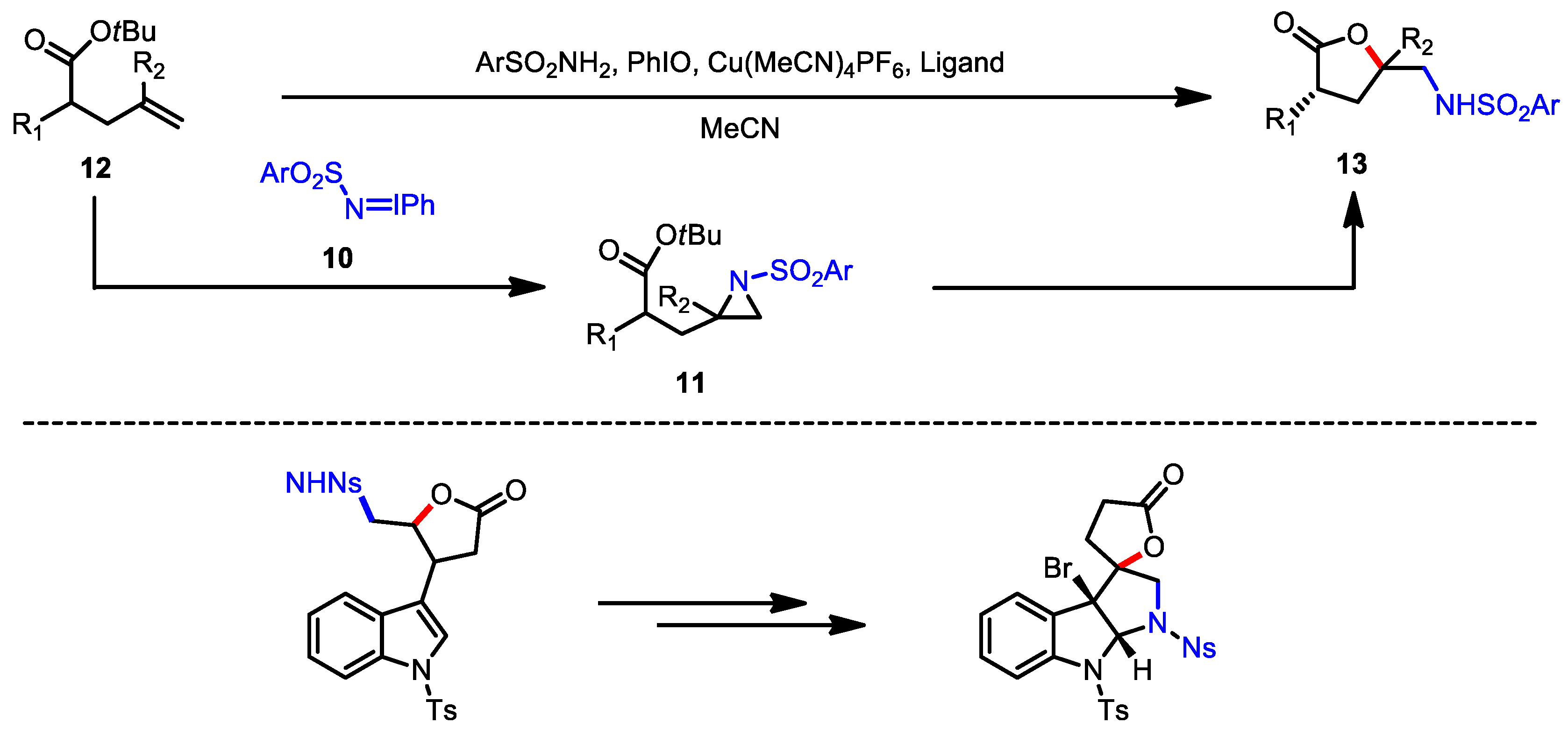
- Karila, D.; Leman, L.; Dodd, R.H. Copper-Catalyzed Iminoiodane-Mediated Aminolactonization of Olefins: Application to the Synthesis of 5,5-Disubstituted Butyrolactones. Org. Lett. 2011, 13, 5830–5833. [Google Scholar] [CrossRef]
- Evans, D.A.; Woerpel, K.A.; Hinman, M.M.; Faul, M.M. Bis(oxazolines) as chiral ligands in metal-catalyzed asymmetric reactions. Catalytic, asymmetric cyclopropanation of olefins. J. Am. Chem. Soc. 1991, 113, 726–728. [Google Scholar] [CrossRef]
- Evans, D.A.; Bilodeau, M.T.; Faul, M.M. Development of the Copper-Catalyzed Olefin Aziridination Reaction. J. Am. Chem. Soc. 1994, 116, 2742–2753. [Google Scholar] [CrossRef]
- Tamaru, Y.; Mizutani, M.; Furukawa, Y.; Kawamura, S.; Yoshida, Z.; Yanagi, K.; Minobe, M. 1,3-Asymmetric induction: Highly stereoselective synthesis of 2,4-trans-disubstituted γ-butyrolactones and γ-butyrothiolactones. J. Am. Chem. Soc. 1984, 106, 1079–1085. [Google Scholar] [CrossRef]
- Evans, D.A.; Ennis, M.D.; Mathre, D.J. Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of α-substituted carboxylic acid derivatives. J. Am. Chem. Soc. 1982, 104, 1737–1739. [Google Scholar] [CrossRef]
- Moriyama, K.; Izumisawa, Y.; Togo, H. Oxidative Intramolecular Bromo-Amination of N-Alkenyl Sulfonamides via Umpolung of Alkali Metal Bromides. J. Org. Chem. 2011, 76, 7249–7255. [Google Scholar] [CrossRef]
- Moriyama, K.; Sugiue, T.; Nishinohara, C.; Togo, H. Divergent Synthesis of α,γ-Disubstituted γ-Butyrolactones through Diastereoselective Bromolactonization with Alkali Metal Bromide: Asymmetric Total Synthesis of (+)-Dubiusamine C. J. Org. Chem. 2015, 80, 9132–9140. [Google Scholar] [CrossRef] [PubMed]
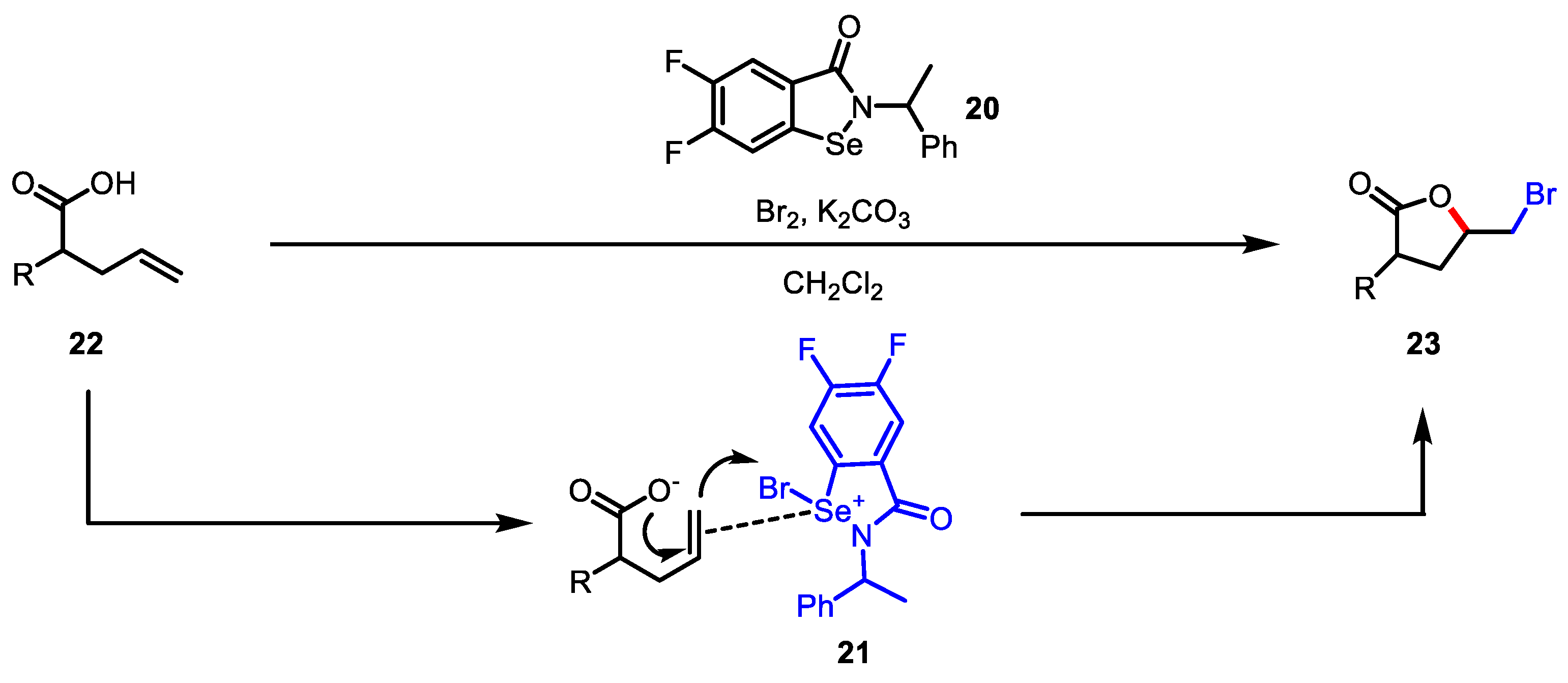
- Balkrishna, S.J.; Prasad, C.D.; Panini, P.; Detty, M.R.; Chopra, D.; Kumar, S. Isoselenazolones as Catalysts for the Activation of Bromine: Bromolactonization of Alkenoic Acids and Oxidation of Alcohols. J. Org. Chem. 2012, 77, 9541–9552. [Google Scholar] [CrossRef]
- Detty, M.R.; Friedman, A.E.; McMillan, M. A Stepwise Mechanism for Oxidative Addition of Bromine to Organoselenium(II) and Organotellurium(II) Compounds. Organometallics 1994, 13, 3338–3345. [Google Scholar] [CrossRef]
- Rosocha, G.; Batey, R.A. Synthesis of 2-bromo-1-aryl-1H-indenes via a Ag(I) promoted domino 2π-electrocyclic ring-opening/4π-electrocyclization reaction of 1,2-diaryl substituted gem-dibromocyclopropanes. Tetrahedron 2013, 69, 8758–8768. [Google Scholar] [CrossRef]
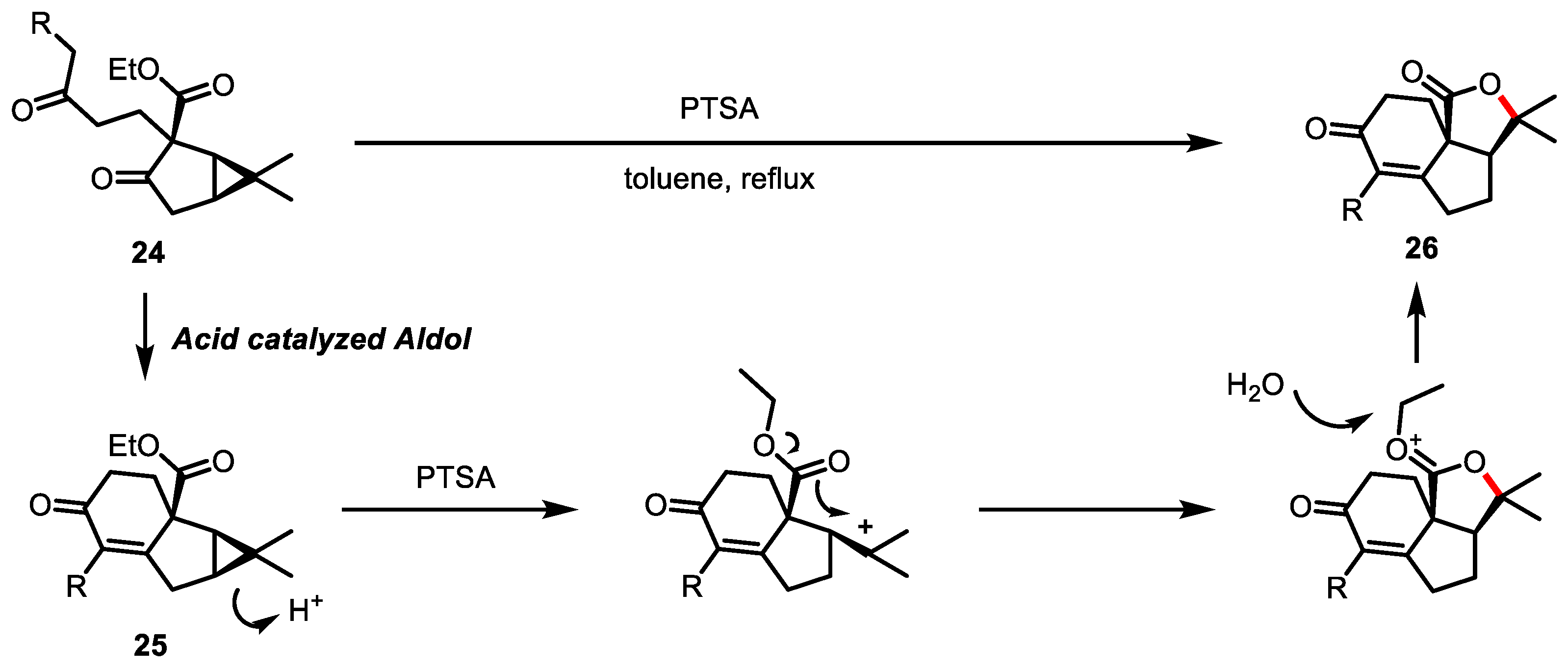
- Kalmode, H.P.; Handore, K.L.; Reddy, D.S. Access to Fused Tricyclic γ-Butyrolactones, A Natural Product-like Scaffold. J. Org. Chem. 2017, 82, 7614–7620. [Google Scholar] [CrossRef]
- Kim, S.J.; Lough, A.J.; Batey, R.A. An Approach to the 9-Oxo-10-oxabicyclo[5.3.0]dec-2-ene Core of the Guaianolide and Pseudoguaianolide Sesquiterpenes via a Domino Electrocyclic Ring-Opening/Carboxylic Acid Trapping of a gem-Dibromocyclopropane. J. Org. Chem. 2018, 83, 13799–13810. [Google Scholar] [CrossRef]
- Bandini, M.; Eichholzer, A. Enantioselective Gold-Catalyzed Allylic Alkylation of Indoles with Alcohols: An Efficient Route to Functionalized Tetrahydrocarbazoles. Angew. Chem. Int. Ed. 2009, 48, 9533–9537. [Google Scholar] [CrossRef]
- Bandini, M.; Eichholzer, A.; Gualandi, A.; Quinto, T.; Savoia, D. Creating Chemical Diversity in Indole Compounds by Merging Au and Ru Catalysis. ChemCatChem 2010, 2, 661–665. [Google Scholar] [CrossRef]
- Marion, N.; Gealageas, R.; Nolan, S.P. [(NHC)AuI]-Catalyzed Rearrangement of Allylic Acetates. Org. Lett. 2007, 9, 2653–2656. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Zhu, L.-L.; Zhang, Y.-X.; Chen, Z. Diastereoselective γ-vinyl butyrolactone synthesis via gold catalyzed cyclization of allylic acetate. Chem. Commun. 2010, 46, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Chiarucci, M.; Locritani, M.; Cera, G.; Bandini, M. Gold(I)-catalyzed synthesis of γ-vinylbutyrolactones by intramolecular oxaallylic alkylation with alcohols. Beilstein J. Org. Chem. 2011, 7, 1198–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
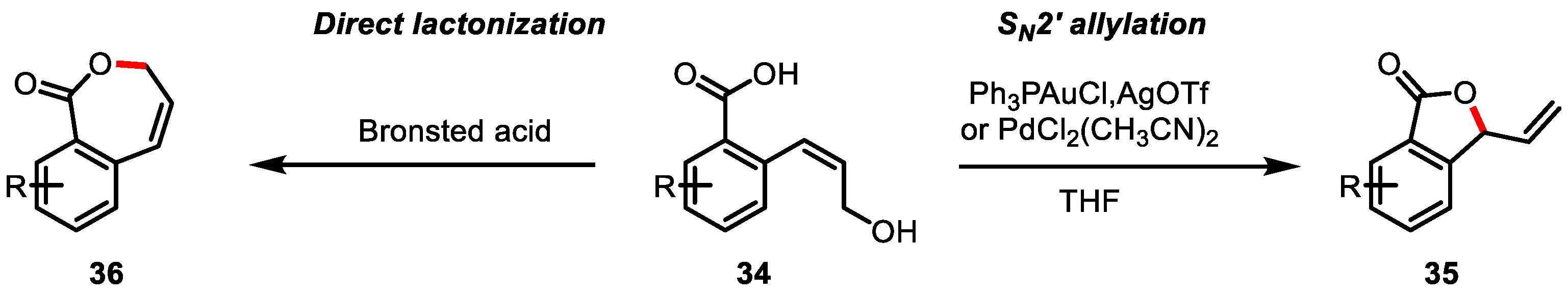
- Liu, J.; Miotto, R.J.; Segard, J.; Erb, A.M.; Aponick, A. Catalytic Dehydrative Lactonization of Allylic Alcohols. Org. Lett. 2018, 20, 3034–3038. [Google Scholar] [CrossRef]
- Okada, T.; Sakaguchi, K.; Shinada, T.; Ohfune, Y. Au-catalyzed cyclization of allenylsilanes. Regioselective conversion to 2-amino-4-silylmethylene γ-butyrolactone. Tetrahedron Lett. 2011, 52, 5740–5743. [Google Scholar] [CrossRef]
- Guo, W.; Cheng, H.-G.; Chen, L.-Y.; Xuan, J.; Feng, Z.-J.; Chen, J.-R.; Lu, L.-Q.; Xiao, W.-J. De Novo Synthesis of γ,γ-Disubstituted Butyrolactones through a Visible Light Photocatalytic Arylation–Lactonization Sequence. Adv. Synth. Catal. 2014, 356, 2787–2793. [Google Scholar] [CrossRef]
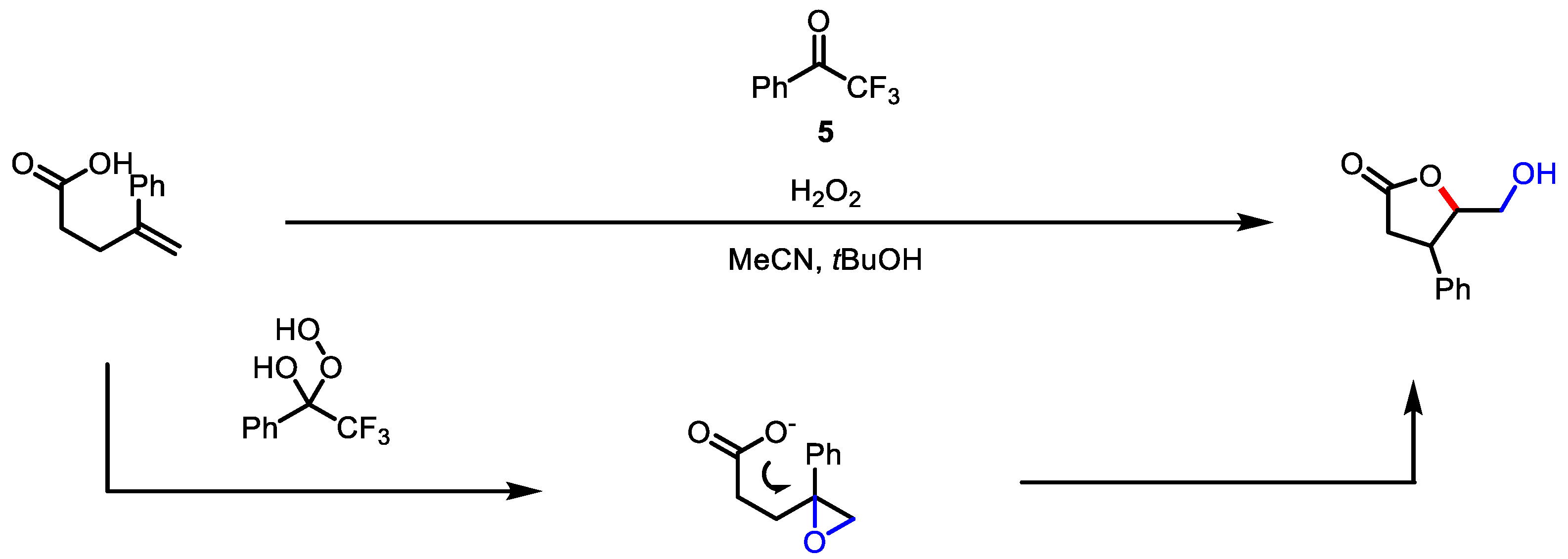
- Yasu, Y.; Arai, Y.; Tomita, R.; Koike, T.; Akita, M. Highly Regio- and Diastereoselective Synthesis of CF3-Substituted Lactones via Photoredox-Catalyzed Carbolactonization of Alkenoic Acids. Org. Lett. 2014, 16, 780–783. [Google Scholar] [CrossRef]
- Sha, W.; Ni, S.; Han, J.; Pan, Y. Access to Alkyl-Substituted Lactone via Photoredox-Catalyzed Alkylation/Lactonization of Unsaturated Carboxylic Acids. Org. Lett. 2017, 19, 5900–5903. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.-J.; Yang, D.-T.; Wang, L.; Song, T.; Wu, L.-Z.; Liu, Q. A Novel Intermolecular Synthesis of γ-Lactones via Visible-Light Photoredox Catalysis. Org. Lett. 2013, 15, 6054–6057. [Google Scholar] [CrossRef]
- Montgomery, T.P.; Hassan, A.; Park, B.Y.; Krische, M.J. Enantioselective conversion of primary alcohols to α-exo-methylene γ-butyrolactones via iridium-catalyzed C-C bond-forming transfer hydrogenation: 2-(Alkoxycarbonyl)allylation. J. Am. Chem. Soc. 2012, 134, 11100–11103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spielmann, K.; Niel, G.; De Figueiredo, R.M.; Campagne, J.M. Catalytic nucleophilic “umpoled” π-allyl reagents. Chem. Soc. Rev. 2018, 47, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.C.; Geary, L.M.; Chen, T.Y.; Zbieg, J.R.; Krische, M.J. Direct, redox-neutral prenylation and geranylation of secondary carbinol C-H bonds: C4-regioselectivity in ruthenium-catalyzed C-C couplings of dienes to α-hydroxy esters. J. Am. Chem. Soc. 2012, 134, 15700–15703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInturff, E.L.; Mowat, J.; Waldeck, A.R.; Krische, M.J. Ruthenium-catalyzed hydrohydroxyalkylation of acrylates with diols and α-hydroxycarbonyl compounds to form spiro- and α-methylene-γ- butyrolactones. J. Am. Chem. Soc. 2013, 135, 17230–17235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Yang, Q.; Zhou, T.; Tian, Q.; Zhang, G. Enantioselective Synthesis of α-exo-Methylene γ-Butyrolactones via Chromium Catalysis. Org. Lett. 2015, 17, 5236–5239. [Google Scholar] [CrossRef]
- McManus, H.A.; Guiry, P.J. Coupling of Bulky, Electron-Deficient Partners in Aryl Amination in the Preparation of Tridentate Bis(oxazoline) Ligands for Asymmetric Catalysis. J. Org. Chem. 2002, 67, 8566–8573. [Google Scholar] [CrossRef]
- Murata, Y.; Takahashi, M.; Yagishita, F.; Sakamoto, M.; Sengoku, T.; Yoda, H. Construction of Spiro-Fused 2-Oxindole/α-Methylene-γ-Butyrolactone Systems with Extremely High Enantioselectivity via Indium-Catalyzed Amide Allylation of N-Methyl Isatin. Org. Lett. 2013, 15, 6182–6185. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.M.R.; Rabe, J. Synthesis and Biological Activity of α-Methylene-γ-butyrolactones. Angew. Chem. Int. Ed. 1985, 24, 94–110. [Google Scholar] [CrossRef]
- Takahashi, M.; Murata, Y.; Yagishita, F.; Sakamoto, M.; Sengoku, T.; Yoda, H. Catalytic enantioselective amide allylation of isatins and its application in the synthesis of 2-oxindole derivatives spiro-fused to the α-methylene-γ-butyrolactone functionality. Chem. Eur. J. 2014, 20, 11091–11100. [Google Scholar] [CrossRef]
- Nair, V.; Vellalath, S.; Babu, B.P. Recent advances in carbon–carbon bond-forming reactions involving homoenolates generated by NHC catalysis. Chem. Soc. Rev. 2008, 37, 2691. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.H.; Shen, L.T.; Ye, S. Highly diastereo- and enantioselective NHC-catalyzed [3+2] annulation of enals and isatins. Chem. Commun. 2011, 47, 10136–10138. [Google Scholar] [CrossRef]
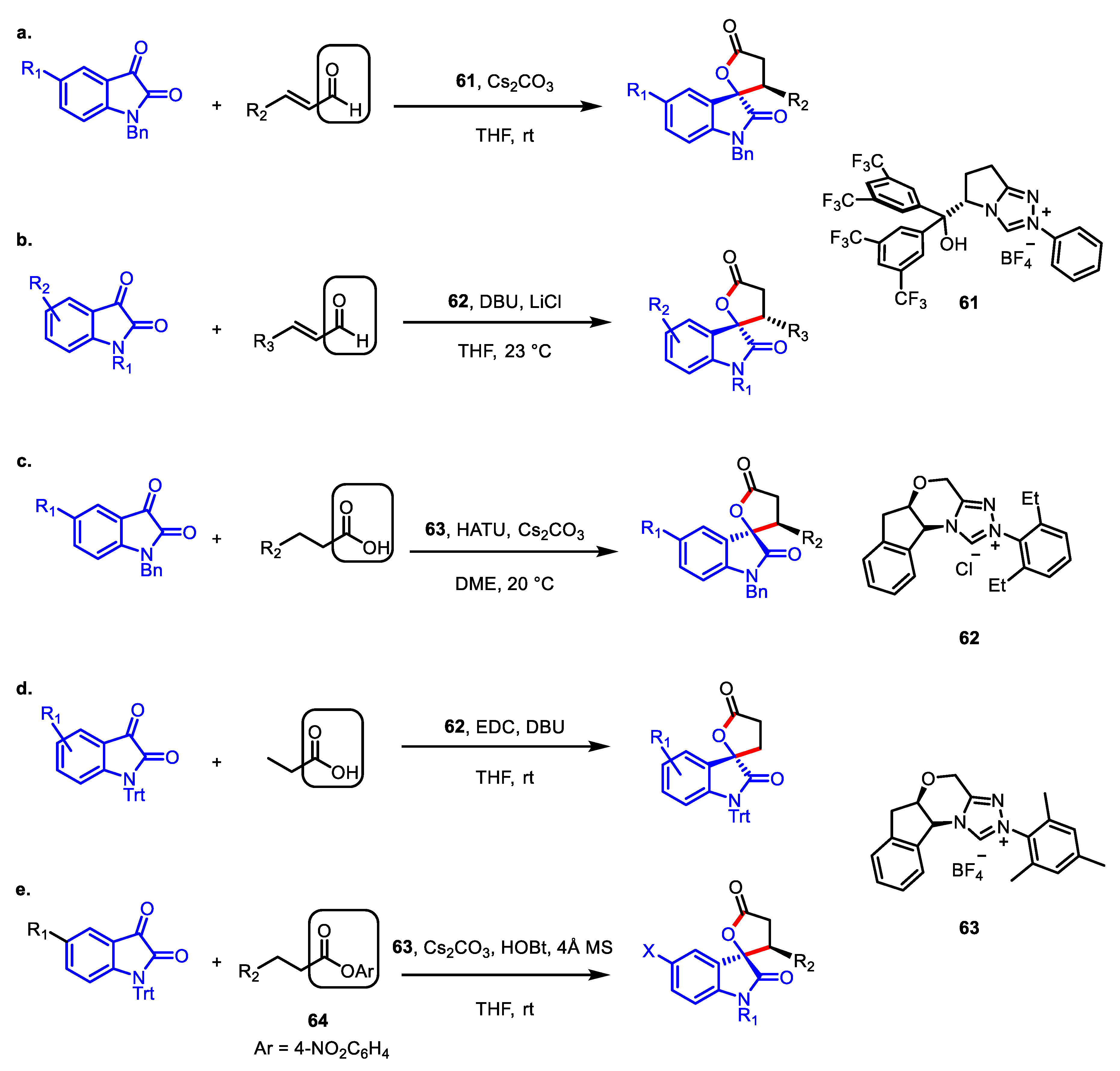
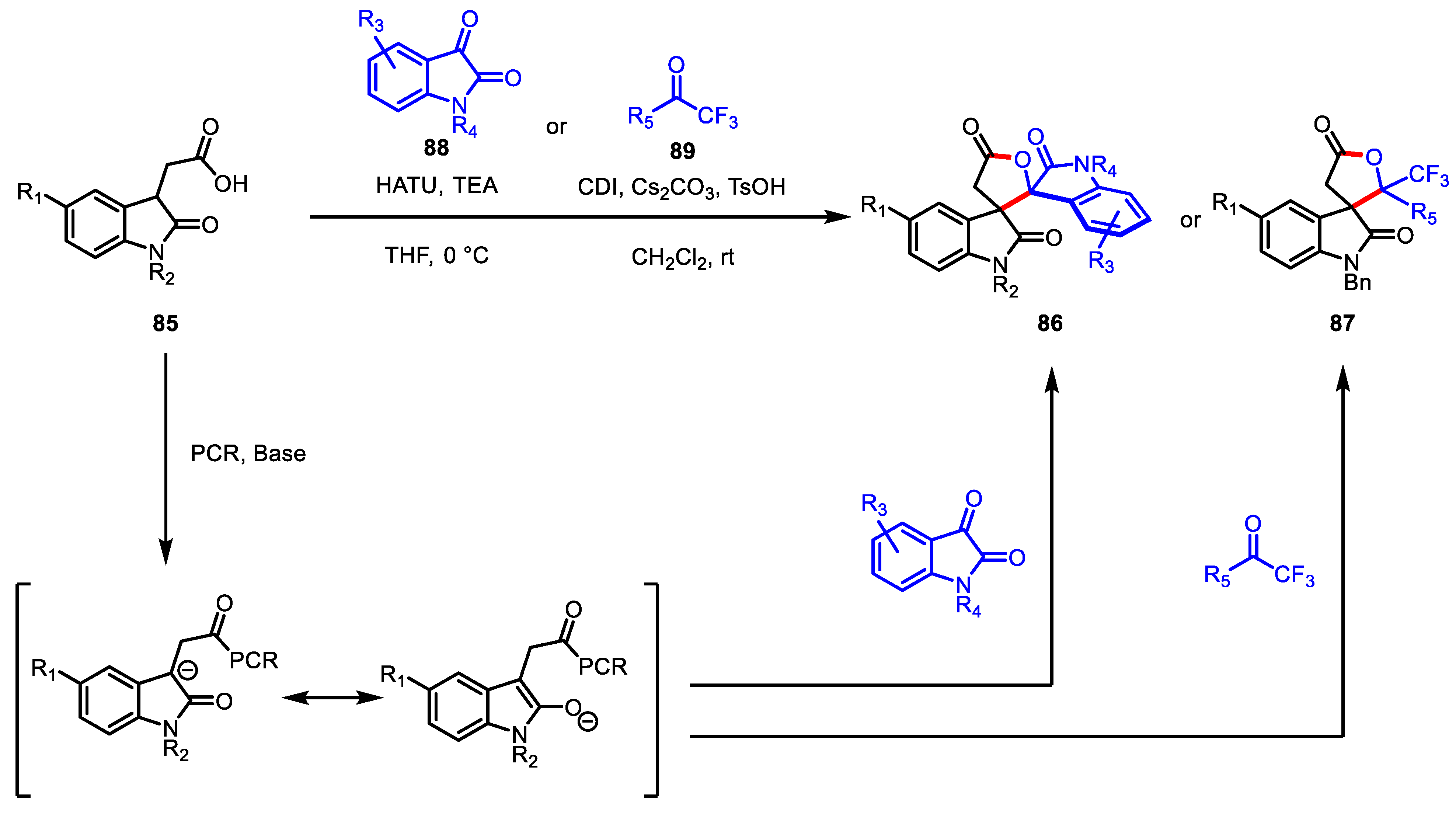
- Dugal-Tessier, J.; O’Bryan, E.A.; Schroeder, T.B.H.; Cohen, D.T.; Scheidt, K.A. An N-heterocyclic carbene/lewis acid strategy for the stereoselective synthesis of spirooxindole lactones. Angew. Chem. Int. Ed. 2012, 51, 4963–4967. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Jiang, K.; Fu, Z.; Torres, J.; Zheng, P.; Yang, S.; Song, B.-A.; Chi, Y.R. Nucleophilic β-Carbon Activation of Propionic Acid as a 3-Carbon Synthon by Carbene Organocatalysis. Chem. Eur. J. 2015, 21, 9360–9363. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Yu, C.; Li, T.; Tu, S.; Yao, C. An NHC-catalyzed in situ activation strategy to β-functionalize saturated carboxylic acid: An enantioselective formal [3+2] annulation for spirocyclic oxindolo-γ-butyrolactones. Chem. Eur. J. 2015, 21, 5355–5359. [Google Scholar] [CrossRef]
- Xu, J.; Yuan, S.; Miao, M.; Chen, Z. 1-Hydroxybenzotriazole-Assisted, N-Heterocyclic Carbene Catalyzed β-Functionalization of Saturated Carboxylic Esters: Access to Spirooxindole Lactones. J. Org. Chem. 2016, 81, 11454–11460. [Google Scholar] [CrossRef]
- Goodman, C.G.; Walker, M.M.; Johnson, J.S. Enantioconvergent synthesis of functionalized γ-butyrolactones via (3+2)-annulation. J. Am. Chem. Soc. 2015, 137, 122–125. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.H.; Twilton, J.; MacMillan, D.W.C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, J.L.; Terrett, J.A.; MacMillant, D.W.C. O-H hydrogen bonding promotes H-atom transfer from α C-H bonds for C-alkylation of alcohols. Science 2015, 349, 1532–1536. [Google Scholar] [CrossRef] [Green Version]
- Kaplaneris, N.; Bisticha, A.; Papadopoulos, G.N.; Limnios, D.; Kokotos, C.G. Photoorganocatalytic synthesis of lactones: Via a selective C-H activation-alkylation of alcohols. Green Chem. 2017, 19, 4451–4456. [Google Scholar] [CrossRef]
- Shono, T.; Ohmizu, H.; Kawakami, S.; Sugiyama, H. Electroreductive hydrocoupling of activated olefins with ketones or aldehydes in the presence of trimethylchlorosilane. Tetrahedron Lett. 1980, 21, 5029–5032. [Google Scholar] [CrossRef]
- Kise, N.; Hamada, Y.; Sakurai, T. Electroreductive coupling of optically active α,β-unsaturated carbonyl compounds with diaryl ketones: Asymmetric synthesis of 4,5,5-trisubstituted γ-butyrolactones. Org. Lett. 2014, 16, 3348–3351. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Dong, S.; Jiang, D.; Zhu, P.; Zhang, H.; Li, R.; Li, Z.; Wang, X.; Tang, W.; Du, D. β-Functionalization of Indolin-2-one-Derived Aliphatic Acids for the Divergent Synthesis of Spirooxindole γ-Butyrolactones. J. Org. Chem. 2017, 82, 4186–4193. [Google Scholar] [CrossRef]
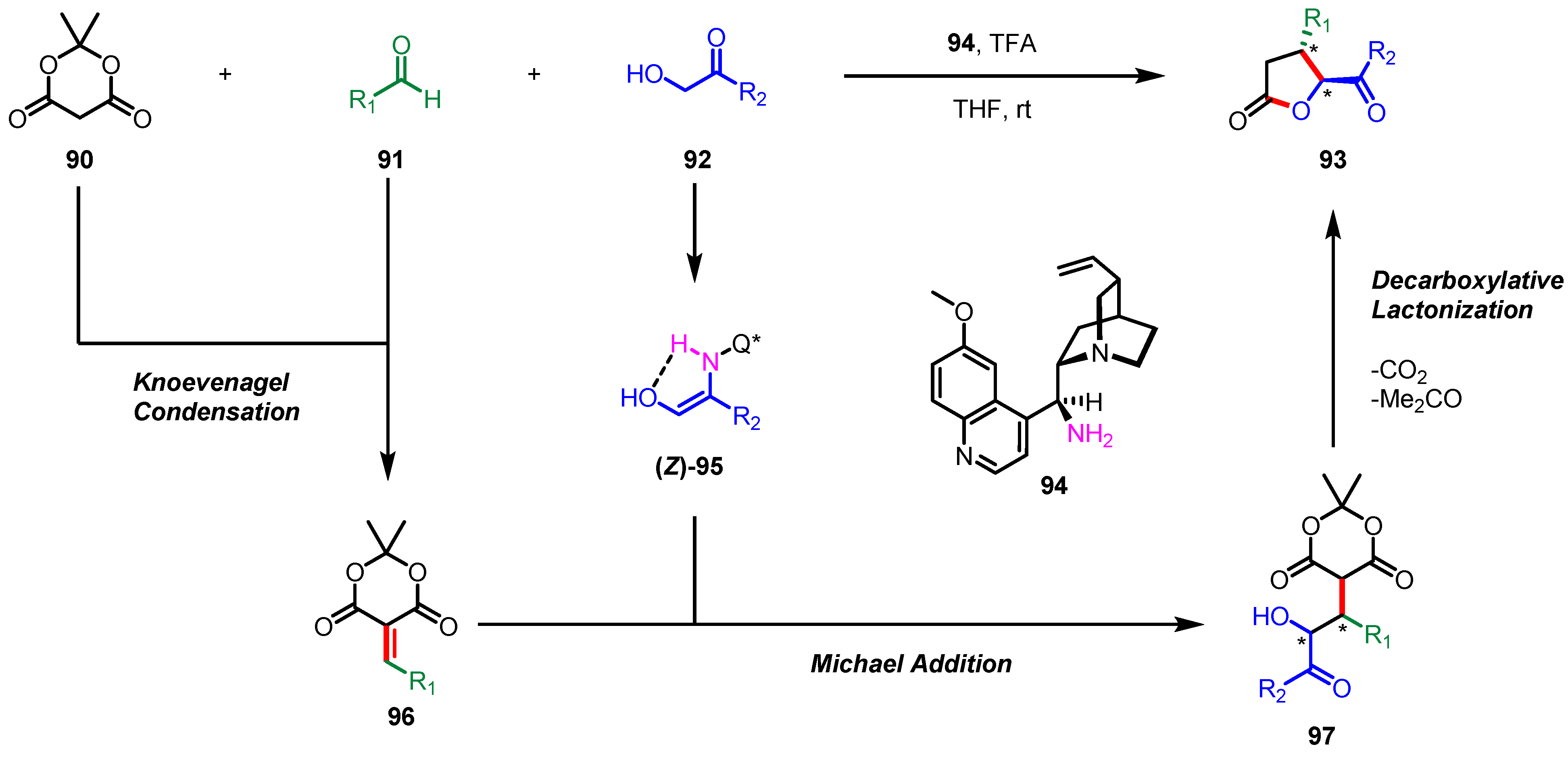
- Khopade, T.M.; Sonawane, A.D.; Arora, J.S.; Bhat, R.G. Direct Organocatalytic Multicomponent Synthesis of Enantiopure γ-Butyrolactones via Tandem Knoevenagel-Michael-Lactonization Sequence. Adv. Synth. Catal. 2017, 359, 3905–3910. [Google Scholar] [CrossRef]
- Hamid, M.H.S.A.; Slatford, P.A.; Williams, J.M.J. Borrowing Hydrogen in the Activation of Alcohols. Adv. Synth. Catal. 2007, 349, 1555–1575. [Google Scholar] [CrossRef]
- Kim, S.W.; Zhang, W.; Krische, M.J. Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res. 2017, 50, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Peña-López, M.; Neumann, H.; Beller, M. Ruthenium pincer-catalyzed synthesis of substituted γ-butyrolactones using hydrogen autotransfer methodology. Chem. Commun. 2015, 51, 13082–13085. [Google Scholar] [CrossRef]
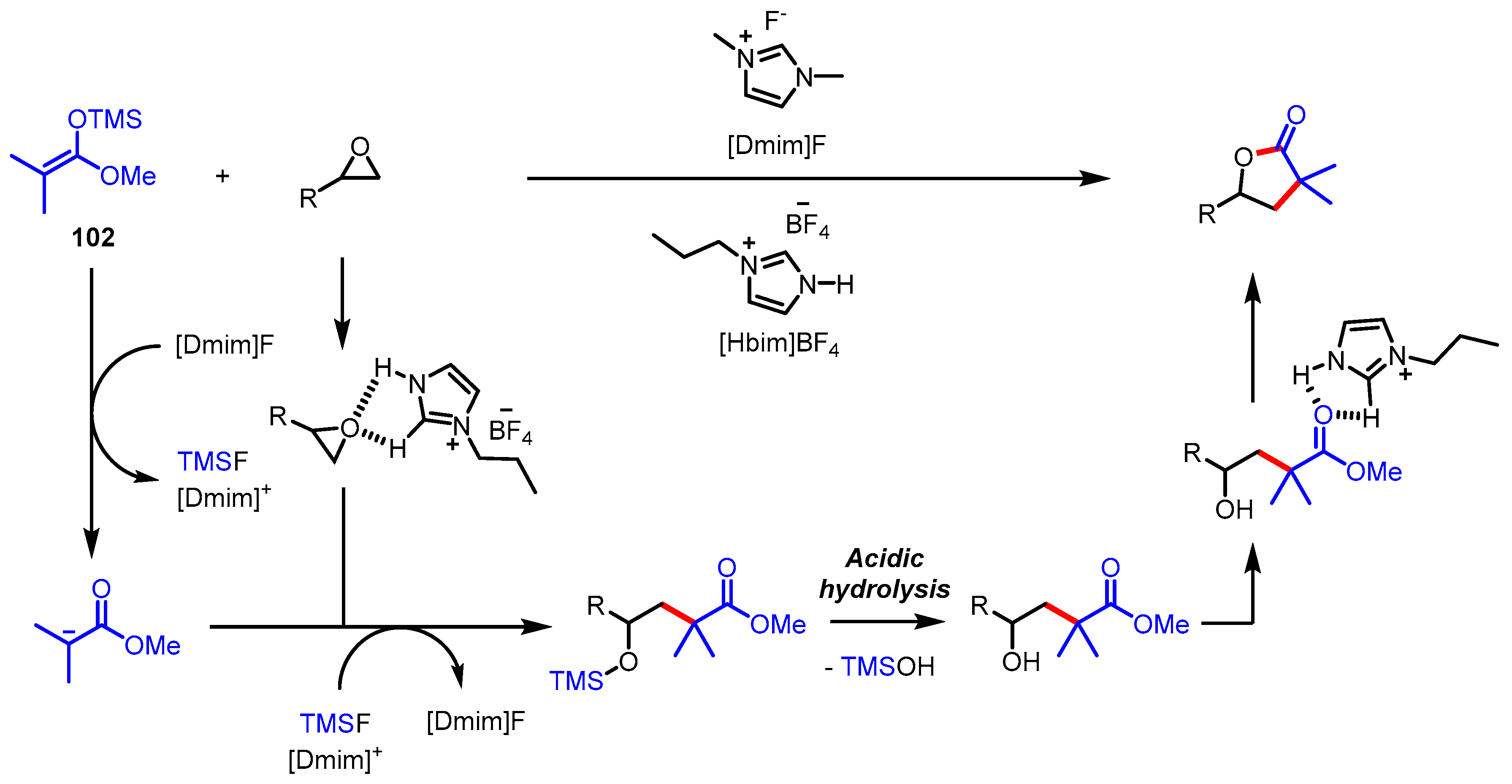
- Keshavarz, M.; Ahmady, A.Z.; Mostoufi, A.; Mohtasham, N. One-pot green regioselesctive synthesis of γ-lactones from epoxides and ketene silyl acetals using 1,3-Dimethylimidazolium fluoride as a recoverable Metal-free catalyst. Molecules 2017, 22, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
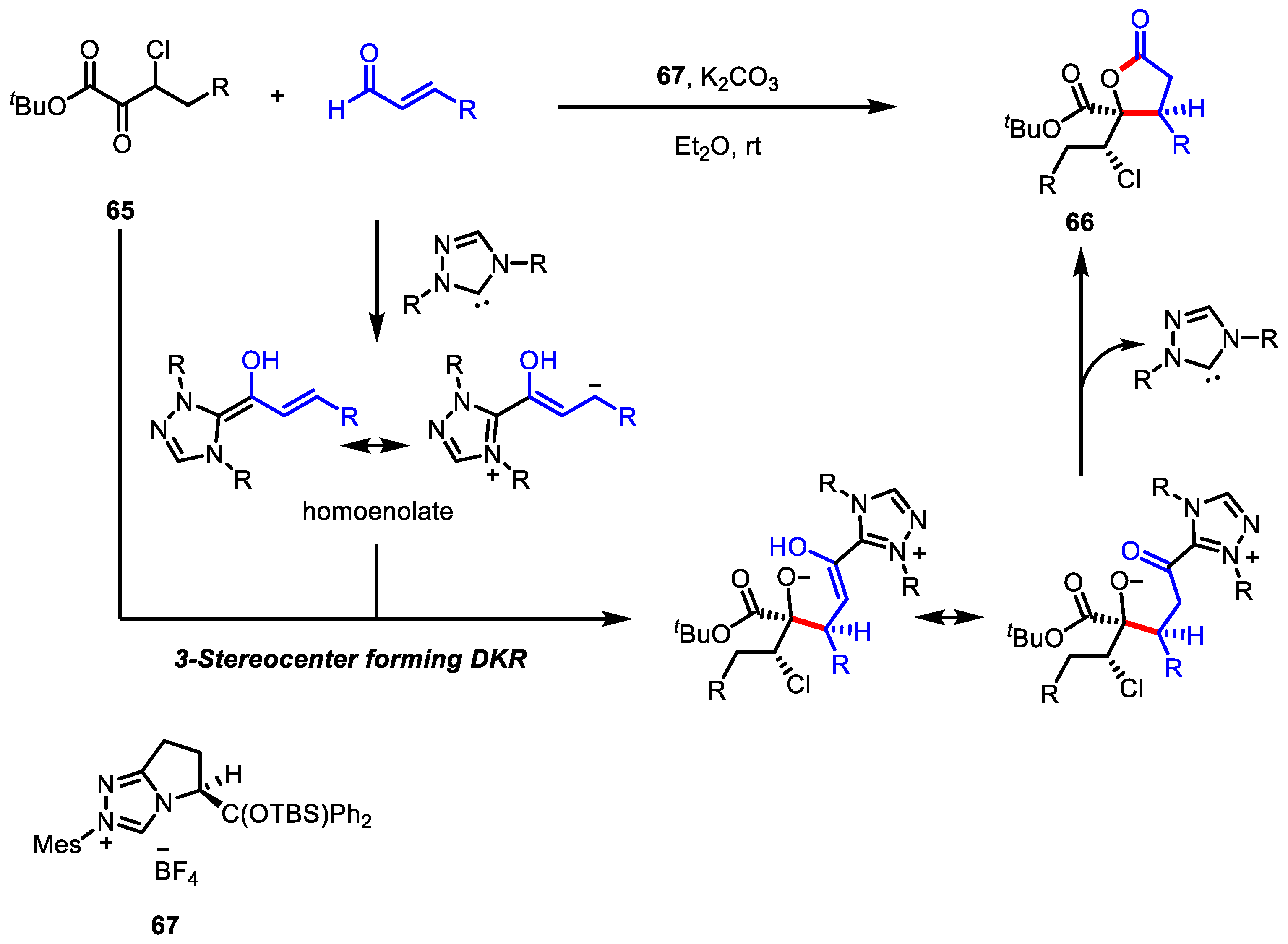
- Zeller, M.A.; Riener, M.; Nicewicz, D.A. Butyrolactone synthesis via polar radical crossover cycloaddition reactions: Diastereoselective syntheses of methylenolactocin and protolichesterinic acid. Org. Lett. 2014, 16, 4810–4813. [Google Scholar] [CrossRef] [PubMed]
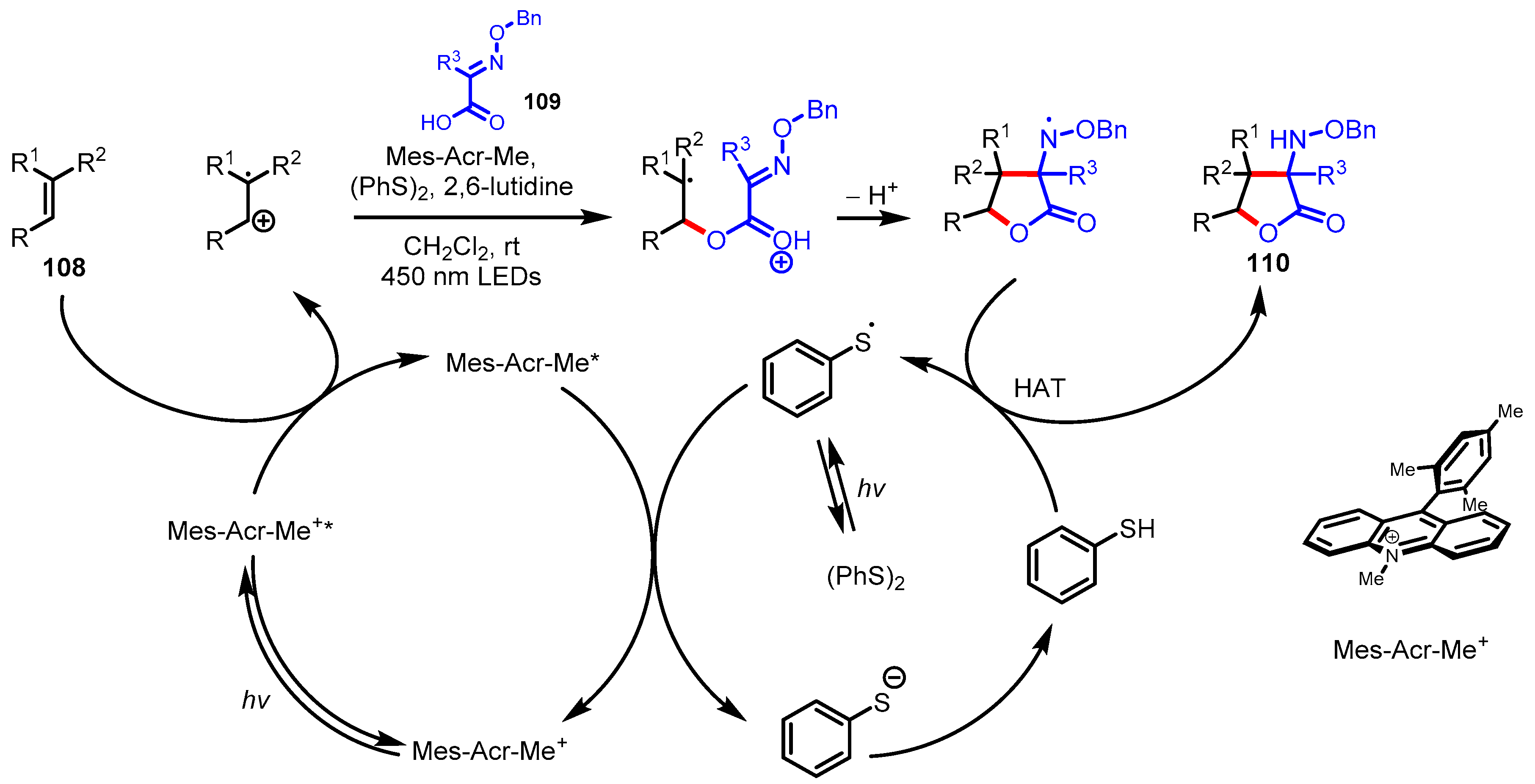
- Cavanaugh, C.L.; Nicewicz, D.A. Synthesis of α-Benzyloxyamino-γ-butyrolactones via a Polar Radical Crossover Cycloaddition Reaction. Org. Lett. 2015, 17, 6082–6085. [Google Scholar] [CrossRef]
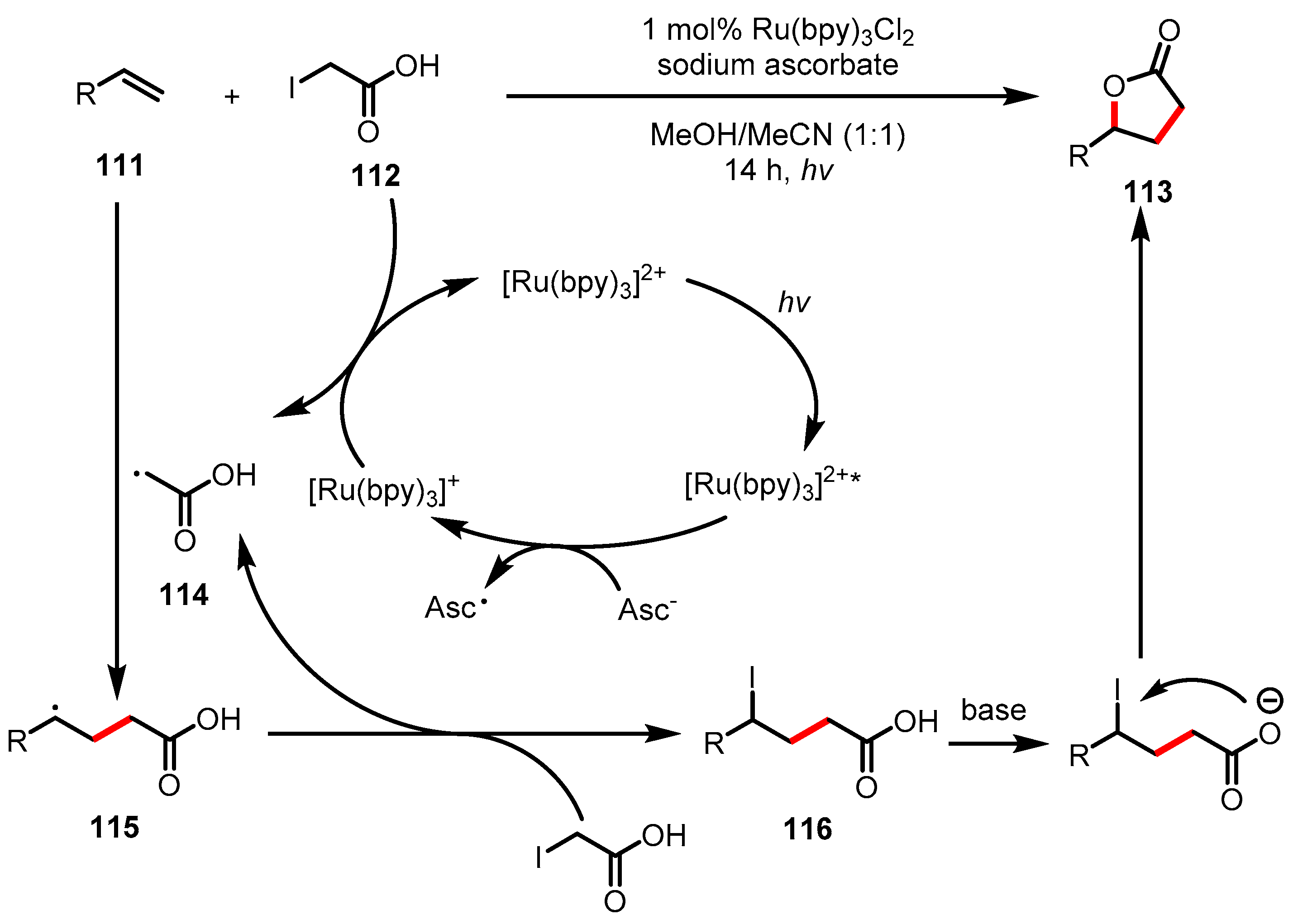
- Triandafillidi, I.; Kokotou, M.G.; Kokotos, C.G. Photocatalytic Synthesis of γ-Lactones from Alkenes: High-Resolution Mass Spectrometry as a Tool to Study Photoredox Reactions. Org. Lett. 2018, 20, 36–39. [Google Scholar] [CrossRef] [PubMed]
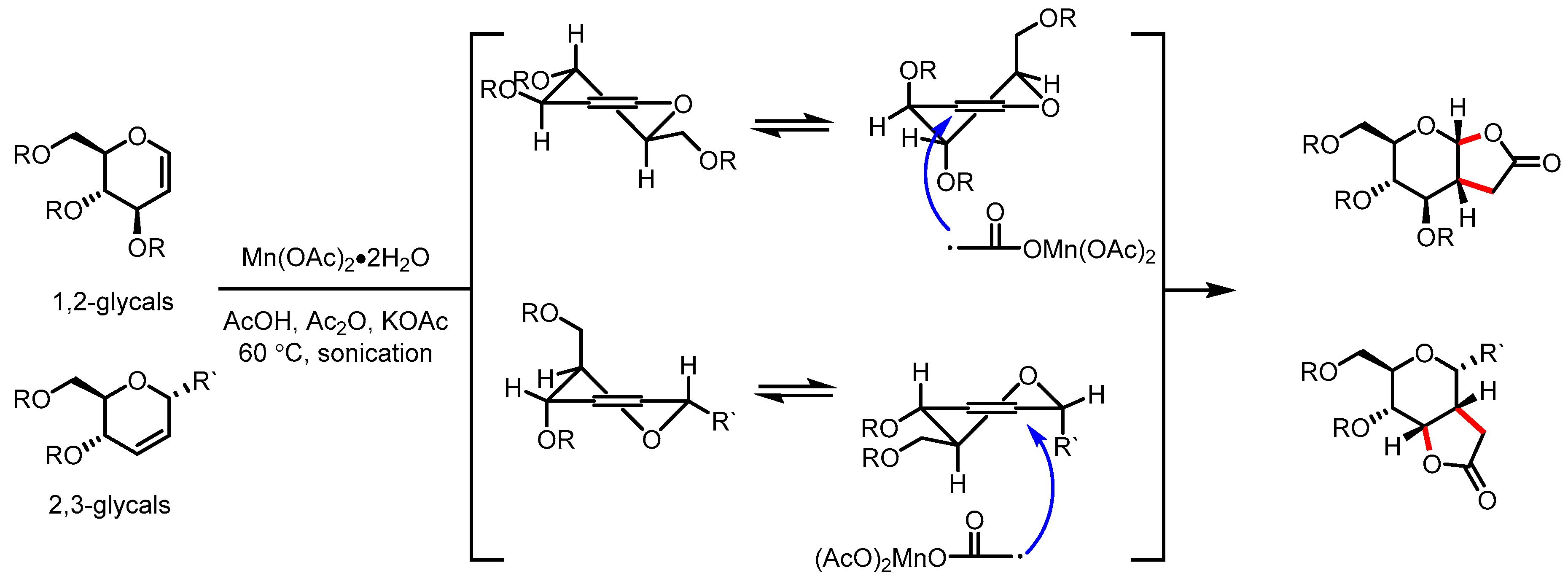
- Yousuf, S.K.; Mukherjee, D.; L, M.; Taneja, S.C. Highly regio-and stereoselective one-pot synthesis of carbohydrate-based butyrolactones. Org. Lett. 2011, 13, 576–579. [Google Scholar] [CrossRef]
- Ye, Z.; Cai, X.; Li, J.; Dai, M. Catalytic Cyclopropanol Ring Opening for Divergent Syntheses of γ-Butyrolactones and δ-Ketoesters Containing All-Carbon Quaternary Centers. ACS Catal. 2018, 8, 5907–5914. [Google Scholar] [CrossRef]
- Grandjean, J.M.M.; Nicewicz, D.A. Synthesis of highly substituted tetrahydrofurans by catalytic polar-radical-crossover cycloadditions of alkenes and alkenols. Angew. Chem. Int. Ed. 2013, 52, 3967–3971. [Google Scholar] [CrossRef]
- Gesmundo, N.J.; Grandjean, J.M.M.; Nicewicz, D.A. Amide and amine nucleophiles in polar radical crossover cycloadditions: Synthesis of γ-lactams and pyrrolidines. Org. Lett. 2015, 17, 1316–1319. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Fang, X.; Wu, L.; Jackstell, R.; Neumann, H.; Beller, M. Transition-metal-catalyzed carbonylation reactions of olefins and alkynes: A personal account. Acc. Chem. Res. 2014, 47, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Babjak, M.; Markovič, M.; Kandríková, B.; Gracza, T. Homogeneous cyclocarbonylation of alkenols with iron pentacarbonyl. Synth. 2014, 46, 809–816. [Google Scholar] [CrossRef]
- Lopatka, P.; Markovič, M.; Koóš, P.; Ley, S.V.; Gracza, T. Continuous Pd-Catalyzed Carbonylative Cyclization Using Iron Pentacarbonyl as a CO Source. J. Org. Chem. 2019, 84, 14394–14406. [Google Scholar] [CrossRef]
- Li, J.; Yang, S.; Wu, W.; Jiang, H. Novel palladium-catalyzed cascade carboxylative annulation to construct functionalized γ-lactones in ionic liquids. Chem. Commun. 2014, 50, 1381–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.-R.; Jiang, H.-F.; Li, Y.-Q.; Chen, H.-J.; Luo, W.; Xu, Y.-B. Protonolysis of the carbon–palladium bond in palladium(II)-catalyzed enyne cyclization in imidazolium-type ionic liquids. Tetrahedron 2008, 64, 2930–2937. [Google Scholar] [CrossRef]
- Higashimae, S.; Tamai, T.; Nomoto, A.; Ogawa, A. Selective Thiolative Lactonization of Internal Alkynes Bearing a Hydroxyl Group with Carbon Monoxide and Organic Disulfides Catalyzed by Transition-Metal Complexes. J. Org. Chem. 2015, 80, 7126–7133. [Google Scholar] [CrossRef] [PubMed]
- Rosa, D.; Nikolaev, A.; Nithiy, N.; Orellana, A. Palladium-Catalyzed Cross-Coupling Reactions of Cyclopropanols. Synlett 2015, 26, 441–448. [Google Scholar]
- Davis, D.C.; Walker, K.L.; Hu, C.; Zare, R.N.; Waymouth, R.M.; Dai, M. Catalytic Carbonylative Spirolactonization of Hydroxycyclopropanols. J. Am. Chem. Soc. 2016, 138, 10693–10699. [Google Scholar] [CrossRef] [Green Version]
- Breit, B. Synthetic aspects of stereoselective hydroformylation. Acc. Chem. Res. 2003, 36, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Ueki, Y.; Ito, H.; Usui, I.; Breit, B. Formation of quaternary carbon centers by highly regioselective hydroformylation with catalytic amounts of a reversibly bound directing group. Chem. Eur. J. 2011, 17, 8555–8558. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, H.; Sun, Y.; Wang, X. Principles and Applications of Enantioselective Hydroformylation of Terminal Disubstituted Alkenes. ACS Catal. 2015, 5, 6828–6837. [Google Scholar] [CrossRef]
- You, C.; Li, S.; Li, X.; Lv, H.; Zhang, X. Enantioselective Rh-Catalyzed Anti-Markovnikov Hydroformylation of 1,1-Disubstituted Allylic Alcohols and Amines: An Efficient Route to Chiral Lactones and Lactams. ACS Catal. 2019, 9, 8529–8533. [Google Scholar] [CrossRef]
- Li, S.; Ma, S. Highly selective nickel-catalyzed methyl-carboxylation of homopropargylic alcohols for α-alkylidene-γ;-butyrolactones. Org. Lett. 2011, 13, 6046–6049. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ma, S. CO 2-activation for γ-butyrolactones and its application in the total synthesis of (±)-heteroplexisolide e. Chem. Asian J. 2012, 7, 2411–2418. [Google Scholar] [CrossRef]
- Lloyd, M.G.; Taylor, R.J.K.; Unsworth, W.P. A One-Pot C–H Insertion/Olefination Sequence for the Formation of α-Alkylidene-γ-butyrolactones. Org. Lett. 2014, 16, 2772–2775. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.G.; D’Acunto, M.; Taylor, R.J.K.; Unsworth, W.P. α-Alkylidene-γ-butyrolactone synthesis via one-pot C–H insertion/olefination: Substrate scope and the total synthesis of (±)-cedarmycins A and B. Tetrahedron 2015, 71, 7107–7123. [Google Scholar] [CrossRef]
- Lloyd, M.G.; D’Acunto, M.; Taylor, R.J.K.; Unsworth, W.P. A selective C–H insertion/olefination protocol for the synthesis of α-methylene-γ-butyrolactone natural products. Org. Biomol. Chem. 2016, 14, 1641–1645. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, W.; Schossig, J.; Rossbacher, R.; Pinkos, R.; Höke, H. Butyrolactone. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Weinheim, Germany, 2019; pp. 1–7. ISBN 9783527306732. [Google Scholar]
- Yim, H.; Haselbeck, R.; Niu, W.; Pujol-Baxley, C.; Burgard, A.; Boldt, J.; Khandurina, J.; Trawick, J.D.; Osterhout, R.E.; Stephen, R.; et al. Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat. Chem. Biol. 2011, 7, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.W.; Kashinathan, P.; Lee, J.M.; Lee, J.H.; Lee, U.; Hwang, J.-S.; Hwang, Y.K.; Chang, J.-S. Production of γ-butyrolactone from biomass-derived 1,4-butanediol over novel copper-silica nanocomposite. Green Chem. 2011, 13, 1672. [Google Scholar] [CrossRef]
- Zhang, B.; Zhu, Y.; Ding, G.; Zheng, H.; Li, Y. Modification of the supported Cu/SiO2 catalyst by alkaline earth metals in the selective conversion of 1,4-butanediol to γ-butyrolactone. Appl. Catal. A Gen. 2012, 443–444, 191–201. [Google Scholar] [CrossRef]
- Reddy, K.H.P.; Anand, N.; Venkateswarlu, V.; Rao, K.S.R.; Burri, D.R. A selective synthesis of 1-phenylethanol and γ-butyrolactone through coupling processes over Cu/MgO catalysts. J. Mol. Catal. A Chem. 2012, 355, 180–185. [Google Scholar] [CrossRef]
- Hu, Q.; Fan, G.; Yang, L.; Cao, X.; Zhang, P.; Wang, B.; Li, F. A gas-phase coupling process for simultaneous production of γ-butyrolactone and furfuryl alcohol without external hydrogen over bifunctional base-metal heterogeneous catalysts. Green Chem. 2016, 18, 2317–2322. [Google Scholar] [CrossRef]
- Prasad, H.; Kannapu, R.; Suh, Y.W.; Narani, A. Coupling of 1,4-Butanediol Dehydrogenation with Nitrobenzene Hydrogenation for Simultaneous Synthesis of γ-Butyrolactone and Aniline over Promoted Cu-MgO Catalysts: Effect of Promoters. Catal. Lett. 2017, 147, 90–101. [Google Scholar]
- Hari, K.; Reddy, P.; Suh, Y.; Anand, N.; David, B.; Seetha, K.; Rao, R. Coupling of ortho-chloronitrobenzene hydrogenation with 1,4-butanediol dehydrogenation over Cu-MgO catalysts: A hydrogen free process. Catal. Commun. 2017, 95, 21–25. [Google Scholar]
- Nagaiah, P.; Venkat Rao, M.; Thirupathaiah, K.; Venkateshwarlu, V.; David Raju, B.; Rama Rao, K.S. Selective vapour phase dehydrogenation of biomass-derived 1,4-butanediol to gamma butyrolactone over Cu/ZrO2 catalysts: Influence of La2O3 promotor. Res. Chem. Intermed. 2018, 44, 5817–5831. [Google Scholar] [CrossRef]
- Bhanushali, J.T.; Prasad, D.; Patil, K.N.; Reddy, K.S.; Rama Rao, K.S.; Jadhav, A.H.; Nagaraja, B.M. Simultaneous dehydrogenation of 1,4- butanediol to γ-butyrolactone and hydrogenation of benzaldehyde to benzyl alcohol mediated over competent CeO2–Al2O3 supported Cu as catalyst. Int. J. Hydrogen Energy 2020, 45, 12874–12888. [Google Scholar] [CrossRef]
- Aellig, C.; Jenny, F.; Scholz, D.; Wolf, P.; Giovinazzo, I.; Kollhoff, F.; Hermans, I. Combined 1,4-butanediol lactonization and transfer hydrogenation/hydrogenolysis of furfural-derivatives under continuous flow conditions. Catal. Sci. Technol. 2014, 4, 2326–2331. [Google Scholar] [CrossRef]
- Huang, L.; Romero, E.; Ressmann, A.K.; Rudroff, F.; Hollmann, F.; Fraaije, M.W.; Kara, S. Nicotinamide Adenine Dinucleotide-Dependent Redox-Neutral Convergent Cascade for Lactonizations with Type II Flavin-Containing Monooxygenase. Adv. Synth. Catal. 2017, 359, 2142–2148. [Google Scholar] [CrossRef] [Green Version]
- Kara, S.; Spickermann, D.; Schrittwieser, J.H.; Weckbecker, A.; Leggewie, C.; Arends, I.W.C.E.; Hollmann, F. Access to Lactone Building Blocks via Horse Liver Alcohol Dehydrogenase-Catalyzed Oxidative Lactonization. ACS Catal. 2013, 3, 2436–2439. [Google Scholar] [CrossRef]
- Li, X.; Zheng, J.; Yang, X.; Dai, W.; Fan, K. Preparation and application of highly efficient Au/SnO2 catalyst in the oxidative lactonization of 1,4-butanediol to γ-butyrolactone. Chin. J. Catal. 2013, 34, 1013–1019. [Google Scholar] [CrossRef]
- Li, X.; Cui, Y.; Yang, X.; Dai, W.; Fan, K. Highly efficient and stable Au/Mn2O3 catalyst for oxidative cyclization of 1,4-butanediol to γ-butyrolactone. Appl. Catal. A Gen. 2013, 458, 63–70. [Google Scholar] [CrossRef]
- Xie, X.; Stahl, S.S. Efficient and Selective Cu/Nitroxyl-Catalyzed Methods for Aerobic Oxidative Lactonization of Diols. J. Am. Chem. Soc. 2015, 137, 3767–3770. [Google Scholar] [CrossRef]
- Chakraborty, S.; Lagaditis, P.O.; Förster, M.; Bielinski, E.A.; Hazari, N.; Holthausen, M.C.; Jones, W.D.; Schneider, S. Well-Defined Iron Catalysts for the Acceptorless Reversible Dehydrogenation-Hydrogenation of Alcohols and Ketones. ACS Catal. 2014, 4, 3994–4003. [Google Scholar] [CrossRef]
- Tang, Y.; Meador, R.I.L.; Malinchak, C.T.; Harrison, E.E.; McCaskey, K.A.; Hempel, M.C.; Funk, T.W. (Cyclopentadienone)iron-Catalyzed Transfer Dehydrogenation of Symmetrical and Unsymmetrical Diols to Lactones. J. Org. Chem. 2020, 85, 1823–1834. [Google Scholar] [CrossRef]
- Coleman, M.G.; Brown, A.N.; Bolton, B.A.; Guan, H. Iron-Catalyzed Oppenauer-Type Oxidation of Alcohols. Adv. Synth. Catal. 2010, 352, 967–970. [Google Scholar] [CrossRef]
- Peña-López, M.; Neumann, H.; Beller, M. Iron(II) Pincer-Catalyzed Synthesis of Lactones and Lactams through a Versatile Dehydrogenative Domino Sequence. ChemCatChem 2015, 7, 865–871. [Google Scholar] [CrossRef]










































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Name | Structure | Target Protein | Disease | Source | Reference |
|---|---|---|---|---|---|---|
| 1 | Pilocarpine |  | Muscarinic receptor | Xerostomia | Natural | [15] |
| 2 | Spironolactone |  | Mineralocorticoid receptor | Heart failure, Hypertension | Synthetic | [16] |
| 3 | Eplerenone |  | Mineralocorticoid receptor | Heart failure, Hypertension | Synthetic | [17] |
| 4 | Drospirenone |  | Progesterone receptor | Oral contraceptive | Synthetic | [18] |
| 5 | Podofilox |  | DNA topoisomerase II | Genital warts | Natural | [19] |
| 6 | Etoposide |  | DNA topoisomerase II | Lung cancer, Leukaemia | Synthetic | [20] |
| 7 | Teniposide |  | DNA topoisomerase II | Lymphoblastic leukaemia | Synthetic | [21] |
| 8 | Vorapaxar |  | Protease-activated receptor | Thrombotic cardiovascular events | Synthetic | [22] |
| Entry | Pharmacological Activity | Structure | Name | Bioassay | Source | Reference |
|---|---|---|---|---|---|---|
| 1 | Anti-inflammation |  | (3aS,9bR)-8-((2-Bromobenzyl)oxy)-6,9-dimethyl-3-methylene-3,3a,4,5-tetrahydronaphtho[1,2-b]furan-2(9bH)-one | UbeH5c binding assay (Kd = 0.283 μM) Therapeutic effect on adjuvant arthritis rat model | Synthetic | [23,24] |
| 2 |  | 3-((4-((4-Fluorobenzyl)oxy)phenyl)(hydroxy)methyl)-5,7-dimethoxyisobenzofuran-1 (3H)-one | Inhibition rate of NO production at 10 µM (95.23 ± 3.21%) Therapeutic effect on adjuvant arthritis rat model | Synthetic | [25,26] | |
| 3 |  | Calcaratarin D | Suppression of NF-κB activation by reducing p65 nuclear translocation Suppression of LPS-induced activation of PI3K/Akt pathway | Natural (Alpinia calcarata) | [27] | |
| 4 |  | (3aR,4R,9aS,9bR)-6,9-Dimethyl-3-methylene-2,7-dioxo-2,3,3a,4,5,7,9a,9b-octahydroazuleno[4,5-b]furan-4-yl methacrylate | NF-κB inhibition (IC100 = 10 μM) | Natural (Viguiera gardneri) | [28] | |
| 5 | Anti-inflammation |  | (1R,3R,4’R,5R,7R)-7-((2,6-Dichloro-7H-purin-7-yl)methyl)-4’-methyl-1-phenyl-4’-vinyldihydro-2’H-spiro[bicyclo[3.2.0]heptane-3,3’-furan]-2’,4-dione (Biyouyanagin analog) | Inhibition of LPS-induced cytokine production | Synthetic | [29] |
| 6 |  | Arctiidilactone | Suppression of LPS-induced NO production | Natural (Arctium lappa L.) | [30] | |
| 7 |  | 2-((2S,4S)-4-Hydroxy-5-oxo-4-(1-tosyl-1H-indol-3-yl)tetrahydrofuran-2-yl)acetonitrile | COX2 inhibition (IC50 < 0.001 uM) | Synthetic | [31] | |
| 8 |  | CD10847 | Caspase-1 inhibition (IC50 = 17 nM) | Synthetic | [32] | |
| 9 |  | Cinatrin C3 | Phospholipase A2 inhibition (IC50 = 70 μM) | Natural (Circinotrichum falcatisporum RF-641) | [33] | |
| 10 | Anticancer |  | Protolichesterinic acid | Cytotoxicity in HeLa cells | Natural (Lichen metabolites) | [34] |
| 11 |  | (1aR,5E,8E,10aS,13aS,14S,14aR)-1a,5,9-Trimethyl-13-methylene-12-oxo-1a,2,3,4,7,10,10a,12,13,13a,14,14a-dodecahydrooxireno[2’,3’:4,5]cyclotetradeca[1,2-b]furan-14-yl acetate | Cytotoxicity in RAW 264.7 cell (IC50 = 5.99 μM) | Natural (Lobophytum sp.) | [35] | |
| 12 |  | Lactoquinoomycin (Medermycin) | AKT inhibition (IC50 = 0.149 μM) Cytotoxicity in MDA468 cells (IC50 = 0.05 μM) | Natural (Streptomyces K73) | [36,37] | |
| 13 |  | Kalafungin | AKT inhibition (IC50 = 0.313 μM) Cytotoxicity in MDA468 cells (IC50 = 0.07 μM) | Natural (Streptomyces tanashiensis) | [36,38] | |
| 14 |  | Frenolicin B | AKT inhibition (IC50 = 0.198 μM) Cytotoxicity in MDA468 cells (IC50 = 0.06 μM) | Natural (Streptomyces roseofulvus strain AM-3867) | [36,39] | |
| 15 | Anticancer |  | 5-((6-Amino-9H-purin-9-yl)methyl)-5-methyl-3-methylenedihydrofuran-2(3H)-one | Cytotoxicity in L1210 cells (ED50 = 0.3 μg/mL) | Synthetic | [40] |
| 16 |  | (E)-N-((2-Amino-6-methylpyrimidin-4-yl)methyl)-3-(((2-oxodihydrofuran-3(2H)-ylidene)methyl)amino)benzenesulfonamide | HSP90 binding (Ki = 1.9 μM) | Synthetic | [41] | |
| 17 | Antibiotic |  | Lactivicin | Inhibition of β-Lactamase in Proteus vulgaris (IC50 = 2.4 μg/mL) | Natural (Bacteria YK-258 and YK-422) | [42,43] |
| 18 |  | (3aS,5S,6aS)-5-Hydroxyhexahydro-2H-cyclopenta[b]furan-2-one | Inhibition of β-lactamase in Klebsiella oxytoca (IC50 = 15 mg/l) | Synthetic | [44] | |
| 19 | Antibiotic |  | N-((3R,3aS,4R,6R,8R,9R,10R,12R,15R,15aS)-9-(((2S,3R,4S,6R)-4-(Dimethylamino)-3-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-15-ethyl-8-methoxy-4,6,8,10,12,15a-hexamethyl-2,5,11,13-tetraoxotetradecahydro-2H-furo[2,3-c][1]oxacyclotetradecin-3-yl)-2-(quinoxalin-2-ylthio)acetamide | Antibacterial activity against erythromycin-susceptible Streptococus pyogenes (MIC = 0.06 μg/mL) | Synthetic | [45] |
| 20 |  | 2-Ethoxycarbonyl-2-[2-(3-p-chlorophenylthiazol-2- yl)hydrazono]propyl-4,4-dimethylbutanolide | Antibacterial activity against Staphylococcus aureus | Synthetic | [46] | |
| 21 |  | (3aS,7aS)-3a,7,7,7a-Tetramethylhexahydrobenzofuran-2(3H)-one | Antibacterial activity against Staphylococcus aureus | Synthetic | [47] | |
| 22 |  | (1aR,10aS,Z)-1a,5-Dimethyl-8-methylene-2,3,6,7,7a,8,10a,10b-octahydrooxireno[2’,3’:9,10]cyclodeca[1,2-b]furan-9(1aH)-one | Antibacterial activity against MRSA USA300 (MIC = 56.7 μM) | Synthetic | [48] | |
| 23 |  | (4S,5S)-5-((S)-1-Iodoethyl)-4-(4-isopropylphenyl)dihydrofuran-2(3H)-one | Antimicrobial activity against Proteus mirabilis (MIC = 0.25 mg/mL) | Synthetic | [49,50] | |
| 24 | Antifungal |  | Carabrone | Fungicidal activity against C. lagenarium (IC50 = 7.10 µg/mL) | Natural (Carpesium abrotanoides) | [51] |
| 25 |  | 4- (3-Fluorophenyl)-2-methylenebutyrolactone | Fungicidal activity against C. lagenarium (IC50 = 57.9 µM) | Synthetic | [52] | |
| 26 |  | 4-[4-(3-Bromobenzoyloxy)phenyl]-2-methylenebutyrolactone | Fungicidal activity against C. lagenarium (IC50 = 8.76 µM) | Synthetic | [53] | |
| 27 |  | Leupyrrins A1 | Fungicidal activity against M. hiemalis (MIC = 0.3 µg/mL) | Natural (Sorangium cellulosum) | [54] | |
| 28 | Immunosuppressive |  | (E)-3-(3,4-Dimethoxyphenyl)-N-(1-oxo-1,3-dihydroisobenzofuran-5-yl)acrylamide | Inhibition of T cells proliferation (IC50 = 0.029 μM) | Synthetic | [57] |
| 29 | Immunosuppressive |  | (4S,5S)-5-((1S,2S)-2-Hydroxy-2-methyl-5-oxocyclopent-3-en-1-yl)-3-methylene-4-(3-oxobutyl)dihydrofuran-2(3H)-one | Inhibition of T lymphocyte proliferation (IC50 = 1.0 μM) | Natural (Artemisia argyi) | [58] |
| 30 |  | (3S,3aS,9bR)-8-((1-(Benzo[d][1,3]dioxol-5-yl)-1H-1,2,3-triazol-5-yl)methoxy)-3,6,9-trimethyl-3a,4,5,9b-tetrahydronaphtho[1,2-b]furan-2(3H)-one (α-Santonin derivative) | Suppression of LPS-induced B-cell proliferation (50% at 10 μM) | Synthetic | [59] | |
| 31 |  | Kinsenoside | VGEFR2 binding Therapeutic effect on autoimmune hepatitis in DCs/Hepa1-6 AIH mouse model | Natural (Anoectochilus roxburghii) | [60,61] | |
| 32 | Neuroprotective |  | (3R,4R)-4-(4-Hydroxy-3-methoxyphenyl)-3-(4-methoxyphenyl)dihydrofuran-2(3H)-one | Neuroprotective activity in SH-SY5Y cells | Natural (Cinnamomum cassia) | [62] |
| 33 | Neuroprotective |  | Japonipene C | Neuroprotective activity in SH-SY5Y cells | Natural (Petasites japonicas) | [63] |
| 34 |  | 3-Benzyl-5-((2-nitrophenoxy)methyl)dihydrofuran-2(3H)-one (3BDO) | PC 12 cell viability assay Alleviation of memory deficits in AβPP/PS1 transgenic mice | Synthetic | [64,65] | |
| 35 | Antioxidant |  | Styraxlignolide E | DPPH Radical-Scavenging Activity (IC50 = 194 µM) | Natural (Styrax japonica) | [66] |
| 36 |  | Norstictic acid | Superoxide scavenging Activity (IC50 = 580 µM) | Natural (Usnea articulate) | [67] | |
| 37 | Hypoglycemic |  | Butyrolactone I | α-Glucosidase inhibition Multiple anti-type 2 diabetic activities in db/db mice | Natural (Aspergillus terreus) | [68] |
| 38 | Hypoglycemic |  | BL-3 | PTP1B Inhibitory Assay | Synthetic | [69] |
| Entry | R | R-RP | PC | Ref |
|---|---|---|---|---|
| 1 | Aryl | ArN2+BF4- | Ru(bpy)3(PF6)2 | [97] |
| 2 | CF3 | Umemoto’s reagent | Ru(bpy)3(PF6)2 | [98] |
| 3 | Alkyl | NHP ester | Ir(ppy)2(dtbbpy)PF6 | [99] |
 | |||
|---|---|---|---|
| Entry | Method | Catalyst | Ref |
| 1 | Vapor phase reaction | Cu-SiO2 nonocomposite | [154] |
| 2 | Vapor phase reaction | SiO2 supported Cu, Ca, Sr or Br promoter | [155] |
| 3 1 | Vapor phase reaction | MgO supported Cu | [156] |
| 4 2 | Vapor phase reaction | CaAlO supported Cu | [157] |
| 5 3 | Vapor phase reaction | MgO supported Cu, Co3O4 promoter | [158] |
| 6 4 | Vapor phase reaction | MgO supported Cu | [159] |
| 7 | Vapor phase reaction | ZrO2 supported Cu, La2O3 promoter | [160] |
| 8 5 | Vapor phase reaction | CeO2-Al2O3 supported Cu | [161] |
| 9 6 | Continuous flow reaction | AlOx supported Cu nanoparticle | [162] |
| 10 | Chemoenzymatic reaction | Type II FMO-E and HLADH | [163] |
| 11 | Chemoenzymatic reaction | HLADH | [164] |
| 12 | Heterogeneous solution phase reaction | SnO2 supported Au | [165] |
| 13 | Heterogeneous solution phase reaction | Mn2O3 supported Au | [166] |
| 15 | Homogeneous solution phase reaction | Cu/nitroxyl | [167] |
| 16 | Homogeneous solution phase reaction | Fe complex 143 | [168] |
| 17 | Homogeneous solution phase reaction | Fe complex 144 | [169] |
| 18 | Homogeneous solution phase reaction | Fe complex 145 | [170] |
| 19 | Homogeneous solution phase reaction | Fe complex 146 | [171] |
| Section | Bond Formation | Reaction | Page |
|---|---|---|---|
| 3.1 |  | Oxidative lactonization | 12 |
| Halolactonization | 14 | ||
| Acid-promoted cyclopropane opening | 15 | ||
| Au-catalyzed oxaallylation | 16 | ||
| Photoredox-catalyzed lactonization | 17 | ||
| 3.2 |  | Transition-metal catalyzed C-C bond coupling | 18 |
| NHC-catalyzed C-C bond coupling | 20 | ||
| Photoredox-catalyzed C-C bond coupling | 23 | ||
| Miscellsious γ-butyrolactone formation | 24 | ||
| 3.3 |  | Ruthenium pincer-catalyzed hydrogen autotransfer | 27 |
| Ionic liquid-assisted epoxide opening and lactonization | 27 | ||
| 3.4 |  | Polar radical crossover cycloaddition (PRCC) | 28 |
| Atom-transfer radical addition (ATRA) | 29 | ||
| Mn(OAc)3-mediated radical lactonization | 30 | ||
| Copper-catalyzed cyclopropanol ring-opening cross-coupling | 30 | ||
| 3.5 |  | Carbonylative lactonization | 31 |
| Hydroformylation-oxidation | 33 | ||
| Carboxylation-lactonization | 35 | ||
| 3.6 |  | C-H insertion | 36 |
| 3.7 |  | Oxidative C2-O1 bond formation | 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hur, J.; Jang, J.; Sim, J. A Review of the Pharmacological Activities and Recent Synthetic Advances of γ-Butyrolactones. Int. J. Mol. Sci. 2021, 22, 2769. https://doi.org/10.3390/ijms22052769
Hur J, Jang J, Sim J. A Review of the Pharmacological Activities and Recent Synthetic Advances of γ-Butyrolactones. International Journal of Molecular Sciences. 2021; 22(5):2769. https://doi.org/10.3390/ijms22052769
Chicago/Turabian StyleHur, Joonseong, Jaebong Jang, and Jaehoon Sim. 2021. "A Review of the Pharmacological Activities and Recent Synthetic Advances of γ-Butyrolactones" International Journal of Molecular Sciences 22, no. 5: 2769. https://doi.org/10.3390/ijms22052769
APA StyleHur, J., Jang, J., & Sim, J. (2021). A Review of the Pharmacological Activities and Recent Synthetic Advances of γ-Butyrolactones. International Journal of Molecular Sciences, 22(5), 2769. https://doi.org/10.3390/ijms22052769