
The Glioblastoma Microenvironment: Morphology, Metabolism, and Molecular Signature of Glial Dynamics to Discover Metabolic Rewiring Sequence

 ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cancer Metabolic Strategies and the Role of the Microenvironment in Glioblastoma
2.1. The Warburg Effect and the Reverse
2.2. The Functional Symbiosis
2.3. The Role of the Microenvironment
3. GBM-Associated Microglia
3.1. Morphology
3.2. Molecular Profile
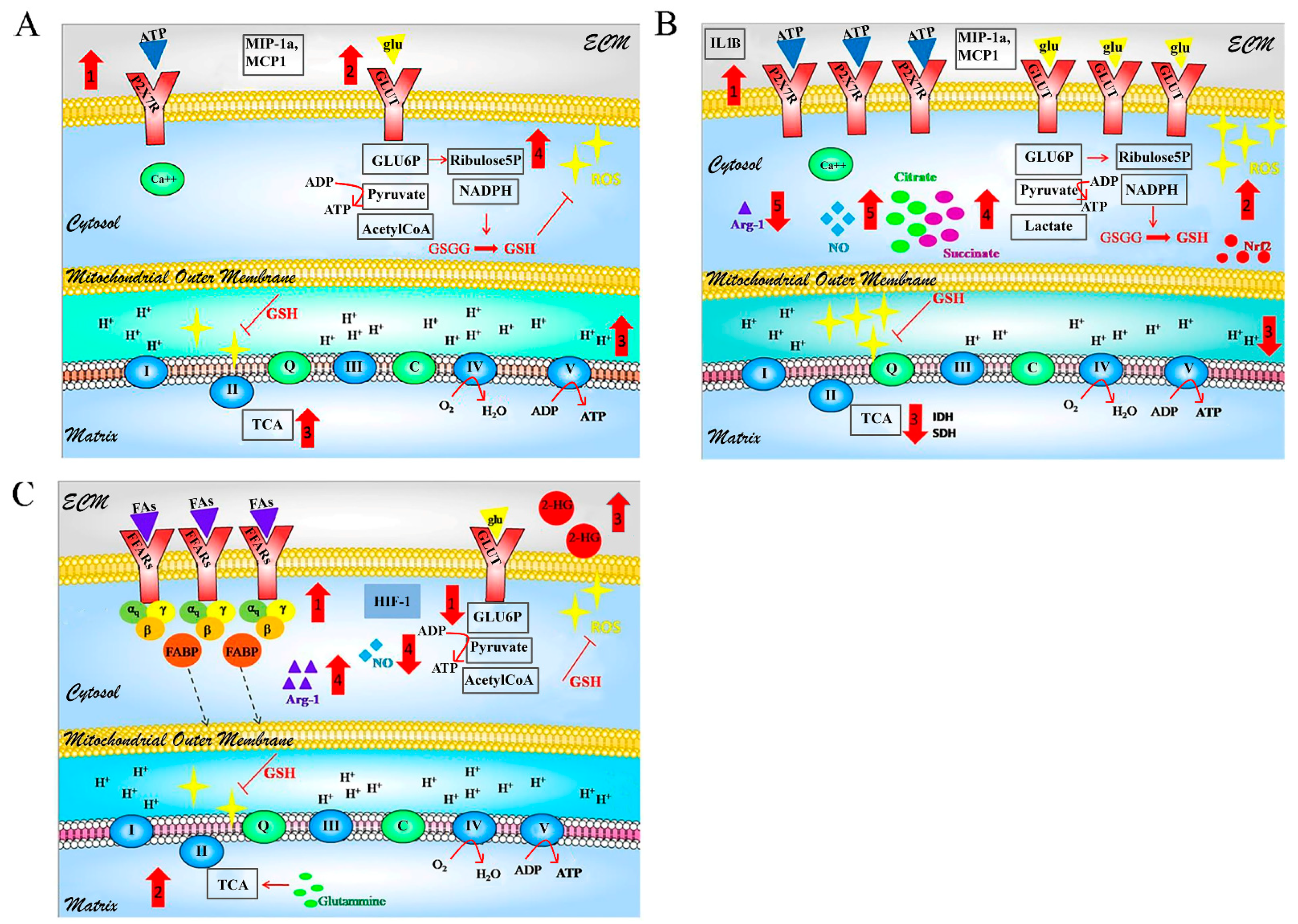
3.3. Metabolism
4. GBM-Associated Astrocytes
4.1. Morphology
4.2. Molecular Profile
4.3. Metabolism
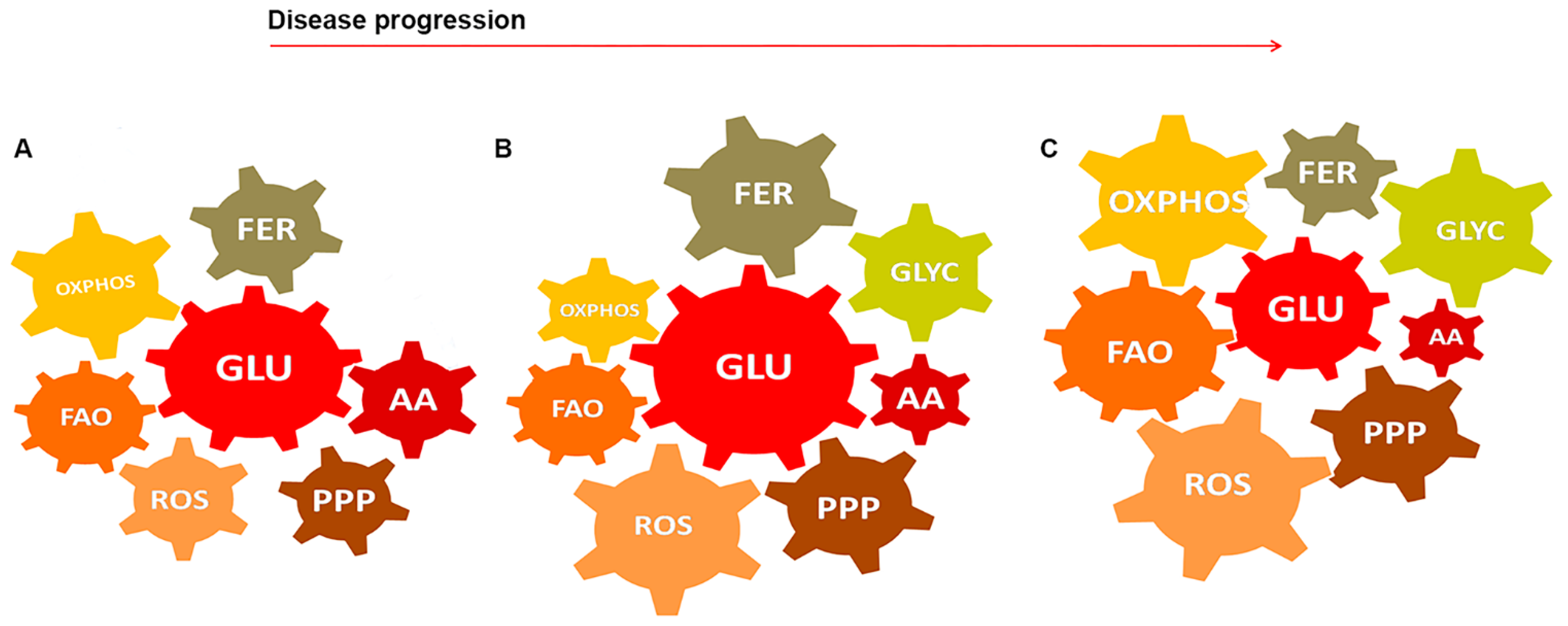
5. GBM-Glial Cells Metabolism as Modules
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- DeAngelis, L.M. Medical progress: Brain tumors. N. Engl. J. Med. 2001, 344, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventero, M.P.; Fuentes-Baile, M.; Quereda, C.; Perez-Valeciano, E.; Alenda, C.; Garcia-Morales, P.; Esposito, D.; Dorado, P.; Manuel Barbera, V.; Saceda, M. Radiotherapy resistance acquisition in Glioblastoma. Role of SOCS1 and SOCS3. PLoS ONE 2019, 14, e0212581. [Google Scholar] [CrossRef] [Green Version]
- Wenger, A.; Vega, S.F.; Kling, T.; Bontell, T.O.; Jakola, A.S.; Carén, H. Intratumor DNA methylation heterogeneity in glioblastoma: Implications for DNA methylation-based classification. Neuro Oncol. 2019. [Google Scholar] [CrossRef]
- De Luca, C.; Colangelo, A.M.; Alberghina, L.; Papa, M. Neuro-Immune Hemostasis: Homeostasis and Diseases in the Central Nervous System. Front. Cell. Neurosci. 2018, 12, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 2012. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.W.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011. [Google Scholar] [CrossRef] [Green Version]
- Alcantara Llaguno, S.; Sun, D.; Pedraza, A.M.; Vera, E.; Wang, Z.; Burns, D.K.; Parada, L.F. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat. Neurosci. 2019, 22, 545–555. [Google Scholar] [CrossRef]
- Antunes, A.R.P.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 2020. [Google Scholar] [CrossRef]
- Richards, L.M.; Whitley, O.K.N.; MacLeod, G.; Cavalli, F.M.G.; Coutinho, F.J.; Jaramillo, J.E.; Svergun, N.; Riverin, M.; Croucher, D.C.; Kushida, M.; et al. Gradient of Developmental and Injury Response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer 2021. [Google Scholar] [CrossRef]
- Placone, A.L.; Quiñones-Hinojosa, A.; Searson, P.C. The role of astrocytes in the progression of brain cancer: Complicating the picture of the tumor microenvironment. Tumor Biol. 2016, 37, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes–Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell. Neurosci. 2018. [Google Scholar] [CrossRef] [Green Version]
- Taheri, B.; Soleimani, M.; Aval, S.F.; Memari, F.; Zarghami, N. C6 glioma-derived microvesicles stimulate the proliferative and metastatic gene expression of normal astrocytes. Neurosci. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Polyzos, A.A.; Lee, D.Y.; Datta, R.; Hauser, M.; Budworth, H.; Holt, A.; Mihalik, S.; Goldschmidt, P.; Frankel, K.; Trego, K.; et al. Metabolic Reprogramming in Astrocytes Distinguishes Region-Specific Neuronal Susceptibility in Huntington Mice. Cell Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef]
- Warburg, O. Injuring of Respiration the Origin of Cancer Cells. Science 1956. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N. Mitochondria: The Ultimate Tumor Suppressor. In Cancer as a Metabolic Disease: On the Origin, Management and Prevention of Cancer; Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Su, Y.T.; Chen, R.; Wang, H.; Song, H.; Zhang, Q.; Chen, L.Y.; Lappin, H.; Vasconcelos, G.; Lita, A.; Maric, D.; et al. Novel targeting of transcription and metabolism in Glioblastoma. Clin. Cancer Res. 2018, 24, 1124–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crunkhorn, S. Targeting cancer cell metabolism in glioblastoma. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef]
- Lau, A.N.; Vander Heiden, M.G. Metabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2020. [Google Scholar] [CrossRef] [Green Version]
- De Luca, C.; Colangelo, A.M.; Virtuoso, A.; Alberghina, L.; Papa, M. Neurons, glia, extracellular matrix and neurovascular unit: A systems biology approach to the complexity of synaptic plasticity in health and disease. Int. J. Mol. Sci. 2020, 21, 1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderone, A.; Formenti, M.; Aprea, F.; Papa, M.; Alberghina, L.; Colangelo, A.M.; Bertolazzi, P. Comparing Alzheimer’s and Parkinson’s diseases networks using graph communities structure. BMC Syst. Biol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Damiani, C.; Gaglio, D.; Sacco, E.; Alberghina, L.; Vanoni, M. Systems metabolomics: From metabolomic snapshots to design principles. Curr. Opin. Biotechnol. 2020, 63, 190–199. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013. [Google Scholar] [CrossRef]
- Griguer, C.E.; Oliva, C.R.; Gillespie, G.Y. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J. Neurooncol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, H.; Ding, Y.; Sharma, J.; Berndt Paetz, M.; Meixensberger, J.; Gaunitz, F.; Birkemeyer, C. Metabolic response of glioblastoma cells associated with glucose withdrawal and pyruvate substitution as revealed by GC-MS. Nutr. Metab. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosc, C.; Selak, M.A.; Sarry, J.E. Resistance Is Futile: Targeting Mitochondrial Energetics and Metabolism to Overcome Drug Resistance in Cancer Treatment. Cell Metab. 2017, 26, 705–707. [Google Scholar] [CrossRef]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017. [Google Scholar] [CrossRef] [Green Version]
- Burns, J.S.; Manda, G. Metabolic pathways of thewarburg effect in health and disease: Perspectives of choice, chain or chance. Int. J. Mol. Sci. 2017, 18, 2755. [Google Scholar] [CrossRef] [Green Version]
- Chinopoulos, C.; Seyfried, T.N. Mitochondrial Substrate-Level Phosphorylation as Energy Source for Glioblastoma: Review and Hypothesis. ASN Neuro 2018. [Google Scholar] [CrossRef]
- Persano, L.; Rampazzo, E.; Della Puppa, A.; Pistollato, F.; Basso, G. The three-layer concentric model of glioblastoma: Cancer stem cells, microenvironmental regulation, and therapeutic implications. Sci. World J. 2011. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018. [Google Scholar] [CrossRef] [Green Version]
- Pistollato, F.; Chen, H.L.; Schwartz, P.H.; Basso, G.; Panchision, D.M. Oxygen tension controls the expansion of human CNS precursors and the generation of astrocytes and oligodendrocytes. Mol. Cell. Neurosci. 2007. [Google Scholar] [CrossRef]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am. J. Pathol. 2010. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007. [Google Scholar] [CrossRef] [Green Version]
- Cuyàs, E.; Corominas-Faja, B.; Menendez, J.A. The nutritional phenome of EMT-induced cancer stem-like cells. Oncotarget 2014. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Luan, Y.; Cai, J.; Wu, S.; Mai, J.; Gu, J.; Zhang, H.; Li, K.; Lin, Y.; Xiao, X.; et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1α Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 2017. [Google Scholar] [CrossRef] [Green Version]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992. [Google Scholar] [CrossRef]
- Xie, T.X.; Xia, Z.; Zhang, N.; Gong, W.; Huang, S. Constitutive NF-κB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol. Rep. 2010. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [Green Version]
- Salem, A.F.; Whitaker-Menezes, D.; Lin, Z.; Martinez-Outschoorn, U.E.; Tanowitz, H.B.; Al-Zoubi, M.S.; Howell, A.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Two-compartment tumor metabolism: Autophagy in the tumor microenvironment, and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavreul, A.; Guette, C.; Faguer, R.; Tétaud, C.; Boissard, A.; Lemaire, L.; Rousseau, A.; Avril, T.; Henry, C.; Coqueret, O.; et al. Glioblastoma-associated stromal cells (GASCs) from histologically normal surgical margins have a myofibroblast phenotype and angiogenic properties. J. Pathol. 2014. [Google Scholar] [CrossRef]
- Civita, P.; Leite, D.M.; Pilkington, G.J. Pre-clinical drug testing in 2d and 3d human in vitro models of glioblastoma incorporating non-neoplastic astrocytes: Tunneling nano tubules and mitochondrial transfer modulates cell behavior and therapeutic respons. Int. J. Mol. Sci. 2019, 20, 6017. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Liu, X.; Wang, B.; Wang, Z.; Liu, Y.; Di, C.; Si, J.; Li, H.; Wu, Q.; Xu, D.; et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 2019. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.; Costalat, R.; Pellerin, L. Determinants of Brain Cell Metabolic Phenotypes and Energy Substrate Utilization Unraveled with a Modeling Approach. PLoS Comput. Biol. 2012. [Google Scholar] [CrossRef]
- Halim, N.D.; Mcfate, T.; Mohyeldin, A.; Okagaki, P.; Korotchkina, L.G.; Patel, M.S.; Jeoung, N.H.; Harris, R.A.; Schell, M.J.; Verma, A. Phosphorylation status of pyruvate dehydrogenase distinguishes metabolic phenotypes of cultured rat brain astrocytes and neurons. Glia 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—New prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Cragnolini, A.; Lampitella, G.; Virtuoso, A.; Viscovo, I.; Panetsos, F.; Papa, M.; Cirillo, G. Regional brain susceptibility to neurodegeneration: What is the role of glial cells? Neural Regen. Res. 2020, 15, 838–842. [Google Scholar] [CrossRef]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Van Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Vinnakota, K.; van Rooijen, N.; Kiwit, J.; Synowitz, M.; Glass, R.; Kettenmann, H. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain. Behav. Immun. 2011. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.; Heppner, F.L.; Tsirka, S.E. Microglia/macrophages promote glioma progression. Glia 2011. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Döring, A.; Zemp, F.J.; Silva, C.; Lun, X.; Wang, X.; Kelly, J.; Hader, W.; Hamilton, M.; Mercier, P.; et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat. Neurosci. 2014. [Google Scholar] [CrossRef]
- McMenamin, P.G. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J. Comp. Neurol. 1999. [Google Scholar] [CrossRef]
- Yang, I.; Han, S.J.; Kaur, G.; Crane, C.; Parsa, A.T. The role of microglia in central nervous system immunity and glioma immunology. J. Clin. Neurosci. 2010, 17, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K. Microglial activation by purines and pyrimidines. Glia 2002, 40, 156–163. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors-redefining innate immunity. Nat. Rev. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 13, 453–460. [Google Scholar] [CrossRef]
- Resende, F.F.B.; Bai, X.; Del Bel, E.A.; Kirchhoff, F.; Scheller, A.; Titze-de-Almeida, R. Evaluation of TgH(CX3CR1-EGFP) mice implanted with mCherry-GL261 cells as an in vivo model for morphometrical analysis of glioma-microglia interaction. BMC Cancer 2016. [Google Scholar] [CrossRef] [Green Version]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When immune cells turn bad—Tumor-associated microglia/macrophages in glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019. [Google Scholar] [CrossRef]
- Bayerl, S.H.; Niesner, R.; Cseresnyes, Z.; Radbruch, H.; Pohlan, J.; Brandenburg, S.; Czabanka, M.A.; Vajkoczy, P. Time lapse in vivo microscopy reveals distinct dynamics of microglia-tumor environment interactions-a new role for the tumor perivascular space as highway for trafficking microglia. Glia 2016. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Silver, J.; Scheffler, B.; Yachnis, A.T.; Smith, G.M.; Steindler, D.A.; Smith, A.A.; Schildts, M.J.; Siebzehnrubl, F.A.; Silver, D.J. Chondroitin Sulfate Proteoglycans Potently Inhibit Invasion and Serve as a Central Organizer of the Brain Tumor Microenvironment. J. Neurosci. 2013. [Google Scholar] [CrossRef] [Green Version]
- Saavedra-López, E.; Roig-Martínez, M.; Cribaro, G.P.; Casanova, P.V.; Gallego, J.M.; Pérez-Vallés, A.; Barcia, C. Phagocytic glioblastoma-associated microglia and macrophages populate invading pseudopalisades. Brain Commun. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banati, R.B.; Egensperger, R.; Maassen, A.; Hager, G.; Kreutzberg, G.W.; Graeber, M.B. Mitochondria in activated microglia in vitro. J. Neurocytol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Zeller, N.; Raasch, J.; Kierdorf, K.; Frenzel, K.; Ketscher, L.; Basters, A.; Staszewski, O.; Brendecke, S.M.; Spiess, A.; et al. USP 18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. 2015. [Google Scholar] [CrossRef] [Green Version]
- Morioka, T.; Baba, T.; Black, K.L.; Streit, W.J. Response of microglial cells to experimental rat glioma. Glia 1992. [Google Scholar] [CrossRef]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; Van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009. [Google Scholar] [CrossRef] [Green Version]
- He, B.P.; Wang, J.J.; Zhang, X.; Wu, Y.; Wang, M.; Bay, B.H.; Chang, A.Y.C. Differential reactions of microglia to brain metastasis of lung cancer. Mol. Med. 2006. [Google Scholar] [CrossRef]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer Immunosurveillance and Immunoediting: The Roles of Immunity in Suppressing Tumor Development and Shaping Tumor Immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar] [CrossRef]
- Wei, J.; Gabrusiewicz, K.; Heimberger, A. The controversial role of microglia in malignant gliomas. Clin. Dev. Immunol. 2013. [Google Scholar] [CrossRef]
- Voisin, P.; Bouchaud, V.; Merle, M.; Diolez, P.; Duffy, L.; Flint, K.; Franconi, J.-M.; Bouzier-Sore, A.-K. Microglia in Close Vicinity of Glioma Cells: Correlation between Phenotype and Metabolic Alterations. Front. Neuroenerget. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, S.L.N.; Abels, E.R.; Van De Haar, L.L.; Zhang, X.; Morsett, L.; Sil, S.; Guedes, J.; Sen, P.; Prabhakar, S.; Hickman, S.E.; et al. Glioblastoma hijacks microglial gene expression to support tumor growth. J. Neuroinflamm. 2020. [Google Scholar] [CrossRef]
- Morrone, F.B.; Horn, A.P.; Stella, J.; Spiller, F.; Sarkis, J.J.F.; Salbego, C.G.; Lenz, G.; Battastini, A.M.O. Increased resistance of glioma cell lines to extracellular ATP cytotoxicity. J. Neurooncol. 2005. [Google Scholar] [CrossRef]
- Fang, K.M.; Wang, Y.L.; Huang, M.C.; Sun, S.H.; Cheng, H.; Tzeng, S.F. Expression of macrophage inflammatory protein-1α and monocyte chemoattractant protein-1 in glioma-infiltrating microglia: Involvement of ATP and P2X7 receptor. J. Neurosci. Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.C.; Maier, L.M.; D’Amico, R.; Mandigo, C.E.; Fontana, E.J.; Waziri, A.; Assanah, M.C.; Canoll, P.; Anderson, R.C.E.; Anderson, D.E.; et al. Dynamics of central and peripheral immunomodulation in a murine glioma model. BMC Immunol. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2015, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Chen, R.; Liu, M.; Feng, J.; Chen, J.; Hu, K. Remodeling the blood–brain barrier microenvironment by natural products for brain tumor therapy. Acta Pharm. Sin. B 2017, 7, 541–553. [Google Scholar] [CrossRef]
- Gjorgjevski, M.; Hannen, R.; Carl, B.; Li, Y.; Landmann, E.; Buchholz, M.; Bartsch, J.W.; Nimsky, C. Molecular profiling of the tumor microenvironment in glioblastoma patients: Correlation of microglia/macrophage polarization state with metalloprotease expression profiles and survival. Biosci. Rep. 2019. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010. [Google Scholar] [CrossRef]
- Sica, A. Role of tumour-associated macrophages in cancer-related inflammation. Exp. Oncol. 2010, 32, 153–158. [Google Scholar]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014. [Google Scholar] [CrossRef] [Green Version]
- Derlindati, E.; Cas, A.D.; Montanini, B.; Spigoni, V.; Curella, V.; Aldigeri, R.; Ardigò, D.; Zavaroni, I.; Bonadonna, R.C. Transcriptomic analysis of human polarized macrophages: More than one role of alternative activation? PLoS ONE 2015. [Google Scholar] [CrossRef] [Green Version]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.L.; Boddeke, H.W.G.M.; et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I.; et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2013, 34, 216–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Dabrowski, M.; Lipko, M.; Sliwa, M.; Maleszewska, M.; Kaminska, B. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia 2013. [Google Scholar] [CrossRef] [PubMed]
- Eder, K.; Kalman, B. The Dynamics of Interactions among Immune and Glioblastoma Cells. NeuroMolecular Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, O.; Achour, S.B.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geric Ivana Metabolic Reprogramming during Microglia Activation. Immunometabolism 2019. [CrossRef] [Green Version]
- Everts, B.; Amiel, E.; Huang, S.C.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; Van Der Windt, G.J.W.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Voloboueva, L.A.; Emery, J.F.; Sun, X.; Giffard, R.G. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. 2013. [Google Scholar] [CrossRef] [Green Version]
- Taetzsch, T.; Levesque, S.; Mcgraw, C.; Brookins, S.; Luqa, R.; Bonini, M.G.; Mason, R.P.; Oh, U.; Block, M.L. Redox regulation of NF-κB p50 and M1 polarization in microglia. Glia 2015. [Google Scholar] [CrossRef] [Green Version]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; Amiel, E.; Van Der Windt, G.J.W.; Freitas, T.C.; Chott, R.; Yarasheski, K.E.; Pearce, E.L.; Pearce, E.J. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 2012. [Google Scholar] [CrossRef] [Green Version]
- Gimeno-Bayón, J.; López-López, A.; Rodríguez, M.J.; Mahy, N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J. Neurosci. Res. 2014. [Google Scholar] [CrossRef] [Green Version]
- Guilarte, T.R.; Loth, M.K.; Guariglia, S.R. TSPO Finds NOX2 in Microglia for Redox Homeostasis. Trends Pharmacol. Sci. 2016, 37, 334–343. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016. [Google Scholar] [CrossRef] [Green Version]
- Finocchiaro, G. TLRgeting Evasion of Immune Pathways in Glioblastoma. Cell Stem Cell 2017, 20, 422–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014. [Google Scholar] [CrossRef] [PubMed]
- Seth, P.; Csizmadia, E.; Hedblom, A.; Vuerich, M.; Xie, H.; Li, M.; Longhi, M.S.; Wegiel, B. Deletion of lactate dehydrogenase-A in myeloid cells triggers antitumor immunity. Cancer Res. 2017. [Google Scholar] [CrossRef] [Green Version]
- Lisi, L.; Laudati, E.; Navarra, P.; Dello Russo, C. The mTOR kinase inhibitors polarize glioma-activated microglia to express a M1 phenotype. J. Neuroinflamm. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisi, L.; Ciotti, G.M.P.; Chiavari, M.; Pizzoferrato, M.; Mangiola, A.; Kalinin, S.; Feinstein, D.L.; Navarra, P. Phospho-mTOR expression in human glioblastoma microglia-macrophage cells. Neurochem. Int. 2019. [Google Scholar] [CrossRef]
- Rodríguez-Prados, J.-C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. J. Immunol. 2010. [Google Scholar] [CrossRef] [Green Version]
- Jha, A.K.; Huang, S.C.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; DI Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017. [Google Scholar] [CrossRef]
- Liu, P.S.; Ho, P.C. Mitochondria: A master regulator in macrophage and T cell immunity. Mitochondrion 2018, 41, 45–50. [Google Scholar] [CrossRef]
- Mieczkowski, J.; Kocyk, M.; Nauman, P.; Gabrusiewicz, K.; Sielska, M.; Przanowski, P.; Maleszewska, M.; Rajan, W.D.; Pszczolkowska, D.; Tykocki, T.; et al. Down-regulation of IKKβ expression in glioma-infiltrating microglia/macrophages is associated with defective inflammatory/immune gene responses in glioblastoma. Oncotarget 2015. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.J.; Zheng, J.Y.; Sun, L.; Yang, H.C.; Cao, Z.Q.; Zhang, X.H.; Zheng, L.T.; Zhen, X.C. The oncometabolite 2-hydroxyglutarate inhibits microglial activation via the AMPK/mTOR/NF-κB pathway. Acta Pharmacol. Sin. 2019. [Google Scholar] [CrossRef]
- Leblond, M.M.; Gérault, A.N.; Corroyer-Dulmont, A.; MacKenzie, E.T.; Petit, E.; Bernaudin, M.; Valable, S. Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Verhaak, R.G.W.; Canolk, P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev. Mol. Diagn. 2012, 12, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soos, J.M.; Morrow, J.; Ashley, T.A.; Szente, B.E.; Bikoff, E.K.; Zamvil, S.S. Astrocytes express elements of the class II endocytic pathway and process central nervous system autoantigen for presentation to encephalitogenic T cells. J. Neuroimmunol. 1998. [Google Scholar] [CrossRef]
- Katz, A.M.; Amankulor, N.M.; Pitter, K.; Helmy, K.; Squatrito, M.; Holland, E.C. Astrocyte-specific expression patterns associated with the PDGF-induced glioma microenvironment. PLoS ONE 2012. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and niche concept. Cancers 2019, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John Lin, C.C.; Yu, K.; Hatcher, A.; Huang, T.W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okolie, O.; Bago, J.R.; Schmid, R.S.; Irvin, D.M.; Bash, R.E.; Miller, C.R.; Hingtgen, S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro Oncol. 2018. [Google Scholar] [CrossRef]
- Lin, C.M.; Yu, C.F.; Huang, H.Y.; Chen, F.H.; Hong, J.H.; Chiang, C.S. Distinct tumor microenvironment at tumor edge as a result of astrocyte activation is associated with therapeutic resistance for brain tumor. Front. Oncol. 2019. [Google Scholar] [CrossRef]
- Papa, M.; De Luca, C.; Petta, F.; Alberghina, L.; Cirillo, G. Astrocyte-neuron interplay in maladaptive plasticity. Neurosci. Biobehav. Rev. 2014, 42, 35–54. [Google Scholar] [CrossRef]
- Silver, J. The glial scar is more than just astrocytes. Exp. Neurol. 2016, 286, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; Cirillo, G.; Riva, G.; Romano, G.; Giussani, C.; Cialdella, A.; Todisco, A.; Virtuoso, A.; Cerrito, M.G.; Bentivegna, A.; et al. Specific expression of a new bruton tyrosine kinase isoform (P65BTK) in the glioblastoma gemistocytic histotype. Front. Mol. Neurosci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Horng, S.; Therattil, A.; Moyon, S.; Gordon, A.; Kim, K.; Argaw, A.T.; Hara, Y.; Mariani, J.N.; Sawai, S.; Flodby, P.; et al. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Investig. 2017. [Google Scholar] [CrossRef]
- Gagliano, N.; Costa, F.; Cossetti, C.; Pettinari, L.; Bassi, R.; Chiriva-Internati, M.; Cobos, E.; Gioia, M.; Pluchino, S. Glioma-astrocyte interaction modifies the astrocyte phenotype in a co-culture experimental model. Oncol. Rep. 2009. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Yan, T.; Zhu, H.; Liang, X.; Leiss, L.; Sakariassen, P.Ø.; Skaftnesmo, K.O.; Huang, B.; Costea, D.E.; Enger, P.Ø.; et al. A co-culture model with brain tumor-specific bioluminescence demonstrates astrocyte-induced drug resistance in glioblastoma. J. Transl. Med. 2014. [Google Scholar] [CrossRef]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mega, A.; Hartmark Nilsen, M.; Leiss, L.W.; Tobin, N.P.; Miletic, H.; Sleire, L.; Strell, C.; Nelander, S.; Krona, C.; Hägerstrand, D.; et al. Astrocytes enhance glioblastoma growth. Glia 2020. [Google Scholar] [CrossRef]
- Lin, Q.; Liu, Z.; Ling, F.; Xu, G. Astrocytes protect glioma cells from chemotherapy and upregulate survival genes via gap junctional communication. Mol. Med. Rep. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Ding, K.; Wang, J.; Li, X.; Zhao, P. Chemoresistance caused by the microenvironment of glioblastoma and the corresponding solutions. Biomed. Pharmacother. 2019, 109, 39–46. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, H.J.; Kim, M.S.; Choi, H.J.; He, J.; Wu, Q.; Aldape, K.; Weinberg, J.S.; Yung, W.K.A.; Conrad, C.A.; et al. Macitentan, a dual endothelin receptor antagonist, in combination with temozolomide leads to glioblastoma regression and long-term survival in mice. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.; Narita, M.; Ikegami, D.; Yamashita, A.; Shimizu, T.; Narita, M.; Niikura, K.; Furuya, M.; Kobayashi, Y.; Miyashita, K.; et al. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain 2013. [Google Scholar] [CrossRef] [Green Version]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W.; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Salhia, B.; Angelov, L.; Roncari, L.; Wu, X.; Shannon, P.; Guha, A. Expression of vascular endothelial growth factor by reactive astrocytes and associated neoangiogenesis. Brain Res. 2000. [Google Scholar] [CrossRef]
- Wanner, I.B.; Anderson, M.A.; Song, B.; Levine, J.; Fernandez, A.; Gray-Thompson, Z.; Ao, Y.; Sofroniew, M.V. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, B.H.; Wahba, A.; Camphausen, K.; Tofilon, P.J. Coculture with astrocytes reduces the radiosensitivity of glioblastoma stem-like cells and identifies additional targets for radiosensitization. Cancer Med. 2015. [Google Scholar] [CrossRef]
- Wurm, J.; Behringer, S.P.; Ravi, V.M.; Joseph, K.; Neidert, N.; Maier, J.P.; Doria-Medina, R.; Follo, M.; Delev, D.; Pfeifer, D.; et al. Astrogliosis releases pro-oncogenic chitinase 3-like 1 causing mapk signaling in glioblastoma. Cancers 2019, 11, 1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.M.; Besson, A.; Fogg, D.K.; Choi, K.S.; Waisman, D.M.; Goodyer, C.G.; Rewcastle, B.; Yong, V.W. Exploitation of astrocytes by glioma cells to facilitate invasiveness: A mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J. Neurosci. 2003. [Google Scholar] [CrossRef]
- Norton, W.T.; Aquino, D.A.; Hozumi, I.; Chiu, F.C.; Brosnan, C.F. Quantitative aspects of reactive gliosis: A review. Neurochem. Res. 1992. [Google Scholar] [CrossRef] [PubMed]
- Duran, R.C.D.; Wang, C.Y.; Zheng, H.; Deneen, B.; Wu, J.Q. Brain region-specific gene signatures revealed by distinct astrocyte subpopulations unveil links to glioma and neurodegenerative diseases. eNeuro 2019. [Google Scholar] [CrossRef]
- Niklasson, M.; Bergström, T.; Jarvius, M.; Sundström, A.; Nyberg, F.; Haglund, C.; Larsson, R.; Westermark, B.; Segerman, B.; Segerman, A. Mesenchymal transition and increased therapy resistance of glioblastoma cells is related to astrocyte reactivity. J. Pathol. 2019. [Google Scholar] [CrossRef]
- Waters, M.R.; Gupta, A.S.; Mockenhaupt, K.; Brown, L.S.N.; Biswas, D.D.; Kordula, T. RelB acts as a molecular switch driving chronic inflammation in glioblastoma multiforme. Oncogenesis 2019. [Google Scholar] [CrossRef] [PubMed]
- Bouzier-Sore, A.K.; Pellerin, L. Unraveling the complex metabolic nature of astrocytes. Front. Cell. Neurosci. 2013, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Mangia, S.; Simpson, I.A.; Vannucci, S.J.; Carruthers, A. The in vivo neuron-to-astrocyte lactate shuttle in human brain: Evidence from modeling of measured lactate levels during visual stimulation. J. Neurochem. 2009. [Google Scholar] [CrossRef] [Green Version]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010. [Google Scholar] [CrossRef]
- Motori, E.; Puyal, J.; Toni, N.; Ghanem, A.; Angeloni, C.; Malaguti, M.; Cantelli-Forti, G.; Berninger, B.; Conzelmann, K.K.; Götz, M.; et al. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 2013. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.G.; Robinson, M.B. Regulation of mitochondrial dynamics in astrocytes: Mechanisms, consequences, and unknowns. Glia 2018, 66, 1213–1234. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.F.; Fischer, S.; Koshkin, A.; Laczko, E.; Fischer, D.; Ogunshola, O.O. Cell-specific metabolomic responses to injury: Novel insights into blood-brain barrier modulation. Sci. Rep. 2020. [Google Scholar] [CrossRef] [PubMed]
- García-Nogales, P.; Almeida, A.; Fernández, E.; Medina, J.M.; Bolaños, J.P. Induction of glucose-6-phosphate dehydrogenase by lipopolysaccharide contributes to preventing nitric oxide-mediated glutathione depletion in cultured rat astrocytes. J. Neurochem. 1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuquet, J.; Quilichini, P.; Nimchinsky, E.A.; Buzsáki, G. Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J. Neurosci. 2010. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Subramanian, A.; Sarkar, R.R. Exploring the differences in metabolic behavior of astrocyte and glioblastoma: A flux balance analysis approach. Syst. Synth. Biol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Dinuzzo, M.; Maraviglia, B.; Giove, F. Why does the brain (not) have glycogen? BioEssays 2011. [Google Scholar] [CrossRef]
- Carpenter, K.L.H.; Jalloh, I.; Hutchinson, P.J. Glycolysis and the significance of lactate in traumatic brain injury. Front. Neurosci. 2015, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Randall, E.C.; Lopez, B.G.C.; Peng, S.; Regan, M.S.; Abdelmoula, W.M.; Basu, S.S.; Santagata, S.; Yoon, H.; Haigis, M.C.; Agar, J.N.; et al. Localized Metabolomic Gradients in Patient-Derived Xenograft Models of Glioblastoma. Cancer Res. 2020, 80, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Patel, S.; Affeck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, G.S.; Moreno, A.P.; Lampe, P.D. Gap junctions between cells expressing connexin 43 or 32 show inverse permselectivity to adenosine and ATP. J. Biol. Chem. 2002. [Google Scholar] [CrossRef] [Green Version]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ávila Rodriguez, M.; Garcia-Segura, L.M.; Cabezas, R.; Torrente, D.; Capani, F.; Gonzalez, J.; Barreto, G.E. Tibolone protects T98G cells from glucose deprivation. J. Steroid Biochem. Mol. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.C.; Jackson, J.G.; Robinson, M.B. Transient oxygen/glucose deprivation causes a delayed loss of mitochondria and increases spontaneous calcium signaling in astrocytic processes. J. Neurosci. 2016. [Google Scholar] [CrossRef] [Green Version]
- Blázquez, C.; Woods, A.; De Ceballos, M.L.; Carling, D.; Guzmán, M. The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes. J. Neurochem. 1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of ketone body metabolism and the role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef] [Green Version]
- Yip, J.; Geng, X.; Shen, J.; Ding, Y. Cerebral gluconeogenesis and diseases. Front. Pharmacol. 2017, 7, 521. [Google Scholar] [CrossRef] [Green Version]
- van der Louw, E.J.T.M.; Olieman, J.F.; van den Bemt, P.M.L.A.; Bromberg, J.E.C.; Oomen-de Hoop, E.; Neuteboom, R.F.; Catsman-Berrevoets, C.E.; Vincent, A.J.P.E. Ketogenic diet treatment as adjuvant to standard treatment of glioblastoma multiforme: A feasibility and safety study. Ther. Adv. Med. Oncol. 2019. [Google Scholar] [CrossRef]
- Arismendi-Morillo, G.; Castellano-Ramírez, A.; Seyfried, T.N. Ultrastructural characterization of the Mitochondria-associated membranes abnormalities in human astrocytomas: Functional and therapeutics implications. Ultrastruct. Pathol. 2017. [Google Scholar] [CrossRef]
- Maurer, G.D.; Brucker, D.P.; Bähr, O.; Harter, P.N.; Hattingen, E.; Walenta, S.; Mueller-Klieser, W.; Steinbach, J.P.; Rieger, J. Differential utilization of ketone bodies by neurons and glioma cell lines: A rationale for ketogenic diet as experimental glioma therapy. BMC Cancer 2011. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Flores, R.; Poff, A.M.; D’Agostino, D.P.; Mukherjee, P. Metabolic therapy: A new paradigm for managing malignant brain cancer. Cancer Lett. 2015. [Google Scholar] [CrossRef]
- Huang, D.; Li, T.; Wang, L.; Zhang, L.; Yan, R.; Li, K.; Xing, S.; Wu, G.; Hu, L.; Jia, W.; et al. Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Sperry, J.; Le Belle, J.E.; Condro, M.C.; Guo, L.; Braas, D.; Vanderveer-Harris, N.; Kim, K.K.O.; Pope, W.B.; Divakaruni, A.S.; Lai, A.; et al. Metabolism of fatty acids and ketone bodies for glioblastoma growth: Implications for Ketogenic Diet Therapy. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.C.; Bockmayr, A. Flux modules in metabolic networks. J. Math. Biol. 2014. [Google Scholar] [CrossRef]
- Çubuk, C.; Hidalgo, M.R.; Amadoz, A.; Rian, K.; Salavert, F.; Pujana, M.A.; Mateo, F.; Herranz, C.; Carbonell-Caballero, J.; Dopazo, J. Differential metabolic activity and discovery of therapeutic targets using summarized metabolic pathway models. NPJ Syst. Biol. Appl. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertz, L. Astrocytic amino acid metabolism under control conditions and during oxygen and/or glucose deprivation. Neurochem. Res. 2003. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Saurty-Seerunghen, M.S.; Bellenger, L.; El-Habr, E.A.; Delaunay, V.; Garnier, D.; Chneiweiss, H.; Antoniewski, C.; Morvan-Dubois, G.; Junier, M.P. Capture at the single cell level of metabolic modules distinguishing aggressive and indolent glioblastoma cells. Acta Neuropathol. Commun. 2019. [Google Scholar] [CrossRef] [Green Version]
- Han, S.J.; Englot, D.J.; Birk, H.; Molinaro, A.M.; Chang, S.M.; Clarke, J.L.; Prados, M.D.; Taylor, J.W.; Berger, M.S.; Butowski, N.A. Impact of timing of concurrent chemoradiation for newly diagnosed glioblastoma: A critical review of current evidence. Neurosurgery 2015. [Google Scholar] [CrossRef] [Green Version]
- Katsigiannis, S.; Krischek, B.; Barleanu, S.; Grau, S.; Galldiks, N.; Timmer, M.; Kabbasch, C.; Goldbrunner, R.; Stavrinou, P. Impact of time to initiation of radiotherapy on survival after resection of newly diagnosed glioblastoma. Radiat. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.T.; Liu, L.; Liu, Y.; Milano, M.T.; Walter, K.A. The impact of timing of concurrent chemoradiation in patients with high-grade glioma in the era of the stupp protocol. Front. Oncol. 2019, 9, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Virtuoso, A.; Giovannoni, R.; De Luca, C.; Gargano, F.; Cerasuolo, M.; Maggio, N.; Lavitrano, M.; Papa, M. The Glioblastoma Microenvironment: Morphology, Metabolism, and Molecular Signature of Glial Dynamics to Discover Metabolic Rewiring Sequence. Int. J. Mol. Sci. 2021, 22, 3301. https://doi.org/10.3390/ijms22073301
Virtuoso A, Giovannoni R, De Luca C, Gargano F, Cerasuolo M, Maggio N, Lavitrano M, Papa M. The Glioblastoma Microenvironment: Morphology, Metabolism, and Molecular Signature of Glial Dynamics to Discover Metabolic Rewiring Sequence. International Journal of Molecular Sciences. 2021; 22(7):3301. https://doi.org/10.3390/ijms22073301
Chicago/Turabian StyleVirtuoso, Assunta, Roberto Giovannoni, Ciro De Luca, Francesca Gargano, Michele Cerasuolo, Nicola Maggio, Marialuisa Lavitrano, and Michele Papa. 2021. "The Glioblastoma Microenvironment: Morphology, Metabolism, and Molecular Signature of Glial Dynamics to Discover Metabolic Rewiring Sequence" International Journal of Molecular Sciences 22, no. 7: 3301. https://doi.org/10.3390/ijms22073301
APA StyleVirtuoso, A., Giovannoni, R., De Luca, C., Gargano, F., Cerasuolo, M., Maggio, N., Lavitrano, M., & Papa, M. (2021). The Glioblastoma Microenvironment: Morphology, Metabolism, and Molecular Signature of Glial Dynamics to Discover Metabolic Rewiring Sequence. International Journal of Molecular Sciences, 22(7), 3301. https://doi.org/10.3390/ijms22073301