
Personalized Development of Antisense Oligonucleotides for Exon Skipping Restores Type XVII Collagen Expression in Junctional Epidermolysis Bullosa

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. A Homozygous COL17A1 Nonsense Mutation Leads to Loss of C17 Expression
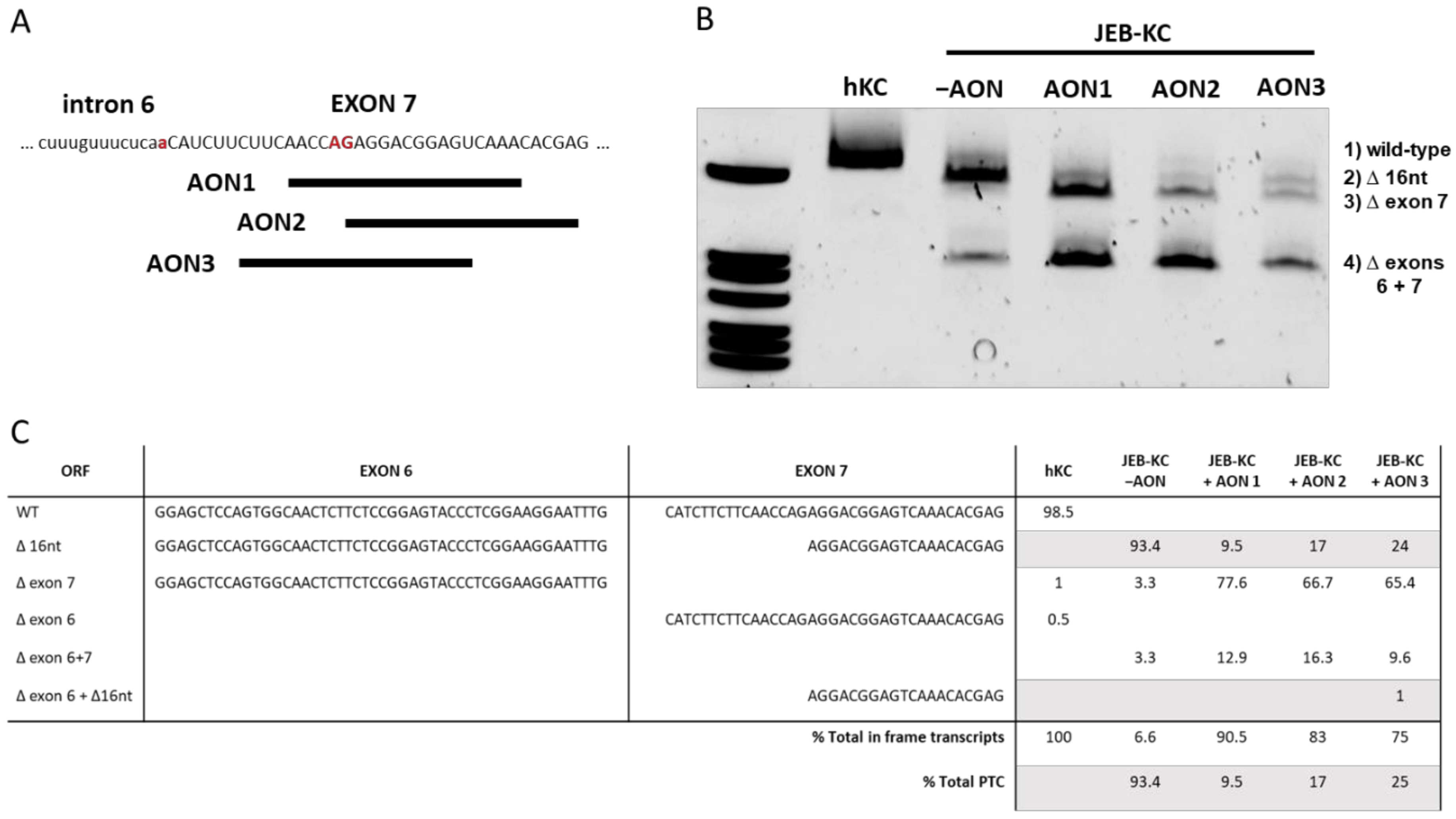
2.2. Design of AONs Capable of Exon 7 Skipping in Combination with A Liposomal Delivery System
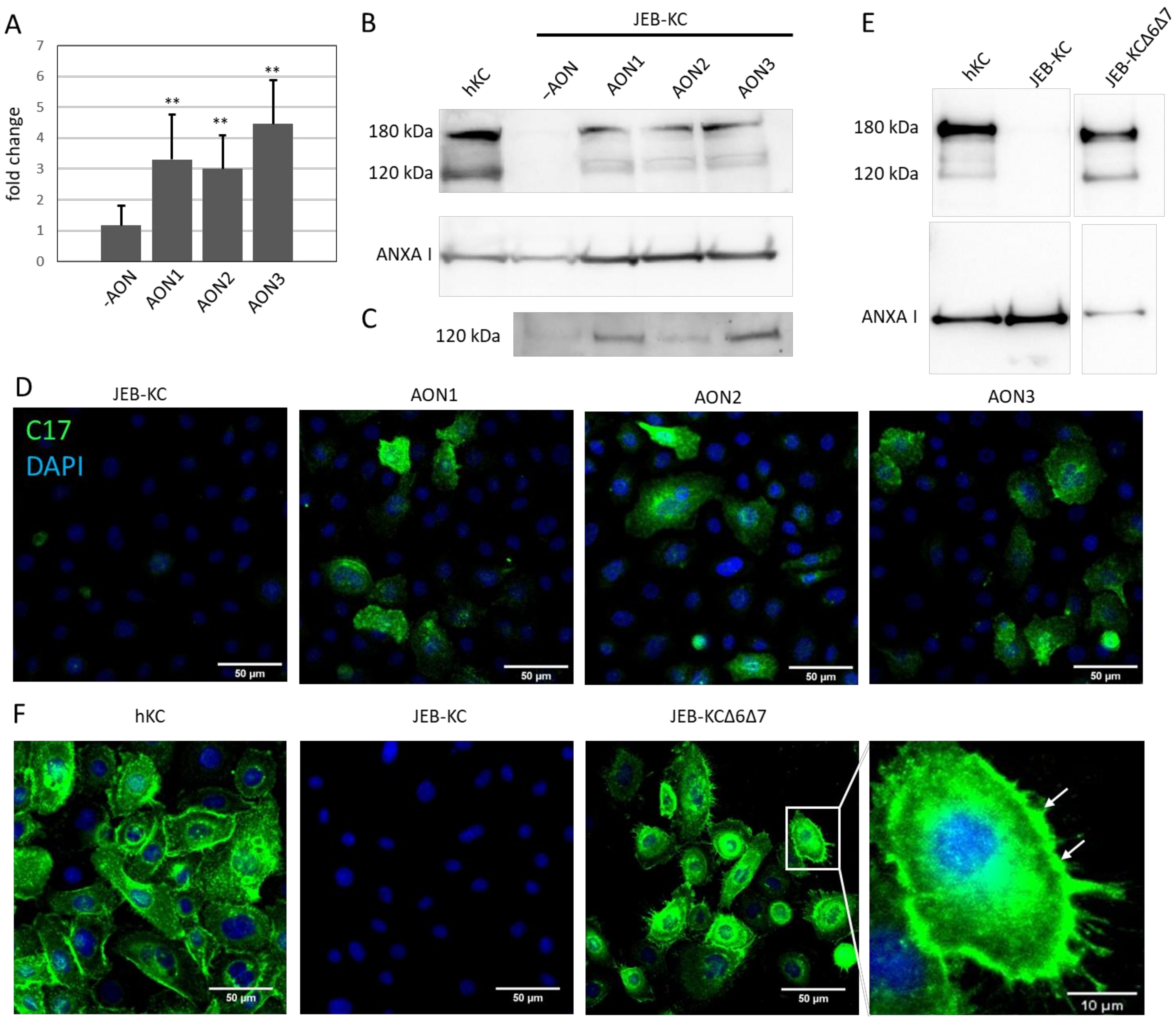
2.3. AON-Induced Exon Skipping Restores Type XVII Collagen Expression
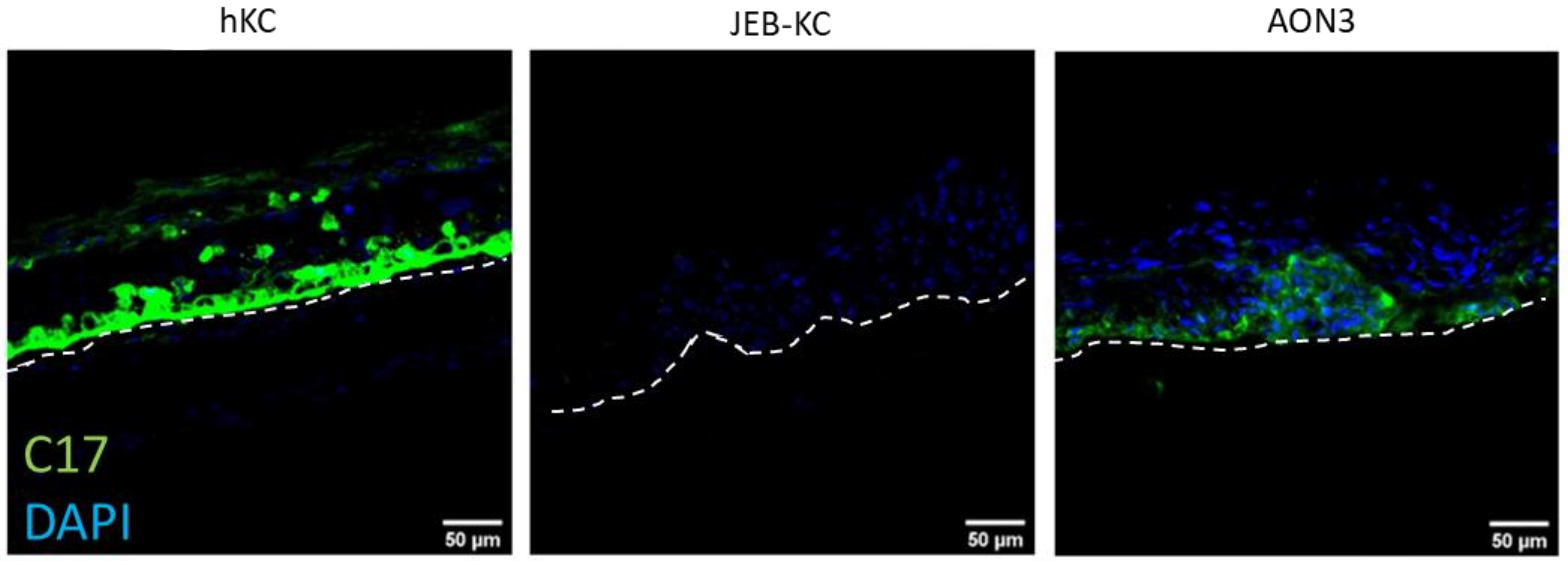
2.4. Treatment of 3D Skin Equivalents with AONs Results in A Stable Epidermis and Correct Localization of Type XVII Collagen
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Generation of A JEB∆6∆7 Cell Line
- F1_ex6+7_fw: 5′-caggcctcgagggccggcgcgccgcATGGATGTAACCAAGAAAAACAAACG-3′
- F1_ex6+7_rv: 5′-cgaatttcactctCTTCATACGCATGGCGGG-3′
- F2_fwd: 5′-cggaaggaatttgAGAGTGAAATTCGAGTTCGAC-3′
- F2_rev: 5′-aatgccaagagccCCTGGAACACCTGGATCAC-3′
- F3_fwd: 5′-caggtgttccaggGGCTCTTGGCATTCCTAG-3′
- F3_rev: 5′-ggaggggggggggcggaatttacgtagcTCACGGCTTGACAGCAATAC-3′
4.3. AON Design
- AON1_C17_20: 5′-TTTGACTCCGTCCTCTGGTT-3′
- AON1_C17_20_Cy5: 5′-Cy5-TTTGACTCCGTCCTCTGGTT-3′
- AON2_C17_20: 5′-TCGTGTTTGACTCCGTCCTC-3′
- AON2_C17_20_Cy5: 5′-Cy5-TCGTGTTTGACTCCGTCCTC-3′
- AON3_C17_20: 5′-CTCCGTCCTCTGGTTGAAGA-3′
- AON3_C17_20_Cy5: 5′-Cy5-CTCCGTCCTCTGGTTGAAGA-3′
4.4. Splice Pattern Evaluation
4.5. DDC642 Liposome Production
4.6. Gel Retardation Assay
4.7. Cell Viability
4.8. Immunofluorescence Microscopy
4.9. Semiquantitative Real-Time PCR (sqRT-PCR)
- C17_FW: 5′-GGAGCGAGCTGATCAGCTACCTCAC-3′
- C17_RV: 5′-GCCATCCCTTGCAGTAGGCCCTG-3′
- GAPDH_FW: 5′-GCCAACGTGTCAGTGGTGGA-3′
- GAPDH_RV: 5′-CACCACCCTGTTGCTGTAGCC-3′
4.10. NGS
4.11. Western Blot Analysis
4.12. Transfection of keratinocytes with AONs
4.13. Conditioning of Cell Culture Supernatants
4.14. 3D Organotypic Skin Equivalents
4.15. Liposomal Delivery of AONs into Human Skin Explants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Has, C.; Bauer, J.; Bodemer, C.; Bolling, M.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef]
- Laimer, M.; Prodinger, C.; Bauer, J.W. Hereditary epidermolysis bullosa. J. Dtsch. Dermatol. Ges. 2015, 13, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Mellerio, J.E. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: Part I. Epithelial associated tissues. J. Am. Acad. Dermatol. 2009, 61, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Mellerio, J.E. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: Part II. Other organs. J. Am. Acad. Dermatol. 2009, 61, 387–402. [Google Scholar] [CrossRef]
- Fine, J.D. Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates from the National Epider-molysis Bullosa Registry. JAMA Dermatol. 2016, 152, 1231–1238. [Google Scholar] [CrossRef]
- Prodinger, C.; Bauer, J.W.; Laimer, M. Translational perspectives to treat Epidermolysis bullosa—Where do we stand? Exp. Dermatol. 2020, 29, 1112–1122. [Google Scholar] [CrossRef]
- Has, C.; South, A.; Uitto, J. Molecular Therapeutics in Development for Epidermolysis Bullosa: Update 2020. Mol. Diagn. Ther. 2020, 24, 299–309. [Google Scholar] [CrossRef]
- Wally, V.; Reisenberger, M.; Kitzmüller, S.; Laimer, M. Small molecule drug development for rare genodermatoses–evaluation of the current status in epidermolysis bullosa. Orphanet J. Rare Dis. 2020, 15, 1–9. [Google Scholar] [CrossRef]
- Wally, V.; Ly, J.; Brunner, V.; Felder, T.K. Diacerein orphan drug development for epidermolysis bullosa simplex: A phase 2/3 randomized, placebo-controlled, double-blind clinical trial. J. Am. Acad. Dermatol. 2018, 78, 892–901.e7. [Google Scholar] [CrossRef] [PubMed]
- Guttmann-Gruber, C.; Tockner, B.; Scharler, C.; Hüttner, C.; Common, J.E.; Tay, A.S.L.; Denil, S.L.I.J.; Klausegger, A.; Trost, A.; Breitenbach, J.; et al. Low-dose calcipotriol can elicit wound closure, anti-microbial, and anti-neoplastic effects in epidermolysis bullosa keratinocytes. Sci. Rep. 2018, 8, 13430. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, M.; Zauner, R.; Ablinger, M.; Piñón-Hofbauer, J.; Guttmann-Gruber, C.; Reisenberger, M.; Lettner, T.; Niklas, N.; Proell, J.; Sajinovic, M.; et al. A cancer stem cell-like phenotype is associated with miR-10b expression in aggressive squamous cell carcinomas. Cell Commun. Signal. 2020, 18, 1–15. [Google Scholar] [CrossRef]
- Marinkovich, M.P.; Tang, J.Y. Gene Therapy for Epidermolysis Bullosa. J. Investig. Dermatol. 2019, 139, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mastaglia, F.L.; Fletcher, S.; Wilton, S.D. Precision Medicine through Antisense Oligonucleotide-Mediated Exon Skipping. Trends Pharmacol. Sci. 2018, 39, 982–994. [Google Scholar] [CrossRef]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Walko, G.; Castanon, M.J.; Wiche, G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015, 360, 529–544. [Google Scholar] [CrossRef]
- Has, C.; Bruckner-Tuderman, L. Collagen XVII and Its Role in Junctional Epidermolysis Bullosa. In Blistering Diseases; Murrell, D., Ed.; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar] [CrossRef]
- Kiritsi, D.; Kern, J.S.; Schumann, H.; Kohlhase, J.; Has, C.; Bruckner-Tuderman, L. Molecular mechanisms of phenotypic variability in junctional epidermolysis bullosa. J. Med. Genet. 2011, 48, 450–457. [Google Scholar] [CrossRef]
- Siva, K.; Covello, G.; Denti, M.A. Exon-Skipping Antisense Oligonucleotides to Correct Missplicing in Neurogenetic Diseases. Nucleic Acid Ther. 2014, 24, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Straub, V.; Haas, M. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Per-spective on the Outstanding Issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef]
- Fairbrother, W.G.; Yeh, R.-F.; Sharp, P.A.; Burge, C.B. Predictive Identification of Exonic Splicing Enhancers in Human Genes. Science 2002, 297, 1007–1013. [Google Scholar] [CrossRef]
- Desmet, E.; Bracke, S.; Forier, K.; Taevernier, L.; Stuart, M.C.A.; De Spiegeleer, B.; Raemdonck, K.; Van Gele, M.; Lambert, J. An elastic liposomal formulation for RNAi-based topical treatment of skin disorders: Proof-of-concept in the treatment of psoriasis. Int. J. Pharm. 2016, 500, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Desmet, E.; Bracke, S.; Forier, K.; Taevernier, L.; Stuart, M.C.; De Spiegeleer, B.; Raemdonck, K.; Van Gele, M.; Lambert, J. Characterization data on the topical carrier DDC642. Data Brief 2016, 7, 1204–1210. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Koster, J.; Geerts, D.; Favre, B.; Borradori, L.; Sonnenberg, A. Analysis of the interactions between BP180, BP230, plectin and the integrin alpha6beta4 important for hemidesmosome assembly. J. Cell Sci. 2003, 116, 387–399. [Google Scholar] [CrossRef]
- Huber, M.; Floeth, M.; Lane, E.B. Deletion of the cytoplasmatic domain of BP180/collagen XVII causes a phenotype with predominant features of epi-dermolysis bullosa simplex. J. Investig. Dermatol. 2002, 118, 185–192. [Google Scholar] [CrossRef]
- Ruzzi, L.; Posteraro, P.; Zambruno, G.; Castiglia, D.; D’Alessio, M.; Pas, H.; Mazzanti, C.; Didona, B.; Owaribe, K.; Meneguzzi, G. A Homozygous Nonsense Mutation in Type XVII Collagen Gene (COL17A1) Uncovers an Alternatively Spliced mRNA Accounting for an Unusually Mild Form of Non-Herlitz Junctional Epidermolysis Bullosa. J. Investig. Dermatol. 2001, 116, 182–187. [Google Scholar] [CrossRef]
- Pasmooij, A.M.G.; Van Zalen, S.; Nijenhuis, A.M.; Kloosterhuis, A.J.; Zuiderveen, J.; Jonkman, M.F.; Pas, H. A very mild form of non-Herlitz junctional epidermolysis bullosa: BP180 rescue by outsplicing of mutated exon 30 coding for the COL15 domain. Exp. Dermatol. 2004, 13, 125–128. [Google Scholar] [CrossRef]
- Hwang, J.; Yokota, T. Recent advancements in exon-skipping therapies using antisense oligonucleotides and genome editing for the treatment of various muscular dystrophies. Expert Rev. Mol. Med. 2019, 21, e5. [Google Scholar] [CrossRef]
- Hahn, J.K.; Neupane, B.; Pradhan, K.; Zhou, Q.; Testa, L.; Pelzl, L.; Maleck, C.; Gawaz, M.; Gramlich, M. The assembly and evaluation of antisense oligonucleotides applied in exon skipping for titin-based mutations in dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 131, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Pijnappel, W.; Van Der Wal, E.; Bergsma, A.; Van Gestel, T.; Pijnenburg, J.; In’t Groen, S.; Zaehres, H.; Araúzo-Bravo, M.; Schöler, H.; Van Der Ploeg, A. Antisense oligonucleotides promote exon inclusion and correct the common c.-32-13T>G (IVS1) GAA splicing variant in iPS-derived skeletal muscle cells from Pompe patients. Neuromuscul. Disord. 2017, 27, S161. [Google Scholar] [CrossRef]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; Born, L.I.V.D.; Khan, M.I.; Msc, S.S.C.; Verheij, J.B.G.M.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Cheok, C.F.; Lane, D.P. Exploiting the p53 Pathway for Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026310. [Google Scholar] [CrossRef]
- Bornert, O.; Nauroy, P.; Tufa, S.F.; Kiritsi, D. QR-313, an Antisense Oligonucleotide, Shows Therapeutic Efficacy for Treatment of Dominant and Recessive Dystrophic Epidermolysis Bullosa: A Preclinical Study. J. Investig. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bremer, J.; Bornert, O.; Nyström, A.; Gostynski, A.; Jonkman, M.F.; Aartsma-Rus, A.; Akker, P.C.V.D.; Pasmooij, A.M. Antisense Oligonucleotide-mediated Exon Skipping as a Systemic Therapeutic Approach for Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 2016, 5, e379. [Google Scholar] [CrossRef]
- Turczynski, S.; Titeux, M.; Tonasso, L.; Décha, A.; Ishida-Yamamoto, A.; Hovnanian, A. Targeted Exon Skipping Restores Type VII Collagen Expression and Anchoring Fibril Formation in an In Vivo RDEB Model. J. Investig. Dermatol. 2016, 136, 2387–2395. [Google Scholar] [CrossRef]
- Kowalewski, C.; Bremer, J.; Gostynski, A.; Wertheim-Tysarowska, K.; Wozniak, K.; Bal, J.; Jonkman, M.; Pasmooij, A. Amelioration of junctional epidermolysis bullosa due to exon skipping. Br. J. Dermatol. 2016, 174, 1375–1379. [Google Scholar] [CrossRef]
- Bremer, J.; Eichhorn, D.S.; Pasmooij, A.M.G. Natural exon skipping sets the stage for exon skipping as therarpy for dystrophic epidermolysis bullosa. Mol. Ther. Nucleic Acids 2019, 18, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Schwieger-Briel, A.; Weibel, L.; Chmel, N.; Leppert, J. A COL7A1 variant leading to in-frame skipping of exon 15 attenuates disease severity in recessive dystrophic epidermolysis bullosa. Br. J. Dermatol. 2015, 173, 1308–1311. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.A.; Ashton, G.H.; Mellerio, J.E.; McMillan, J.R.; Eady, R.A.; Salas-Alanis, J.C.; Swensson, O. Moderation of Phenotypic Severity in Dystrophic and Junctional Forms of Epidermolysis Bullosa Through In-Frame Skipping of Exons Containing Non-Sense or Frameshift Mutations. J. Investig. Dermatol. 1999, 113, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Carita, A.C.; Eloy, J.O.; Chorilli, M.; Lee, R.J.; Leonardi, G.R. Recent Advances and Perspectives in Liposomes for Cutaneous Drug Delivery. Curr. Med. Chem. 2018, 25, 606–635. [Google Scholar] [CrossRef]
- Peking, P.; Muss, W.H.; Kocher, T.; Koller, U. An ex vivo RNA trans-splicing strategy to correct human generalized severe epidermolysis bullosa simplex. Br. J. Dermatol. 2019, 180, 141–148. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ablinger, M.; Lettner, T.; Friedl, N.; Potocki, H.; Palmetzhofer, T.; Koller, U.; Illmer, J.; Liemberger, B.; Hainzl, S.; Klausegger, A.; et al. Personalized Development of Antisense Oligonucleotides for Exon Skipping Restores Type XVII Collagen Expression in Junctional Epidermolysis Bullosa. Int. J. Mol. Sci. 2021, 22, 3326. https://doi.org/10.3390/ijms22073326
Ablinger M, Lettner T, Friedl N, Potocki H, Palmetzhofer T, Koller U, Illmer J, Liemberger B, Hainzl S, Klausegger A, et al. Personalized Development of Antisense Oligonucleotides for Exon Skipping Restores Type XVII Collagen Expression in Junctional Epidermolysis Bullosa. International Journal of Molecular Sciences. 2021; 22(7):3326. https://doi.org/10.3390/ijms22073326
Chicago/Turabian StyleAblinger, Michael, Thomas Lettner, Nicole Friedl, Hannah Potocki, Theresa Palmetzhofer, Ulrich Koller, Julia Illmer, Bernadette Liemberger, Stefan Hainzl, Alfred Klausegger, and et al. 2021. "Personalized Development of Antisense Oligonucleotides for Exon Skipping Restores Type XVII Collagen Expression in Junctional Epidermolysis Bullosa" International Journal of Molecular Sciences 22, no. 7: 3326. https://doi.org/10.3390/ijms22073326
APA StyleAblinger, M., Lettner, T., Friedl, N., Potocki, H., Palmetzhofer, T., Koller, U., Illmer, J., Liemberger, B., Hainzl, S., Klausegger, A., Reisenberger, M., Lambert, J., Van Gele, M., Desmet, E., Van Maelsaeke, E., Wimmer, M., Zauner, R., Bauer, J. W., & Wally, V. (2021). Personalized Development of Antisense Oligonucleotides for Exon Skipping Restores Type XVII Collagen Expression in Junctional Epidermolysis Bullosa. International Journal of Molecular Sciences, 22(7), 3326. https://doi.org/10.3390/ijms22073326