
Deletion of Trpm4 Alters the Function of the Nav1.5 Channel in Murine Cardiac Myocytes

 , , and
, , and
Abstract
:1. Introduction
2. Results
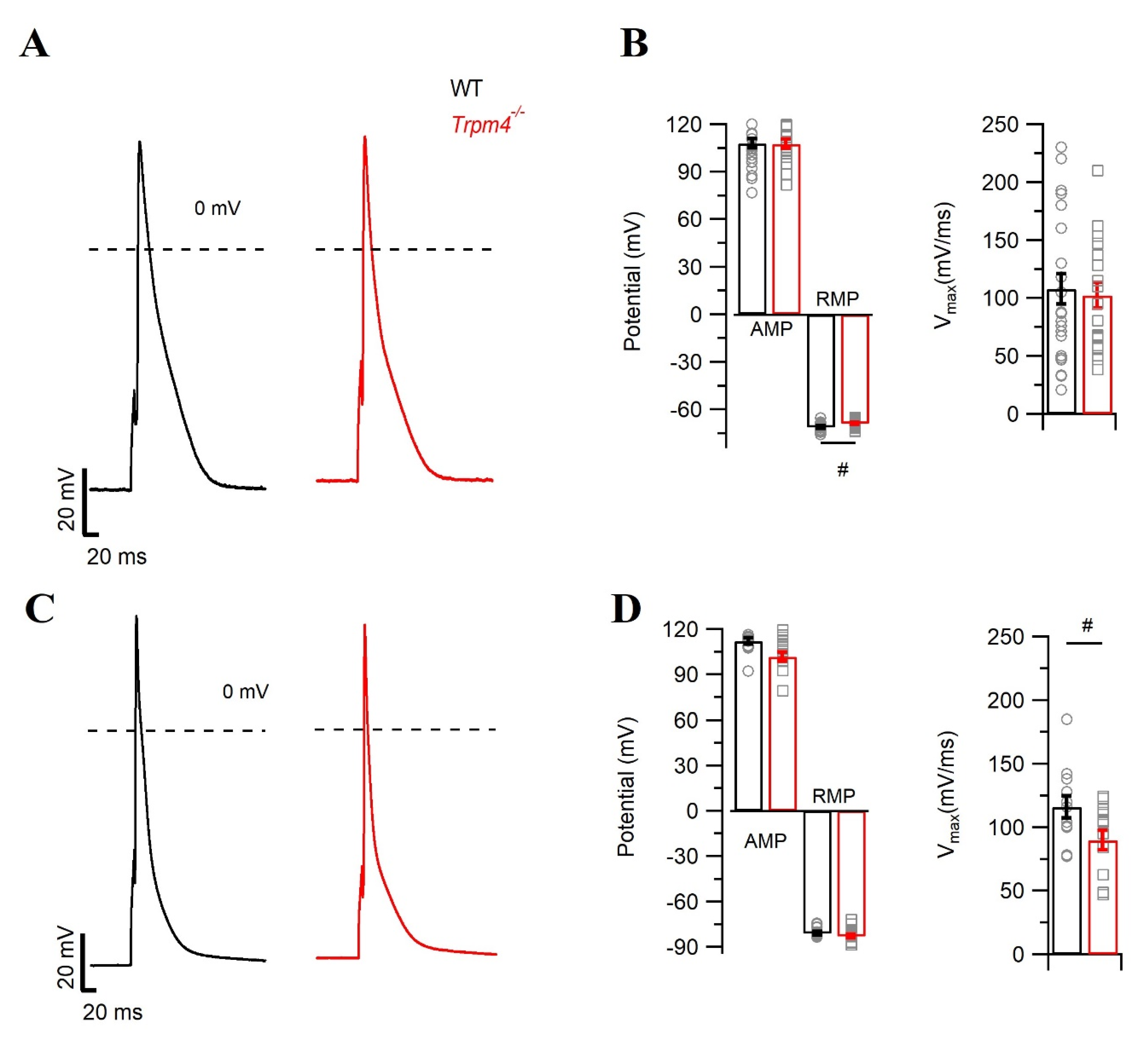
2.1. Deletion of Trpm4 Affects the Mouse Cardiac Action Potential
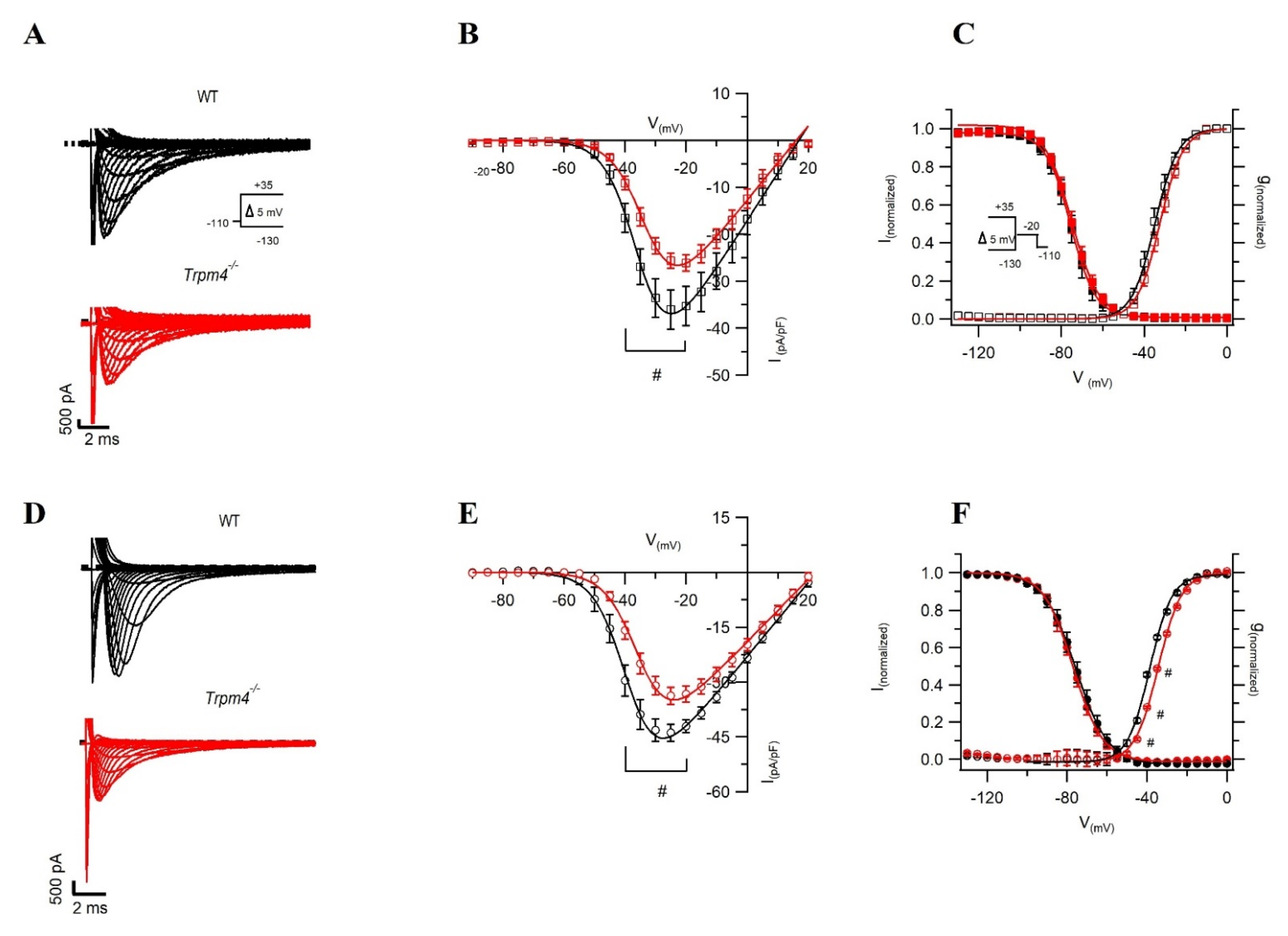
2.2. Deletion of Trpm4 Affects Na+ Currents in Atrial and Ventricular Myocytes
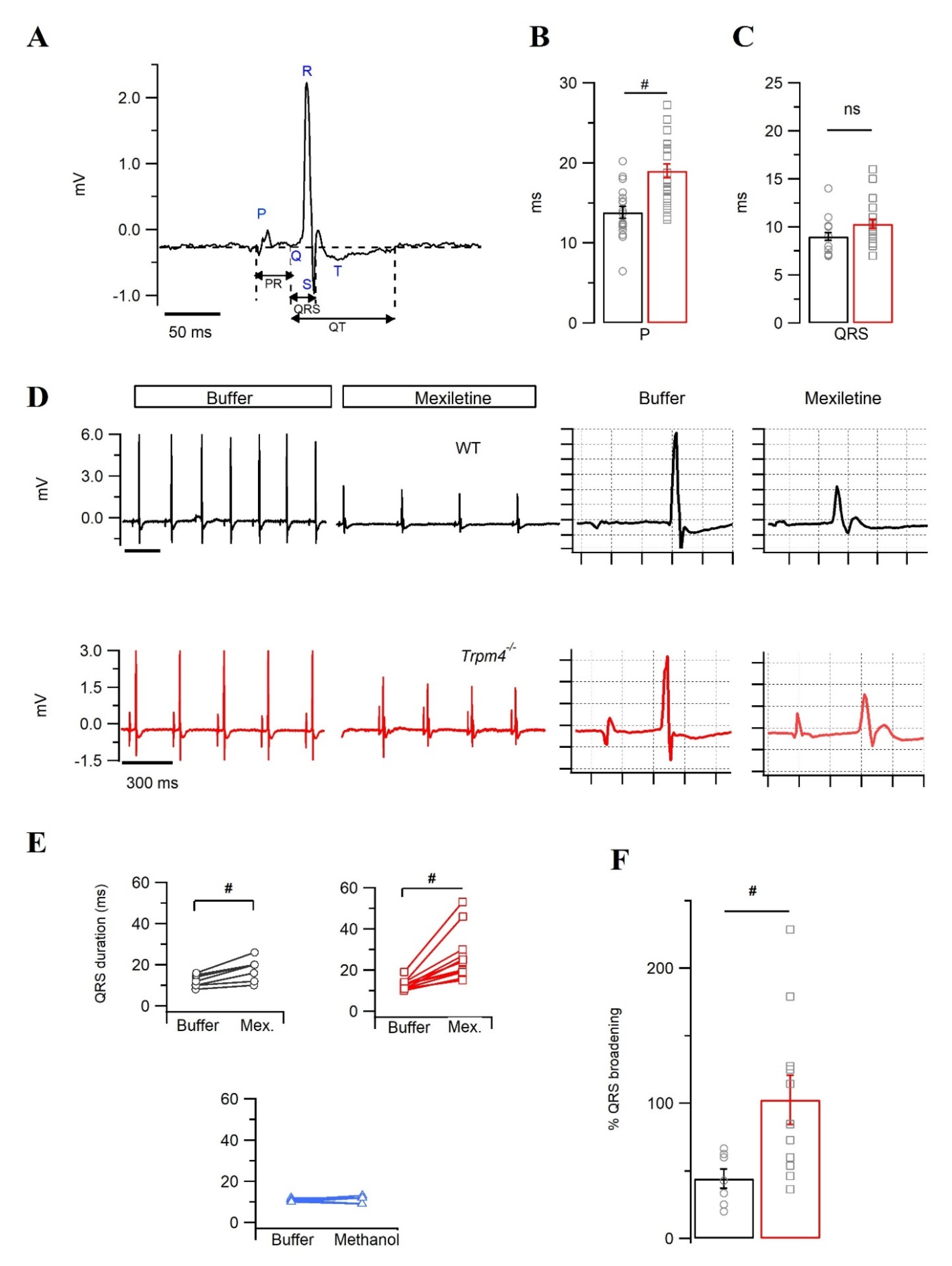
2.3. Trpm4−/− Mouse Hearts Display an Increased Sensitivity towards the Na+ Channel Inhibitor Mexiletine
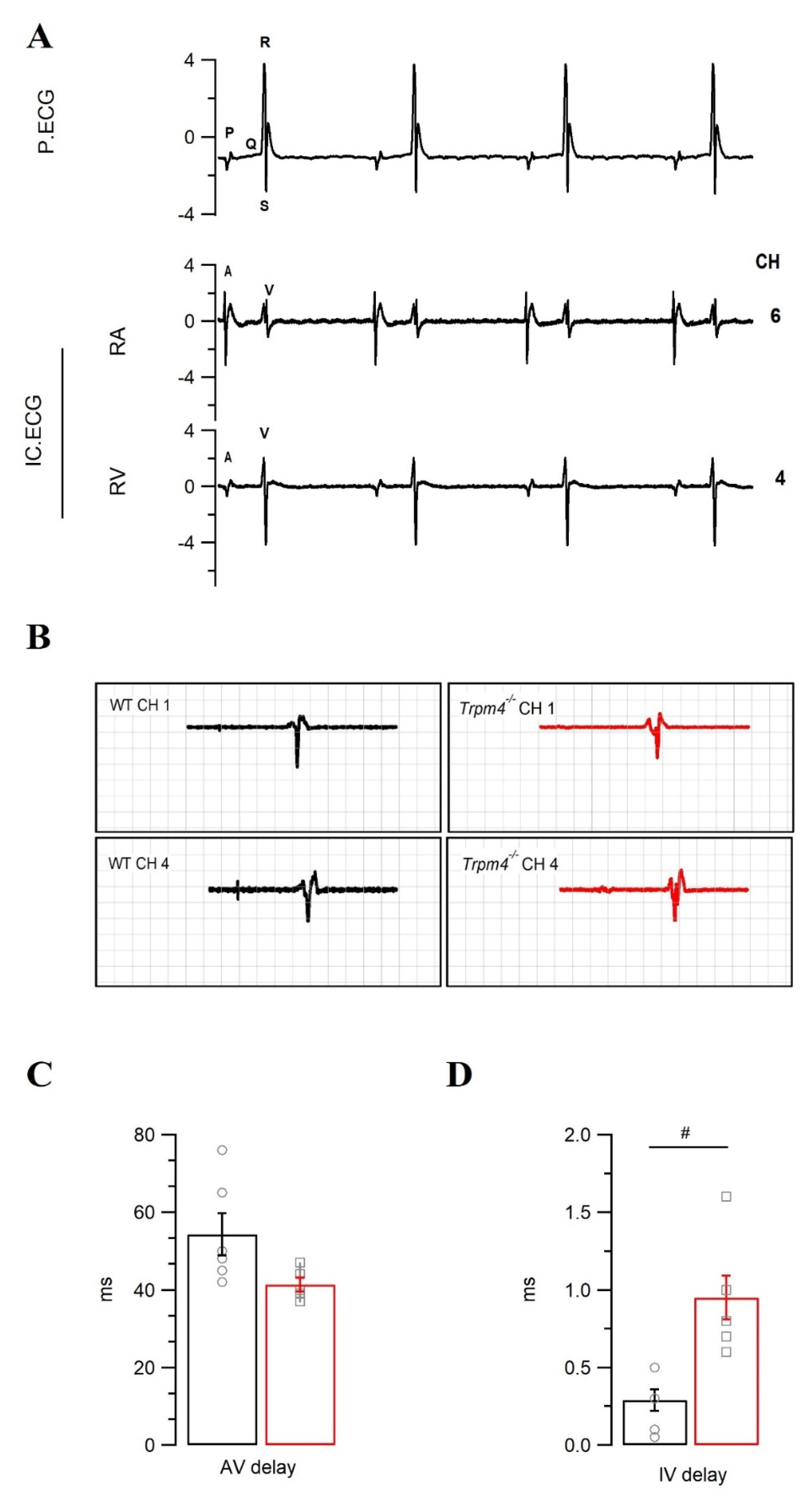
2.4. Intraventricular Conduction Is Slower in Trpm4−/− Mouse Hearts Than in WT Mouse Hearts
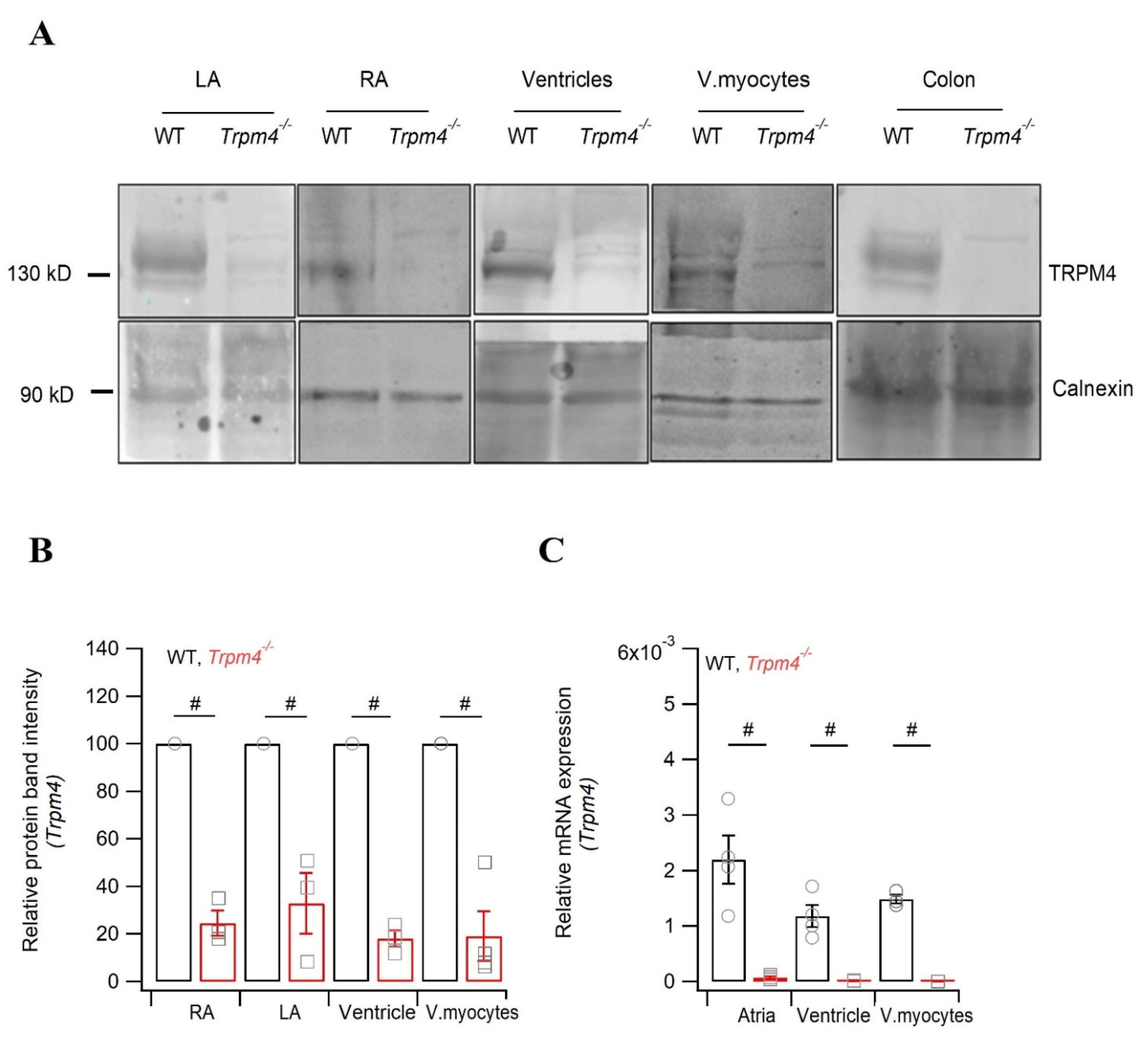
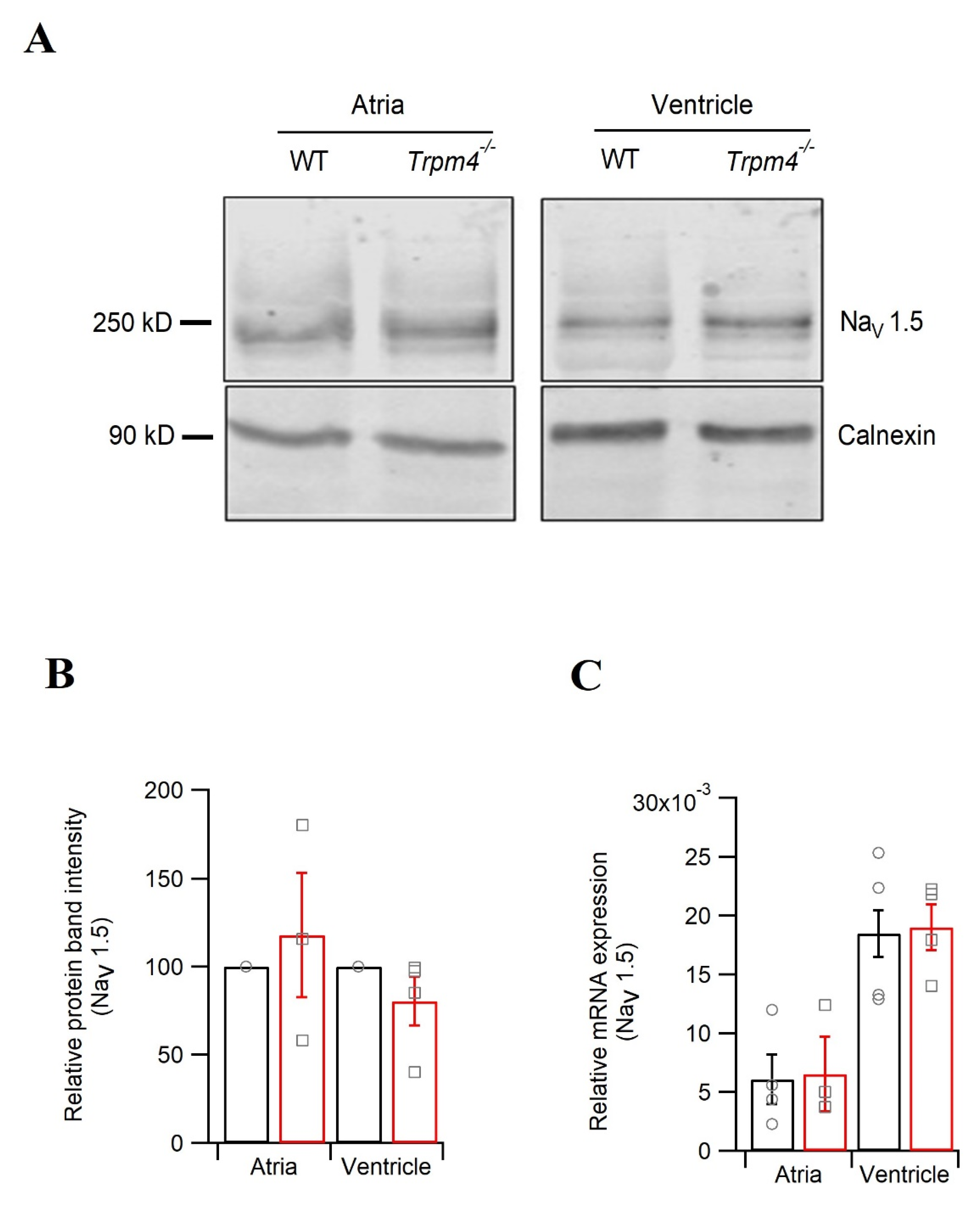
2.5. Knockdown of Trpm4 Does Not Alters Nav1.5 Expression in Mouse Ventricles
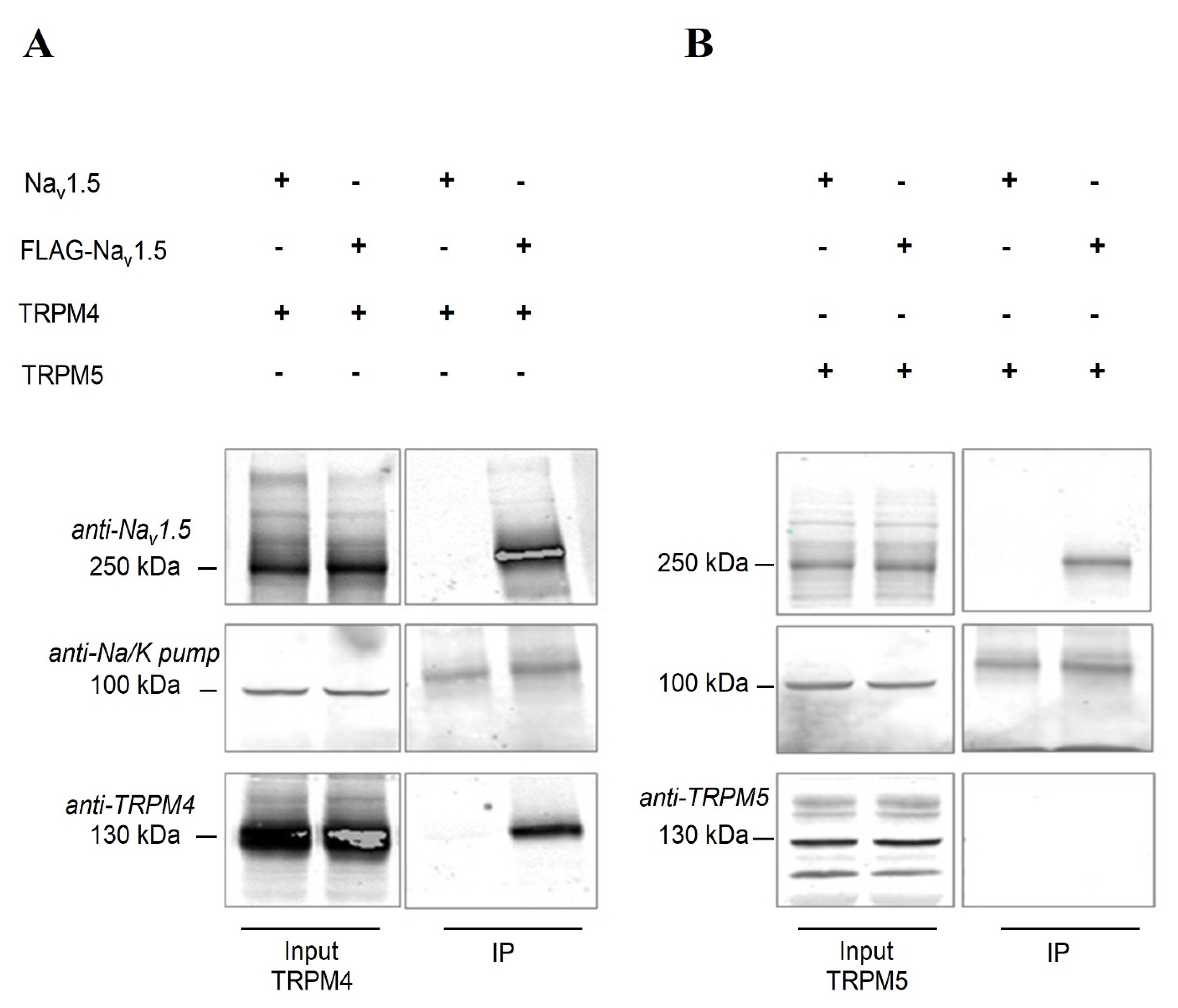
2.6. Nav1.5 Interacts with TRPM4 in an Heterologous Expression System
3. Discussion
4. Limitations
5. Materials and Methods
5.1. Trpm4−/− C57BL6/N Mice
5.2. Isolation of Atrial and Ventricular Myocytes
5.3. Single-Cell Myocyte Electrophysiology
5.3.1. Data Acquisition and Analysis
5.3.2. Action Potential Measurements
5.3.3. Voltage Clamp Recordings
5.4. Surface ECG Recordings
5.5. Pseudo- and Intracardiac ECG Recordings on Langendorff-Perfused Hearts
5.6. RNA Preparation and Quantitative RT-PCR
5.7. Western Blot
5.8. Immunoprecipitation Studies
5.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colquhoun, D.; Neher, E.; Reuter, H.; Stevens, C.F. Inward Current Channels Activated by Intracellular Ca in Cultured Cardiac Cells. Nature 1981, 294, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Launay, P.; Fleig, A.; Perraud, A.-L.; Scharenberg, A.M.; Penner, R.; Kinet, J.-P. TRPM4 Is a Ca2+-Activated Nonselective Cation Channel Mediating Cell Membrane Depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Prenen, J.; Voets, T.; Droogmans, G. Intracellular Nucleotides and Polyamines Inhibit the Ca2+-Activated Cation Channel TRPM4b. Pflug. Arch. 2004, 448, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Xu, F.; Miyoshi, I.; Sato, E.; Ono, K.; Iijima, T. Identification and Characterization of the Murine TRPM4 Channel. Biochem. Biophys. Res. Commun. 2003, 307, 522–528. [Google Scholar] [CrossRef]
- Yoo, J.C.; Yarishkin, O.V.; Hwang, E.M.; Kim, E.; Kim, D.-G.; Park, N.; Hong, S.-G.; Park, J.-Y. Cloning and Characterization of Rat Transient Receptor Potential-Melastatin 4 (TRPM4). Biochem. Biophys. Res. Commun. 2010, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Guinamard, R.; Chatelier, A.; Demion, M.; Potreau, D.; Patri, S.; Rahmati, M.; Bois, P. Functional Characterization of a Ca(2+)-Activated Non-Selective Cation Channel in Human Atrial Cardiomyocytes. J. Physiol. 2004, 558, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Amarouch, M.-Y.; Syam, N.; Abriel, H. Biochemical, Single-Channel, Whole-Cell Patch Clamp, and Pharmacological Analyses of Endogenous TRPM4 Channels in HEK293 Cells. Neurosci. Lett. 2013, 541, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Prenen, J.; Droogmans, G.; Voets, T.; Vennekens, R.; Freichel, M.; Wissenbach, U.; Flockerzi, V. Voltage Dependence of the Ca2+-Activated Cation Channel TRPM4. J. Biol. Chem. 2003, 278, 30813–30820. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Prenen, J.; Tang, J.; Wang, C.; Owsianik, G.; Janssens, A.; Voets, T.; Zhu, M.X. Regulation of the Ca2+ Sensitivity of the Nonselective Cation Channel TRPM4. J. Biol. Chem. 2005, 280, 6423–6433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, M.; Schulze-Bahr, E.; Corfield, V.; Beckmann, A.; Stallmeyer, B.; Kurtbay, G.; Ohmert, I.; Schulze-Bahr, E.; Brink, P.; Pongs, O. Impaired Endocytosis of the Ion Channel TRPM4 Is Associated with Human Progressive Familial Heart Block Type I. J. Clin. Investig. 2009, 119, 2737–2744. [Google Scholar] [CrossRef]
- Liu, H.; El Zein, L.; Kruse, M.; Guinamard, R.; Beckmann, A.; Bozio, A.; Kurtbay, G.; Mégarbané, A.; Ohmert, I.; Blaysat, G.; et al. Gain-of-Function Mutations in TRPM4 Cause Autosomal Dominant Isolated Cardiac Conduction Disease. Circ. Cardiovasc. Genet 2010, 3, 374–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Chatel, S.; Simard, C.; Syam, N.; Salle, L.; Probst, V.; Morel, J.; Millat, G.; Lopez, M.; Abriel, H.; et al. Molecular Genetics and Functional Anomalies in a Series of 248 Brugada Cases with 11 Mutations in the TRPM4 Channel. PLoS ONE 2013, 8, e54131. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, B.; Ozhathil, L.C.; Medeiros-Domingo, A.; Gollob, M.H.; Abriel, H. Four TRPM4 Cation Channel Mutations Found in Cardiac Conduction Diseases Lead to Altered Protein Stability. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Daumy, X.; Amarouch, M.-Y.; Lindenbaum, P.; Bonnaud, S.; Charpentier, E.; Bianchi, B.; Nafzger, S.; Baron, E.; Fouchard, S.; Thollet, A.; et al. Targeted Resequencing Identifies TRPM4 as a Major Gene Predisposing to Progressive Familial Heart Block Type I. Int. J. Cardiol. 2016, 207, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Nakamura, K.; Nishi, N.; Igawa, O.; Yoshida, M.; Miyoshi, T.; Watanabe, A.; Morita, H.; Ito, H. TRPM4 Mutation in Patients With Ventricular Noncompaction and Cardiac Conduction Disease. Circ. Genom. Precis. Med. 2018, 11, e002103. [Google Scholar] [CrossRef] [Green Version]
- Mathar, I.; Kecskes, M.; der Mieren, G.V.; Jacobs, G.; Londoño, J.E.C.; Uhl, S.; Flockerzi, V.; Voets, T.; Freichel, M.; Nilius, B.; et al. Increased β-Adrenergic Inotropy in Ventricular Myocardium From Trpm4−/− Mice. Circ. Res. 2014, 114, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Demion, M.; Thireau, J.; Gueffier, M.; Finan, A.; Khoueiry, Z.; Cassan, C.; Serafini, N.; Aimond, F.; Granier, M.; Pasquié, J.-L.; et al. Trpm4 Gene Invalidation Leads to Cardiac Hypertrophy and Electrophysiological Alterations. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Simard, C.; Hof, T.; Keddache, Z.; Launay, P.; Guinamard, R. The TRPM4 Non-Selective Cation Channel Contributes to the Mammalian Atrial Action Potential. J. Mol. Cell. Cardiol. 2013, 59, 11–19. [Google Scholar] [CrossRef]
- Hof, T.; Sallé, L.; Coulbault, L.; Richer, R.; Alexandre, J.; Rouet, R.; Manrique, A.; Guinamard, R. TRPM4 Non-Selective Cation Channels Influence Action Potentials in Rabbit Purkinje Fibres. J. Physiol. 2016, 594, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Simard, C.; Sallé, L.; Rouet, R.; Guinamard, R. Transient Receptor Potential Melastatin 4 Inhibitor 9-Phenanthrol Abolishes Arrhythmias Induced by Hypoxia and Re-Oxygenation in Mouse Ventricle. Br. J. Pharm. 2012, 165, 2354–2364. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Fei, Y.; Li, W.; Chen, Y.; Wang, Q.; Xiao, Y.; Wang, Y.; Li, Y. The Transient Receptor Potential Melastatin 4 Channel Inhibitor 9-Phenanthrol Modulates Cardiac Sodium Channel. Br. J. Pharmacol. 2018, 175, 4325–4337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veress, R.; Baranyai, D.; Hegyi, B.; Kistamás, K.; Dienes, C.; Magyar, J.; Bányász, T.; Nánási, P.P.; Szentandrássy, N.; Horváth, B. Transient Receptor Potential Melastatin 4 Channel Inhibitor 9-Phenanthrol Inhibits K+ but Not Ca2+ Currents in Canine Ventricular Myocytes. Can. J. Physiol. Pharm. 2018, 96, 1022–1029. [Google Scholar] [CrossRef]
- Burris, S.K.; Wang, Q.; Bulley, S.; Neeb, Z.P.; Jaggar, J.H. 9-Phenanthrol Inhibits Recombinant and Arterial Myocyte TMEM16A Channels. Br. J. Pharm. 2015, 172, 2459–2468. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Naruse, K.; Takahashi, K. Role of the TRPM4 Channel in Cardiovascular Physiology and Pathophysiology. Cells 2018, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Mathar, I.; Vennekens, R.; Meissner, M.; Kees, F.; Van der Mieren, G.; Camacho Londoño, J.E.; Uhl, S.; Voets, T.; Hummel, B.; van den Bergh, A.; et al. Increased Catecholamine Secretion Contributes to Hypertension in TRPM4-Deficient Mice. J. Clin. Investig. 2010, 120, 3267–3279. [Google Scholar] [CrossRef] [Green Version]
- Vennekens, R.; Olausson, J.; Meissner, M.; Bloch, W.; Mathar, I.; Philipp, S.E.; Schmitz, F.; Weissgerber, P.; Nilius, B.; Flockerzi, V.; et al. Increased IgE-Dependent Mast Cell Activation and Anaphylactic Responses in Mice Lacking the Calcium-Activated Nonselective Cation Channel TRPM4. Nat. Immunol. 2007, 8, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Sunami, A.; Fan, Z.; Sawanobori, T.; Hiraoka, M. Use-Dependent Block of Na+ Currents by Mexiletine at the Single Channel Level in Guinea-Pig Ventricular Myocytes. Br. J. Pharm. 1993, 110, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Gutstein, D.E.; Morley, G.E.; Tamaddon, H.; Vaidya, D.; Schneider, M.D.; Chen, J.; Chien, K.R.; Stuhlmann, H.; Fishman, G.I. Conduction Slowing and Sudden Arrhythmic Death in Mice With Cardiac-Restricted Inactivation of Connexin43. Circ. Res. 2001, 88, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Guinamard, R.; Demion, M.; Magaud, C.; Potreau, D.; Bois, P. Functional Expression of the TRPM4 Cationic Current in Ventricular Cardiomyocytes from Spontaneously Hypertensive Rats. Hypertension 2006, 48, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Grand, T.; Demion, M.; Norez, C.; Mettey, Y.; Launay, P.; Becq, F.; Bois, P.; Guinamard, R. 9-Phenanthrol Inhibits Human TRPM4 but Not TRPM5 Cationic Channels. Br. J. Pharm. 2008, 153, 1697–1705. [Google Scholar] [CrossRef] [Green Version]
- Guinamard, R.; Hof, T.; Del Negro, C.A. The TRPM4 Channel Inhibitor 9-Phenanthrol. Br. J. Pharm. 2014, 171, 1600–1613. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.M. Rudy Yoram Ionic Mechanisms of Propagation in Cardiac Tissue. Circ. Res. 1997, 81, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Kléber, A.G.; Rudy, Y. Basic Mechanisms of Cardiac Impulse Propagation and Associated Arrhythmias. Physiol. Rev. 2004, 84, 431–488. [Google Scholar] [CrossRef] [Green Version]
- Gavillet, B.; Rougier, J.S.; Domenighetti, A.A.; Behar, R.; Boixel, C.; Ruchat, P.; Lehr, H.A.; Pedrazzini, T.; Abriel, H. Cardiac Sodium Channel Nav1.5 Is Regulated by a Multiprotein Complex Composed of Syntrophins and Dystrophin. Circ. Res. 2006, 99, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hof, T.; Simard, C.; Rouet, R.; Sallé, L.; Guinamard, R. Implication of the TRPM4 Nonselective Cation Channel in Mammalian Sinus Rhythm. Heart Rhythm. 2013, 10, 1683–1689. [Google Scholar] [CrossRef]
- Mironov, S.L.; Skorova, E.Y. Stimulation of Bursting in Pre-Bötzinger Neurons by Epac through Calcium Release and Modulation of TRPM4 and K-ATP Channels. J. Neurochem. 2011, 117, 295–308. [Google Scholar] [CrossRef]
- Guinamard, R.; Bouvagnet, P.; Hof, T.; Liu, H.; Simard, C.; Sallé, L. TRPM4 in Cardiac Electrical Activity. Cardiovasc. Res. 2015, 108, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponce-Balbuena, D.; Guerrero-Serna, G.; Valdivia, C.R.; Caballero, R.; Diez-Guerra, F.J.; Jiménez-Vázquez, E.N.; Ramírez, R.J.; Monteiro da Rocha, A.; Herron, T.J.; Campbell, K.F.; et al. Cardiac Kir2.1 and NaV1.5 Channels Traffic Together to the Sarcolemma to Control Excitability. Circ. Res. 2018, 122, 1501–1516. [Google Scholar] [CrossRef]
- Matamoros, M.; Pérez-Hernández, M.; Guerrero-Serna, G.; Amorós, I.; Barana, A.; Núñez, M.; Ponce-Balbuena, D.; Sacristán, S.; Gómez, R.; Tamargo, J.; et al. Nav1.5 N-Terminal Domain Binding to A1-Syntrophin Increases Membrane Density of Human Kir2.1, Kir2.2 and Nav1.5 Channels. Cardiovasc. Res. 2016, 110, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Utrilla, R.G.; Nieto-Marín, P.; Alfayate, S.; Tinaquero, D.; Matamoros, M.; Pérez-Hernández, M.; Sacristán, S.; Ondo, L.; de Andrés, R.; Díez-Guerra, F.J.; et al. Kir2.1-Nav1.5 Channel Complexes Are Differently Regulated than Kir2.1 and Nav1.5 Channels Alone. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- A Microtranslatome Coordinately Regulates Sodium and Potassium Currents in the Human Heart|ELife. Available online: https://elifesciences.org/articles/52654 (accessed on 14 February 2020).
- Duthoit, G.; Fressart, V.; Hidden-Lucet, F.; Simon, F.; Kattygnarath, D.; Charron, P.; Himbert, C.; Aouate, P.; Guicheney, P.; Lecarpentier, Y.; et al. Brugada ECG Pattern: A Physiopathological Prospective Study Based on Clinical, Electrophysiological, Angiographic, and Genetic Findings. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, G.F.; Jeron, A.; Koren, G. Measurement of Heart Rate and Q-T Interval in the Conscious Mouse. Am. J. Physiol. Heart Circ. Physiol. 1998, 274, H747–H751. [Google Scholar] [CrossRef] [PubMed]
- Boukens, B.J.; Hoogendijk, M.G.; Verkerk, A.O.; Linnenbank, A.; van Dam, P.; Remme, C.-A.; Fiolet, J.W.; Opthof, T.; Christoffels, V.M.; Coronel, R. Early Repolarization in Mice Causes Overestimation of Ventricular Activation Time by the QRS Duration. Cardiovasc. Res. 2013, 97, 182–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, B.; Smith, P.A.; Abriel, H. The Ion Channel TRPM4 in Murine Experimental Autoimmune Encephalomyelitis and in a Model of Glutamate-Induced Neuronal Degeneration. Mol. Brain 2018, 11, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atrial | Ventricular | |||
|---|---|---|---|---|
| WT | Trpm4−/− | WT | Trpm4−/− | |
| RMP | −71.0 ± 3.6 | −68.9 ± 2.5 # | −81.1 ± 4.6 | −82.9 ± 5.1 |
| AMP | 107.0 ± 14.2 | 106.5 ± 12.3 | 112.0 ± 7.8 | 101.0 ± 11.5 |
| Vmax (mV/ms) | 107.7 ± 64.0 | 102.2 ± 48.0 | 115.8 ± 30.0 | 89.7 ± 28.3 # |
| APD 30 | 8.8 ± 4.3 | 9.0 ± 5.7 | 4.3 ± 3.2 | 4.7 ± 2.2 |
| APD 50 | 18.0 ± 8.7 | 18.5 ± 11.8 | 8.5 ± 6.0 | 8.6 ± 3.9 |
| APD 90 | 71.5 ± 19.7 | 77.8 ± 26.9 | 58.1 ± 30.3 | 52.7 ± 12.4 |
| Atrial | Ventricular | |||
|---|---|---|---|---|
| WT | Trpm4−/− | WT | Trpm4−/− | |
| Cm (pF) | 62.2 ± 3.8 | 50.8 ± 2.4 # | 187.7 ± 16.7 | 164.5 ± 10.9 |
| INa-peak density (pA/pF) | −48.0 ± 2.3 | −36.3 ± 2.6 # | −36.0 ± 4.4 | −25.6 ± 1.6 # |
| V1/2 of activation (mV) | −38.6 ± 1.4 | −34.3 ± 0.9 # | −34.7 ± 0.1 | −32.4 ± 0.1 |
| k of activation | 4.0 ± 0.2 | 5.2 ± 0.2 # | 5.6 ± 0.1 | 5.5 ± 0.1 |
| V1/2 of inactivation (mV) | −75.0 ± 1.9 | −76.2 ± 1.4 | −75.5 ± 0.1 | −74.9 ± 0.9 |
| k of inactivation | 6.2 ± 0.1 | 6.1 ± 0.1 | 6.0 ± 0.1 | 6.6 ± 0.1 |
| Surface ECG | Pseudo-ECG | |||
|---|---|---|---|---|
| WT | Trpm4−/− | WT | Trpm4−/− | |
| P dur. (ms) | 12.1 ± 2.1 | 14.1 ±2.0 | 13.8 ± 0.7 | 19.0 ± 0.9 # |
| PR (ms) | 47.1 ± 0.9 | 43.3 ± 2.6 | 45.4 ± 2.2 | 48.8 ± 3.0 |
| QRS (ms) | 10.2 ± 0.2 | 10.4 ± 0.5 | 9.1 ± 0.4 | 10.3 ± 0.5 |
| QT (ms) | 56.3 ± 2.1 | 65.1 ± 4.1 | 80.5 ± 12.4 | 99.1 ± 10.3 |
| QTc (ms) | 43.7 ± 2.9 | 44.2 ± 3.3 | 52.0 ± 7.3 | 61.8 ± 6.6 |
| HR (bpm) | 361 ± 20.0 | 280 ± 19.0 # | 246 ± 11.9 | 230 ± 8.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozhathil, L.C.; Rougier, J.-S.; Arullampalam, P.; Essers, M.C.; Ross-Kaschitza, D.; Abriel, H. Deletion of Trpm4 Alters the Function of the Nav1.5 Channel in Murine Cardiac Myocytes. Int. J. Mol. Sci. 2021, 22, 3401. https://doi.org/10.3390/ijms22073401
Ozhathil LC, Rougier J-S, Arullampalam P, Essers MC, Ross-Kaschitza D, Abriel H. Deletion of Trpm4 Alters the Function of the Nav1.5 Channel in Murine Cardiac Myocytes. International Journal of Molecular Sciences. 2021; 22(7):3401. https://doi.org/10.3390/ijms22073401
Chicago/Turabian StyleOzhathil, Lijo Cherian, Jean-Sébastien Rougier, Prakash Arullampalam, Maria C. Essers, Daniela Ross-Kaschitza, and Hugues Abriel. 2021. "Deletion of Trpm4 Alters the Function of the Nav1.5 Channel in Murine Cardiac Myocytes" International Journal of Molecular Sciences 22, no. 7: 3401. https://doi.org/10.3390/ijms22073401
APA StyleOzhathil, L. C., Rougier, J. -S., Arullampalam, P., Essers, M. C., Ross-Kaschitza, D., & Abriel, H. (2021). Deletion of Trpm4 Alters the Function of the Nav1.5 Channel in Murine Cardiac Myocytes. International Journal of Molecular Sciences, 22(7), 3401. https://doi.org/10.3390/ijms22073401