
DFT Studies of Selected Epoxies with Mesogenic Units–Impact of Molecular Structure on Electro-Optical Response


Abstract
:1. Introduction
2. Results and Discussion
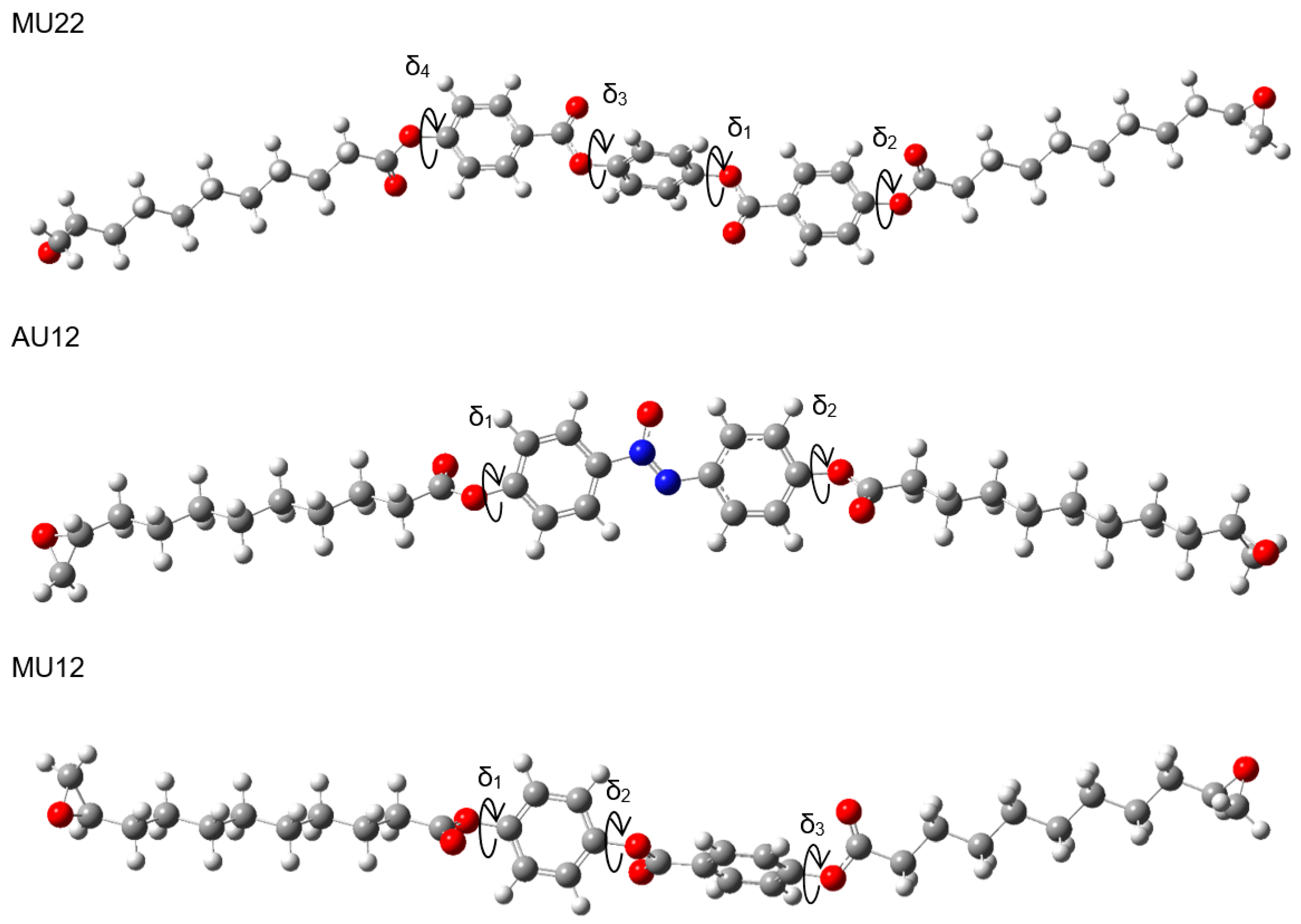
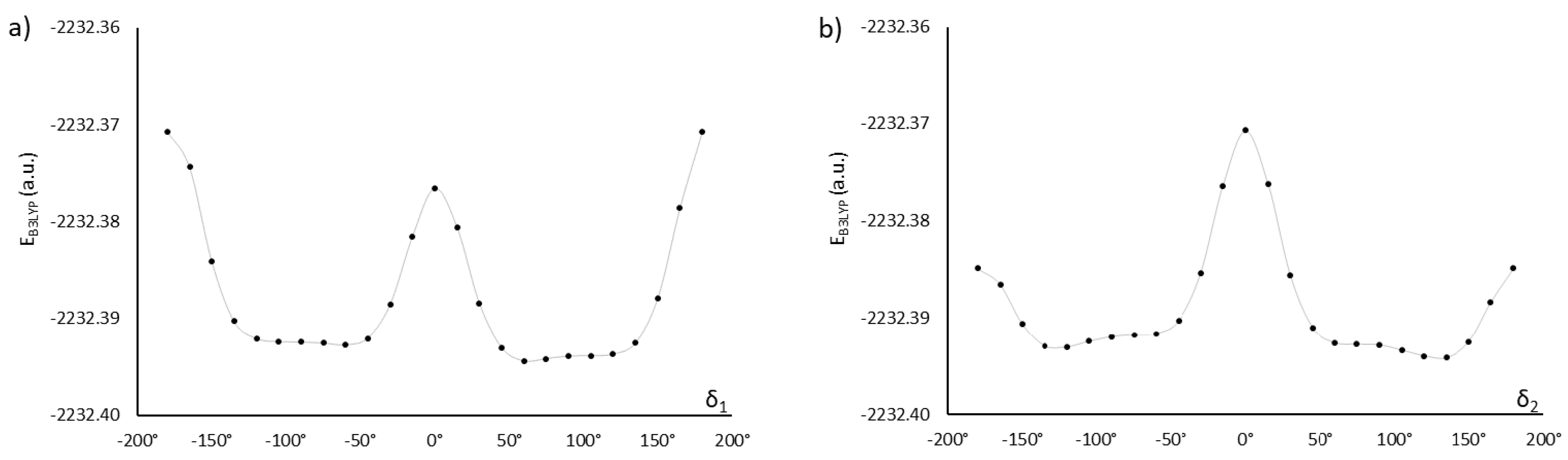
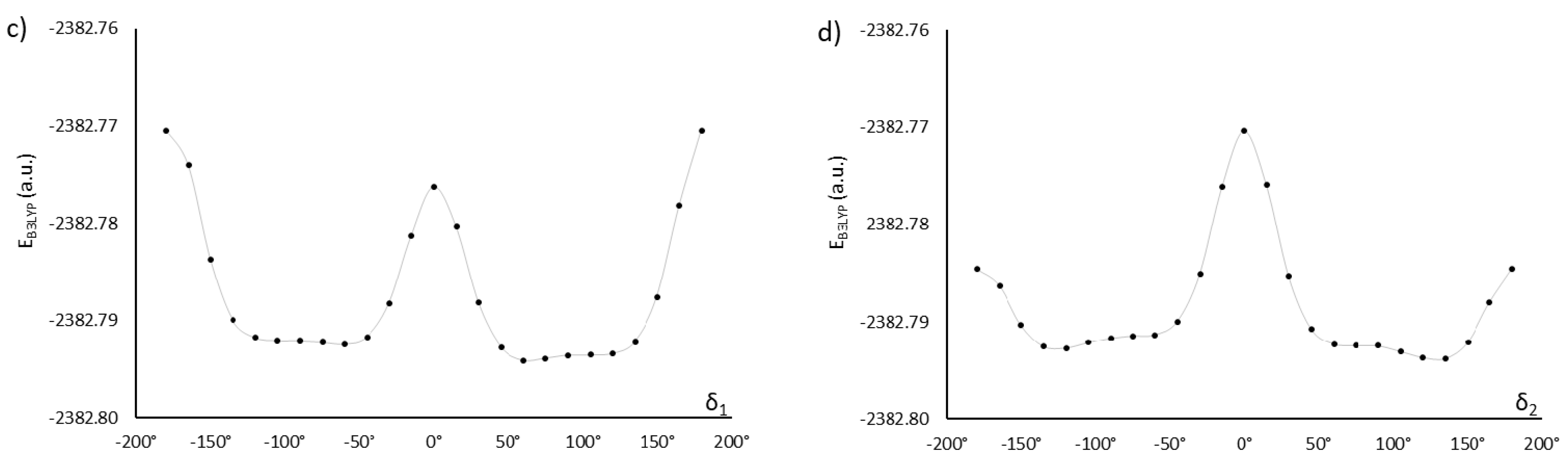
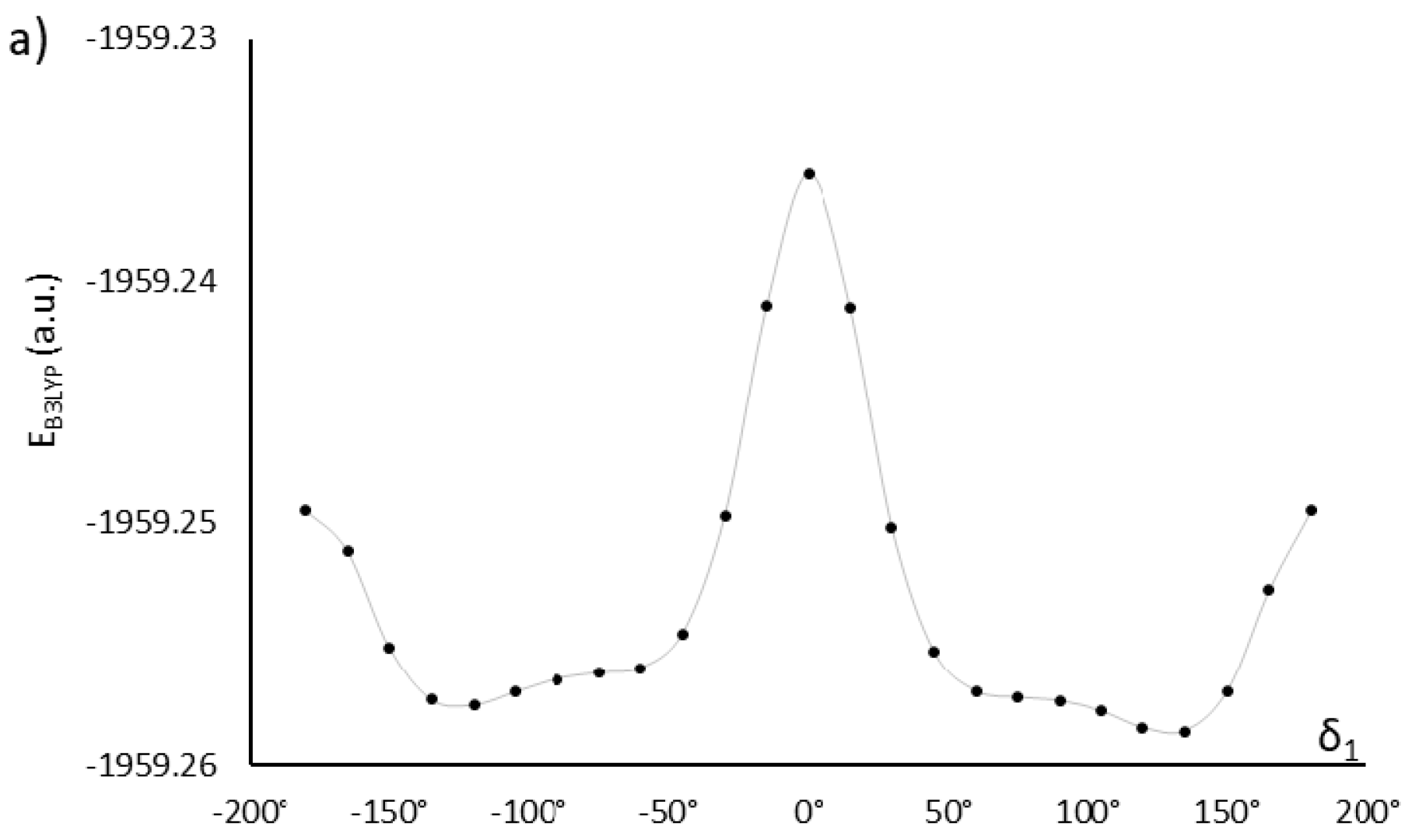
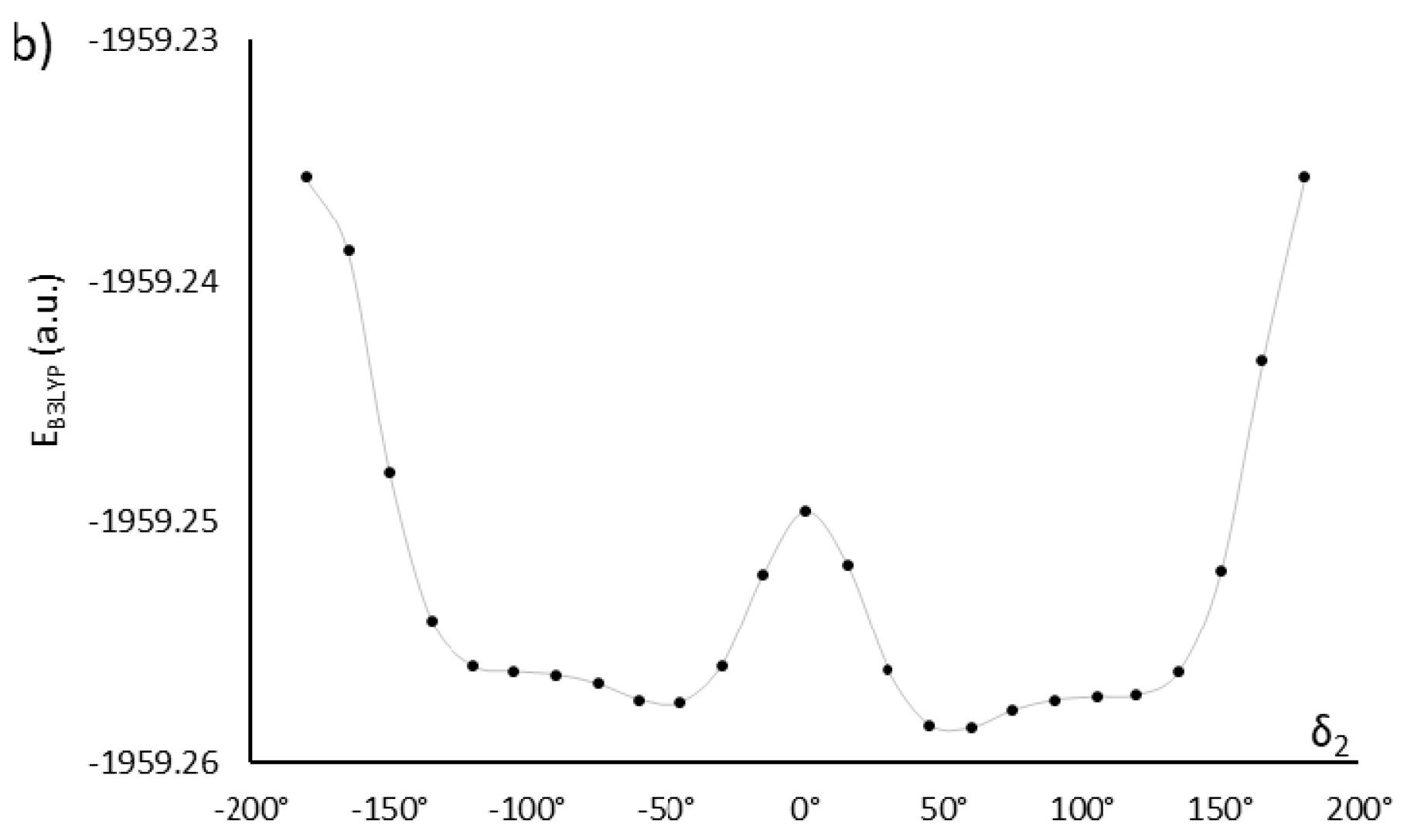
2.1. Choosing the Best Conformation—DFT Optimization
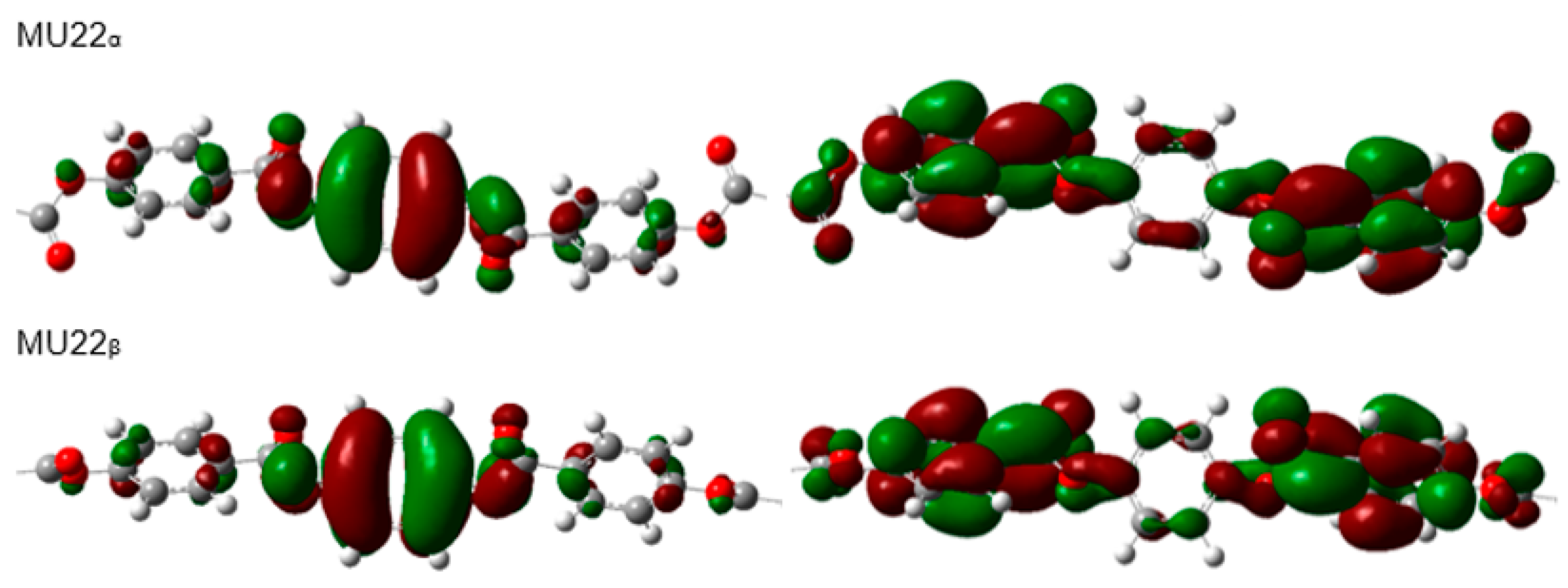
2.2. Electronic Structure and Stability
2.3. Non-Linear Properties
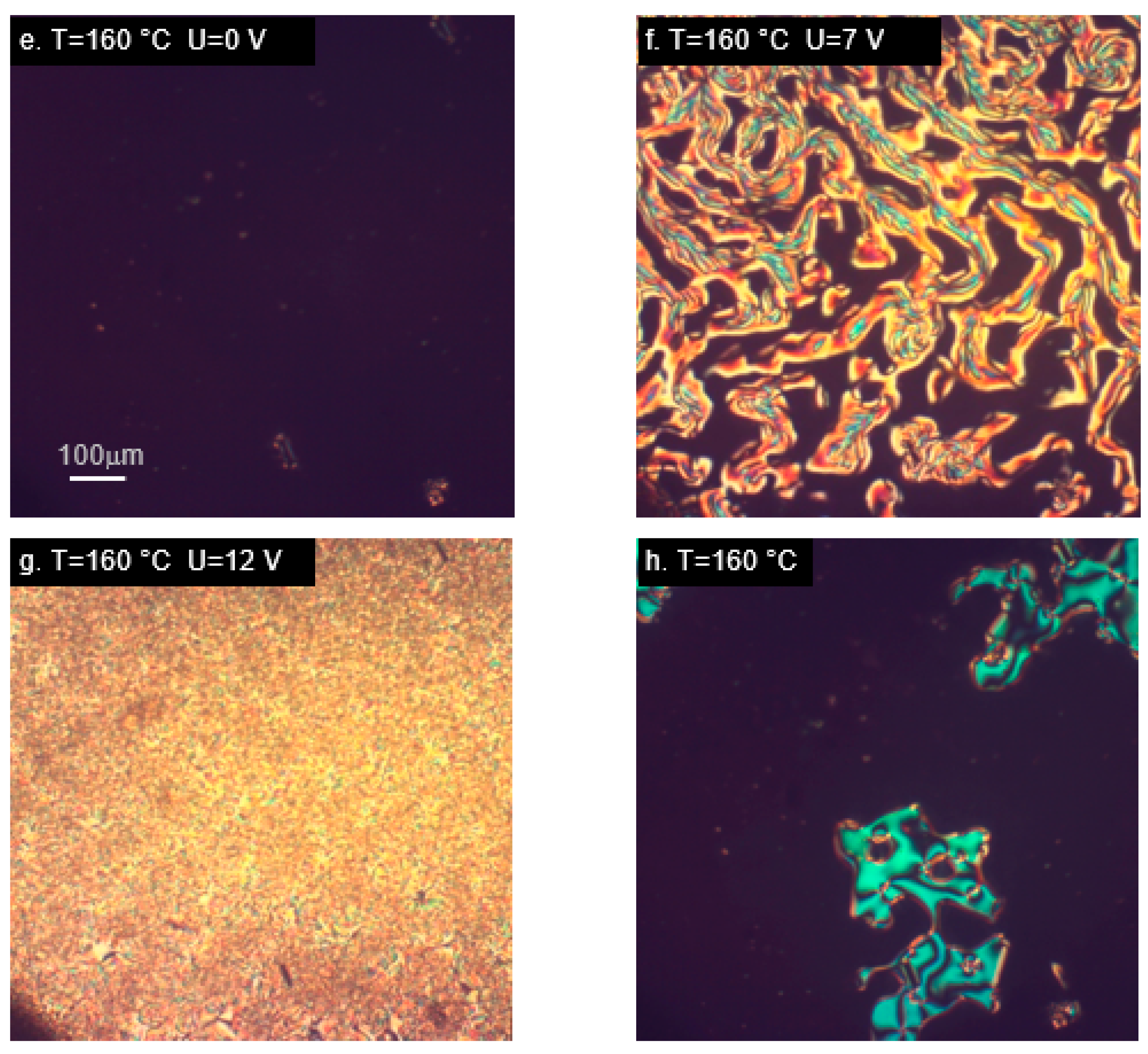
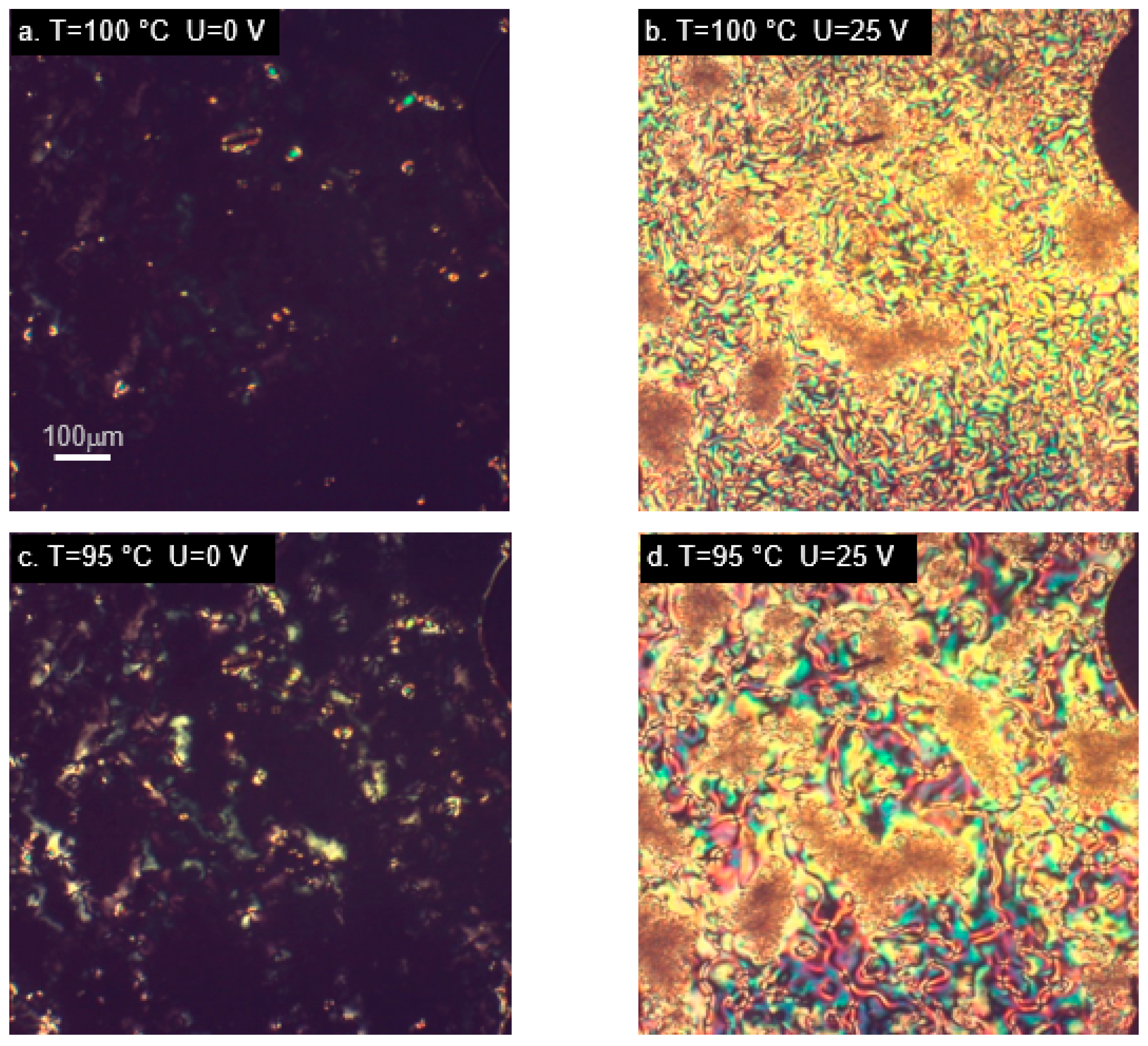
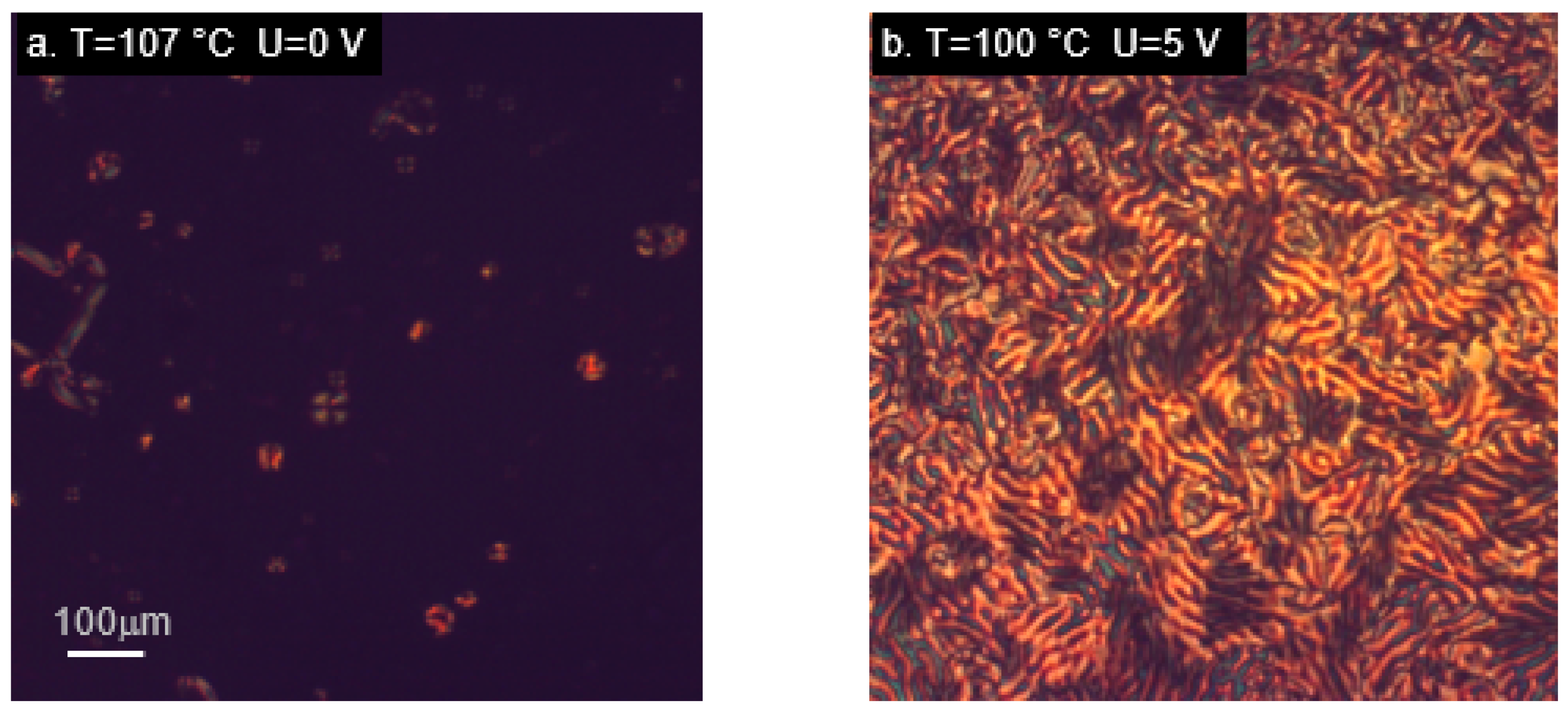
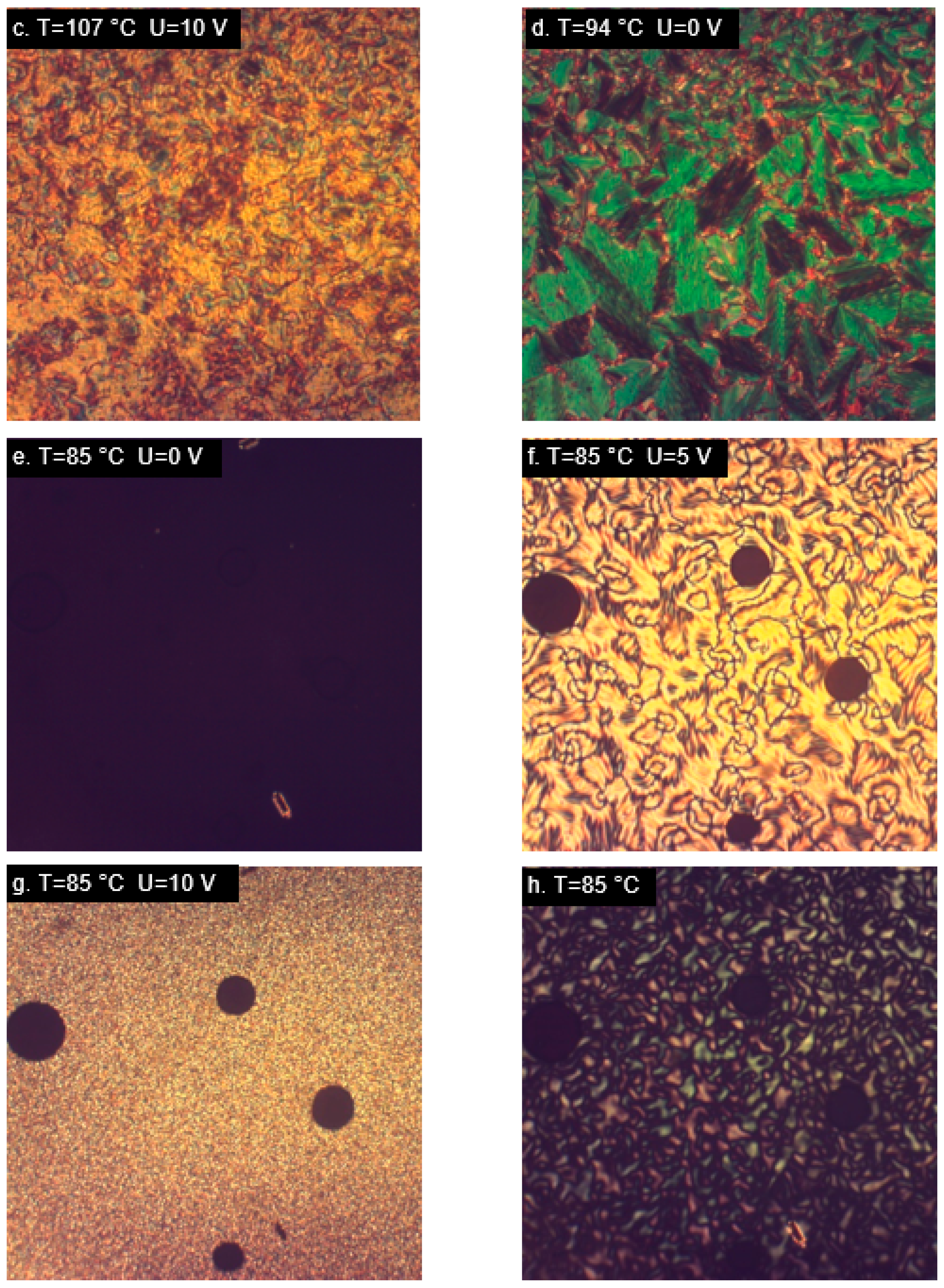
2.4. Orientation in the Electric Field—Experimental Results
3. Materials and Methods
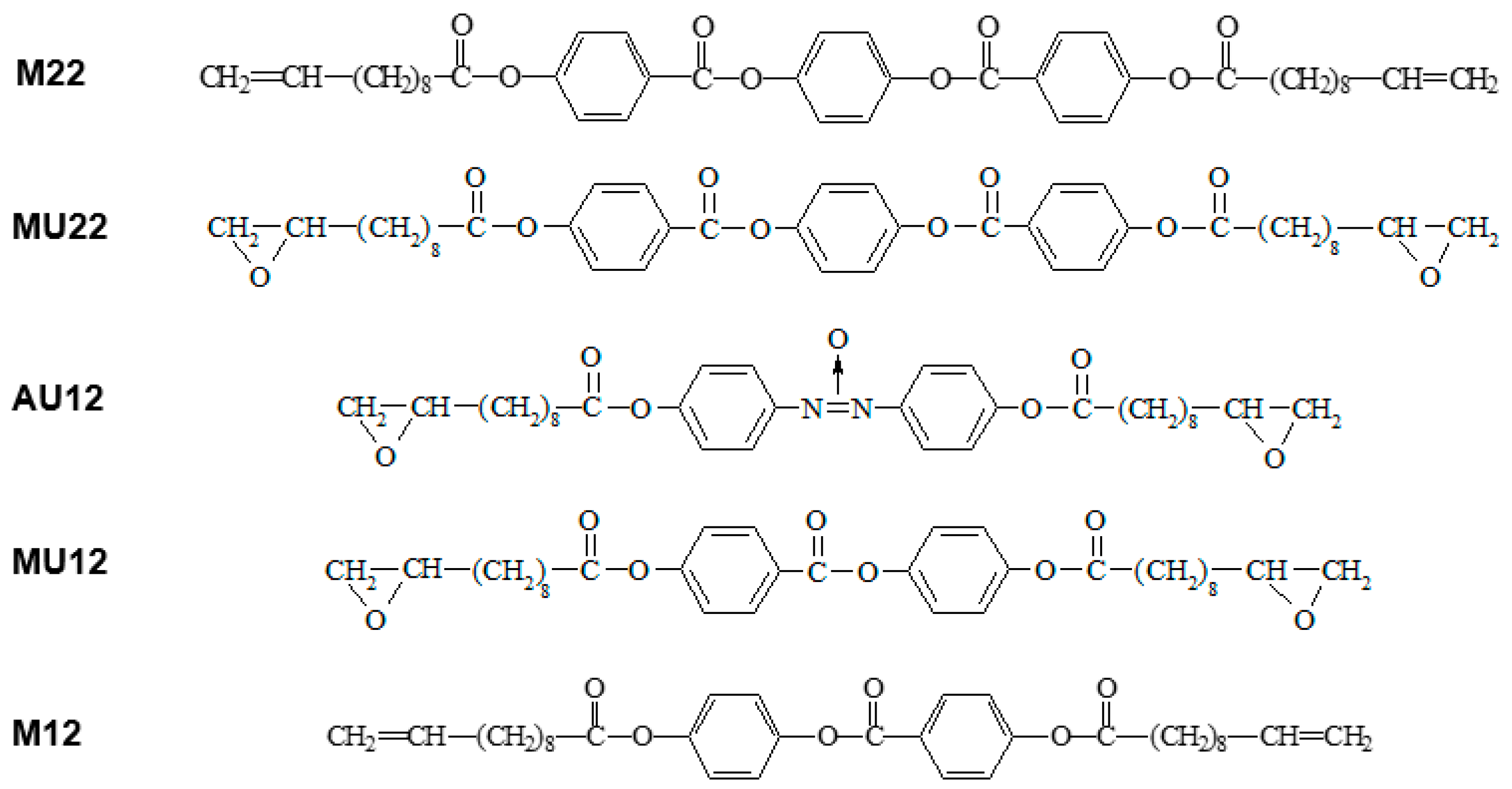
3.1. Materials
3.2. Analytic Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pintre, I.C.; Serrano, J.L.; Ros, M.B.; Martinez-Perdiguero, J.; Alonso, I.; Ortega, J.; Folcia, C.L.; Etxebarria, J.; Alicante, R.; Villacampa, B. Bent-core liquid crystals in a route to efficient organic nonlinear optical materials. J. Mater. Chem. 2010, 20, 2965. [Google Scholar] [CrossRef]
- Miniewicz, A.; Girones, J.; Karpinski, P.; Mossety-Leszczak, B.; Galina, H.; Dutkiewicz, M. Photochromic and nonlinear optical properties of azo-functionalized POSS nanoparticles dispersed in nematic liquid crystals. J. Mater. Chem. C 2014, 2, 432. [Google Scholar] [CrossRef]
- Chen, H.W.; Lee, J.H.; Lin, B.Y.; Chen, S.; Wu, S.T. Liquid crystal display and organic light-emitting diode display: Present status and future perspectives. Light Sci. Appl. 2018, 7, 17168. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.K.; Chen, W.H.; Li, C.Y.; Cheng, K.T. High-Contrast and Scattering-Type Transflective Liquid Crystal Displays Based on Polymer-Network Liquid Crystals. Polymers 2020, 12, 739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossety-Leszczak, B.; Włodarska, M. Liquid-crystalline epoxy thermosets as matrices for ordered nanocomposites—a summary of experimental studies. Polym. Compos. 2017, 38, 277. [Google Scholar] [CrossRef]
- Mossety-Leszczak, B.; Kisiel, M.; Szałański, P.; Włodarska, M.; Szeluga, U.; Pusz, S. The Influence of a Magnetic Field on the Morphology and Thermomechanical Properties of a Liquid Crystalline Epoxy Carbon Composite. Polym. Compos. 2018, 39, E2573. [Google Scholar] [CrossRef]
- Miniewicz, A.; Tomkowicz, M.; Karpinski, P.; Sznitko, L.; Mossety-Leszczak, B.; Dutkiewicz, M. Light sensitive polymer obtained by dispersion of azo-functionalized POSS nanoparticles. Chem. Phys. 2015, 456, 65. [Google Scholar] [CrossRef]
- Gray, G.W.; Winsor, P.A. Liquid Crystals & Plastic Crystals; Ellis Horwood Ltd.: Chichester, UK, 1974; Volume 1. [Google Scholar]
- De Jeu, W.H. Physical Properties of Liquid Crystalline Materials; Gordon and Breach Science Publishers: London, UK, 1980. [Google Scholar]
- De Gennes, P.G. The Physics of Liquid Crystals; Clarendon Press: Oxford, UK, 1974. [Google Scholar]
- Melnyk, O.; Garbovskiy, Y.; Bueno- Baques, D.; Glushchenko, A. Electro-Optical Switching of Dual-Frequency Nematic Liquid Crystals: Regimes of Thin and Thick Cells. Crystals 2019, 9, 314. [Google Scholar] [CrossRef] [Green Version]
- Vimal, T.; Agrahari, K.; Sonker, R.K.; Manohar, R. Investigation of thermodynamical, dielectric and electro-optical parameters of nematic liquid crystal doped with polyaniline and silver nanoparticles. J. Mol. Liquids 2019, 290, 111241. [Google Scholar] [CrossRef]
- Khalid, M.; Hussain, R.; Hussain, A.; Ali, B.; Jaleel, F.; Imran, M.; Assiri, M.A.; Khan, M.U.; Ahmed, S.; Abid, S.S.; et al. Electron Donor and Acceptor Influence on the Nonlinear Optical Response of Diacetylene-Functionalized Organic Materials (DFOMs): Density Functional Theory Calculations. Molecules 2019, 24, 2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiran, A.J.; Kim, H.C.; Kim, K.; Rotermund, F.; Ravindra, H.J.; Dharmaprakash, S.M.; Lim, H. Superior characteristics of organic chalcone single crystals as efficient nonlinear optical material. Appl. Phys. Lett. 2008, 92, 113307. [Google Scholar] [CrossRef]
- Kawamata, J.; Inoue, K.; Inabe, T. Salient nonlinear optical properties of novel organic crystals comprising π-conjugated ketones. Appl. Phys. Lett. 1995, 66, 3102. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Q.; Zhang, W.; Li, Y.; Huang, J.; Wei, C.; Tang, Z.; Wang, L.; Yu, G. Synthesis, properties, and charge transport characteristics of the conjugated copolymers containing the azaisoindigo and benzothiadiazole units. Dyes Pigment. 2020, 180, 108438. [Google Scholar] [CrossRef]
- Włodarska, M. Dipole moment calculation in solution for some liquid crystalline molecules. J. Mol. Struct. 2014, 1059, 44. [Google Scholar] [CrossRef]
- Johri, G.K. A New Method of Calculation of Dipole Moment and Relaxation Time for Dilute Solutions at Microwave Frequency. J. Phys. Soc. 1984, 53, 802. [Google Scholar] [CrossRef]
- Smyth, C.P. Dielectric Behaviour and Structure; McGraw Hill Book Co., Inc.: New York, NY, USA, 1955. [Google Scholar]
- Kuze, N.; Ebizuka, M.; Fujiwara, H.; Takeuchi, H.; Egawa, T.; Konaka, S.; Fogarasi, G. Molecular Structure of p-Azoxyanisole, a Mesogen, Determined by Gas-Phase Electron Diffraction Augmented by ab Initio Calculations. J. Phys. Chem. A 1998, 102, 2080. [Google Scholar] [CrossRef]
- Janbon, S.; Davey, R.J.; Shankland, K. The crystal structure of a metastable polymorph of para-azoxyanisole. Cryst. Eng. Comm. 2008, 10, 279. [Google Scholar] [CrossRef]
- Scridonesi, V.C. The MO-SCF Quantum approach of the p-azoxyanisole (PPA) molecule I. Quantum energetic molecular characteristics. An. Univ. Bucuresti Chim. 2005, I–II, 407. [Google Scholar]
- Dwivedi, A.K.; Pal, B.; Kumar, N.; Kumar, A.; Singh, P.; Thapa, K.B.; Singh, D. Spectroscopic Analysis of PAA (paraazoxy anisole) liquid crystal molecule studied by DFT Methodology. Infokara Res. 2019, 8, 76. [Google Scholar]
- Włodarska, M.; Mossety-Leszczak, B. Phase transitions and dielectric properties in a symmetric liquid crystalline compound with central triaromatic group. Eur. Phys. J. Appl. Phys. 2017, 79, 10202. [Google Scholar] [CrossRef]
- Mossety-Leszczak, B.; Galina, H.; Włodarska, M. Synthesis and phase transitions of mesogenic compounds with functional groups in the tail. Phase Transit. 2011, 84, 15. [Google Scholar] [CrossRef]
- Mossety-Leszczak, B.; Włodarska, M.; Galina, H.; Bak, G.W. Comparing liquid crystalline properties of two epoxy compounds based on the same azoxy group. Mol. Cryst. Liq. Cryst. 2008, 490, 52. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results Obtained with the Correlation Energy Density Functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comp. Chem. 1982, 3, 214. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial Conformation | Optimized Structure | ||||
|---|---|---|---|---|---|
| δ1, δ2, δ3, δ4 (°) | EB3LYP (Ha) | μ (D) | Θ (°) | Symbol | |
| μM (D) | μeo (D) | ||||
| 75, 120, 105, −120 | −2232.41195087 | 0.3 | 88 | M22α | |
| 0.0 | 0.4 | ||||
| 75, 120, 75, 120 | −2232.41187634 | 0.3 | 90 | M22 | |
| 75, −120, 105, 120 | −2232.41184511 | 0.4 | 78 | M22 | |
| 75, −120, −105, −120 | −2232.41181277 | 0.1 | 90 | M22 | |
| 75, −120, 75, −120 | −2232.41170039 | 2.6 | 90 | M22β | |
| 2.2 | 0.4 | ||||
| 75, 120, −105, 120 | −2232.41165861 | 2.1 | 89 | M22 | |
| 75, 120, −75, −120 | −2232.41149737 | 2.2 | 89 | M22 | |
| 75, −120, −75, 120 | −2232.41147492 | 2.3 | 89 | M22 | |
| 75, 120, 105, −120 | −2382.81374195 | 0.7 | 89 | MU22α | |
| 0.0 | 2.1 | ||||
| 75, 120, 75, 120 | −2382.81366538 | 3.6 | 90 | MU22 | |
| 75, −120, 105, 120 | −2382.81363725 | 1.0 | 86 | MU22 | |
| 75, −120, −105, −120 | −2382.81359203 | 3.3 | 90 | MU22 | |
| 75, −120, 75, −120 | −2382.81348624 | 2.2 | 90 | MU22β | |
| 2.2 | 2.1 | ||||
| 75, 120, −105, 120 | −2382.81350061 | 2.3 | 88 | MU22 | |
| 75, 120, −75, −120 | −2382.81329249 | 3.6 | 89 | MU22 | |
| 75, −120, −75, 120 | −2382.81324045 | 3.2 | 88 | MU22 | |
| Initial Conformation | Optimized Structures | ||||
|---|---|---|---|---|---|
| δ1, δ2 (°) | EB3LYP (Ha) | μ (D) | Θ (°) | Symbol | |
| μm (D) | μeo (D) | ||||
| 60, −120 | −1959.27603366 | 3.9 | 67 | AU12α | |
| 1.6 | 2.1 | ||||
| 60, −60 | −1959.27597586 | 1.3 | 50 | AU12 | |
| 1.6 | 2.1 | ||||
| 60, 120 | −1959.27593243 | 5.1 | 73 | AU12β | |
| 2.5 | 2.1 | ||||
| 60, 60 | −1959.27593164 | 3.1 | 75 | AU12 | |
| 120, −120 | −1959.27573355 | 2.3 | 25 | AU12 | |
| 120, 60 | −1959.27565102 | 6.6 | 79 | AU12 | |
| 120, 120 | −1959.27564991 | 4.7 | 66 | AU12 | |
| 120, −60 | −1959.27559634 | 4.3 | 74 | AU12 | |
| Initial Conformation | Optimized Structure | ||||
|---|---|---|---|---|---|
| δ1, δ2, δ3 (°) | EB3LYP (Ha) | μ (D) | Θ (°) | Symbol | |
| μm (D) | μeo (D) | ||||
| 60, 120, 45 | −1812.79456691 | 2.0 | 38 | M12α | |
| 2.0 | 0.4 | ||||
| 60, 120, −45 | −1812.79453283 | 2.0 | 37 | M12 | |
| 1.9 | 0.4 | ||||
| 60, −120, −45 | −1812.79452681 | 2.9 | 51 | M12β | |
| 2.9 | 0.4 | ||||
| 60, 60, −45 | −1812.79450621 | 1.8 | 11 | M12 | |
| 60, 60, 45 | −1812.79449519 | 2.8 | 52 | M12 | |
| 60, −120, 45 | −1812.79448290 | 1.9 | 19 | M12 | |
| 60, −60, 45 | −1812.79422750 | 2.9 | 46 | M12 | |
| 60, −60, −45 | −1812.79417161 | 2.8 | 43 | M12 | |
| 60, 120, 45 | −1963.19638534 | 4.7 | 69 | MU12α | |
| 2.0 | 2.1 | ||||
| 60, 120, −45 | −1963.19638204 | 3.8 | 65 | MU12 | |
| 1.9 | 2.1 | ||||
| 60, −120, −45 | −1963.19635377 | 5.5 | 70 | MU12β | |
| 2.9 | 2.1 | ||||
| 60, 60, −45 | −1963.19633532 | 2.3 | 35 | MU12 | |
| 60, −120, 45 | −1963.19633065 | 2.7 | 47 | MU12 | |
| 60, 60, 45 | −1963.19630662 | 5.0 | 67 | MU12 | |
| 60, −60, 45 | −1963.19602995 | 2.7 | 32 | MU12 | |
| 60, −60, −45 | −1963.19598906 | 4.2 | 57 | MU12 | |
| Molecule | HOMO (Ha) | LUMO (Ha) | Band Gap (eV) | Dipole (D) |
|---|---|---|---|---|
| MU22α | −0.233 | −0.056 | 4.82 | 0.7 |
| MU22β | −0.234 | −0.055 | 4.87 | 2.2 |
| M22α | −0.232 | −0.054 | 4.84 | 0.3 |
| M22β | −0.234 | −0.055 | 4.87 | 2.6 |
| AU12α | −0.217 | −0.079 | 3.76 | 3.9 |
| AU12β | −0.217 | −0.079 | 3.76 | 5.1 |
| MU12α | −0.233 | −0.054 | 4.87 | 4.7 |
| MU12β | −0.232 | −0.054 | 4.84 | 5.5 |
| M12α | −0.232 | −0.053 | 4.87 | 2.0 |
| M12β | −0.231 | −0.053 | 4.84 | 2.9 |
| Molecule | αxx | αxy | αyy | αxz | αyz | αzz |
|---|---|---|---|---|---|---|
| MU22α | 799 | −14 | 412 | −7 | −8 | 294 |
| MU22β | 793 | −26 | 359 | 0 | 0 | 349 |
| M22α | 812 | −8 | 401 | −6 | −9 | 288 |
| M22β | 807 | −21 | 343 | 0 | 0 | 346 |
| AU12α | 722 | 21 | 357 | 21 | 1 | 251 |
| AU12β | 720 | −18 | 355 | −1 | −25 | 256 |
| MU12α | 621 | 1 | 345 | −2 | 7 | 262 |
| MU12β | 622 | −3 | 334 | −3 | −23 | 273 |
| M12α | 635 | −1 | 328 | −10 | 5 | 261 |
| M12β | 635 | −6 | 320 | 6 | −28 | 270 |
| Molecule | βxxx | βxxy | βxyy | βyyy | βxxz | βxyz | βyyz | βxzz | βyzz | βzzz |
|---|---|---|---|---|---|---|---|---|---|---|
| MU22α | −4 | 7 | 7 | 7 | 0 | −14 | −9 | −7 | −12 | 28 |
| MU22β | 0 | 0 | 0 | 0 | 373 | −132 | 30 | 0 | 0 | −37 |
| M22α | 0 | −3 | −9 | 5 | 29 | 17 | −6 | 7 | −2 | −11 |
| M22β | 0 | 0 | 0 | 0 | 408 | −108 | 25 | 0 | 0 | −53 |
| AU12α | 750 | 771 | 77 | 16 | 85 | 117 | 9 | 2 | 36 | −2 |
| AU12β | −753 | 741 | −60 | 20 | −267 | 23 | −7 | −15 | 27 | 51 |
| MU12α | −664 | 382 | 60 | 40 | −121 | 4 | −4 | −3 | 44 | 26 |
| MU12β | 660 | −295 | −54 | −49 | 208 | 86 | 22 | −69 | −53 | −41 |
| M12α | −688 | 283 | 62 | −1 | −129 | 21 | 2 | −3 | −7 | 7 |
| M12β | 662 | −223 | −69 | 1 | 230 | 96 | 5 | −57 | −10 | −1 |
| Compound | Temperatures of Phase Transitions (°C) | Route |
|---|---|---|
| M22 | Cr1 → 130 (Cr2) 136 (SmC) → 168 (N) → 192 (I) Cr → 125 (SmC) → 166 (N) → 192 (I) | heating cooling |
| MU22 | Cr1 → 78 (Cr2) → 138 (N) → 189 (I) Cr1 → 125 (N) → 185 (I) | heating cooling |
| AU12 | Cr → 90 (SmA) → 97 (N) → 110.5 (I) Cr → 80 (SmA) → 97 (N) → 110 (I) | heating cooling |
| M12 | Cr → 78 (SmA) → 90 (N) → 97 (I) Cr → 71 (SmF) → 74 (SmC) → 90 (N) → 97 (I) | heating cooling |
| MU12 | Cr → 79 (N) → 89 (I) Cr → 67(N) → 90 (I) | heating cooling |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Włodarska, M.; Mossety-Leszczak, B. DFT Studies of Selected Epoxies with Mesogenic Units–Impact of Molecular Structure on Electro-Optical Response. Int. J. Mol. Sci. 2021, 22, 3424. https://doi.org/10.3390/ijms22073424
Włodarska M, Mossety-Leszczak B. DFT Studies of Selected Epoxies with Mesogenic Units–Impact of Molecular Structure on Electro-Optical Response. International Journal of Molecular Sciences. 2021; 22(7):3424. https://doi.org/10.3390/ijms22073424
Chicago/Turabian StyleWłodarska, Magdalena, and Beata Mossety-Leszczak. 2021. "DFT Studies of Selected Epoxies with Mesogenic Units–Impact of Molecular Structure on Electro-Optical Response" International Journal of Molecular Sciences 22, no. 7: 3424. https://doi.org/10.3390/ijms22073424
APA StyleWłodarska, M., & Mossety-Leszczak, B. (2021). DFT Studies of Selected Epoxies with Mesogenic Units–Impact of Molecular Structure on Electro-Optical Response. International Journal of Molecular Sciences, 22(7), 3424. https://doi.org/10.3390/ijms22073424