
Switching Ion Binding Selectivity of Thiacalix[4]arene Monocrowns at Liquid–Liquid and 2D-Confined Interfaces

 , , and
, , and
Abstract
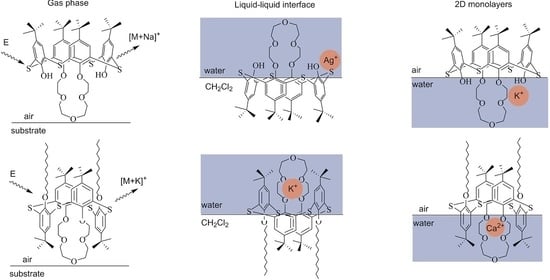
:
1. Introduction
2. Results and Discussion
2.1. Design, Synthesis, and Characterization of Thiacalix[4]monocrown-Ethers
- (1)
- Ionizable cone thiacalixcrowns, with four to six oxygen atoms in the crown-ether ring (type I);
- (2)
- Amphiphilic 1,3-alternate thiacalixcrowns bearing C12-chain at the lower rim (type II);
- (3)
- Conformationally flexible thiacalixcrowns bearing OMe groups at the lower rim (type III).
2.2. Complexation of Metal Ions at Liquid–Liquid Interface
2.3. Aggregation of Crown-Ether Ligands with and without Metal Ions
2.4. Langmuir Monolayer Formation
2.5. Metal Ion–Monolayer Interactions at the Air–Water Interface
2.6. Mechanism of Metal Ion Binding by Thiacalixcrowns at Different Interfaces
3. Materials and Methods
Synthesis of Compounds 5a–c
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MALDI | Matrix-assisted laser desorption–ionization |
| CS | Chemical shift |
| NMR | Nuclear magnetic resonance |
| PC | Pinched cone |
| DC | Distorted cone |
| IR | Infrared |
| DLS | Dynamic light scattering |
| FT-PGSE | Fourier transform pulsed-gradient spin-echo |
| PSD | Particle size distribution |
| DFT | Density functional theory |
| TR | Transfer ratio |
| AFM | Atomic force microscopy |
| SPOT | Surface potential |
| UVRAS | UV/visible reflection–absorption spectroscopy |
| BPP-STE-LED | Bipolar pulse pair–stimulated echo-longitudinal eddy current delay |
| PBE | Perdew–Burke–Ernzerhof |
| B3LYP | Becke, 3-parameter, Lee–Yang–Parr |
| PCM | Polarizable continuum model |
| SMD | Solvation model based on density |
| TZVP | Triple-zeta valence polarization |
References
- Huskens, J.; Prins, L.J.; Haag, R.; Ravoo, B.J. Multivalency: Concepts, Research & Applications; Wiley: Chichester, UK, 2018. [Google Scholar] [CrossRef]
- Yamada, S. Cation–π interactions in organic crystals. Coord. Chem. Rev. 2020, 415, 213301. [Google Scholar] [CrossRef]
- Neri, P.; Sessler, J.L.; Wang, M.-X. Calixarenes and Beyond; Springer: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Schneider, H.-J. Strain effects determine the performance of artificial allosteric systems: Calixarenes as models. Chem. Commun. 2019, 55, 3433–3444. [Google Scholar] [CrossRef]
- Atanassova, M.; Kurteva, V. Synergism as a phenomenon in solvent extraction of 4f-elements with calixarenes. RSC Adv. 2016, 6, 11303–11324. [Google Scholar] [CrossRef]
- Kumar, R.; Sharma, A.; Singh, H.; Suating, P.; Kim, H.S.; Sunwoo, K.; Shim, I.; Gibb, B.C.; Kim, J.S. Revisiting fluorescent calixarenes: From molecular sensors to smart materials. Chem. Rev. 2019, 119, 9657–9721. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, M.H.; Mutihac, L.; Vicens, J.; Kim, J.S. Host–guest sensing by calixarenes on the surfaces. Chem. Soc. Rev. 2012, 41, 1173–1190. [Google Scholar] [CrossRef]
- Ovsyannikov, A.; Solovieva, S.; Antipin, I.; Ferlay, S. Coordination polymers based on calixarene derivatives: Structures and properties. Coord. Chem. Rev. 2017, 352, 151–186. [Google Scholar] [CrossRef]
- Casnati, A.; Pochini, R.; Ungaro, R.; Bocchi, C.; Ugozzoli, F.; Egberink, R.J.M.; Struijk, H.; Lugtengerg, R.; de Jong, F.; Reinhoudt, D.N. 1,3-Alternate calix[4]arenecrown-5 conformers: New synthetic ionophores with better K+/Na+ selectivity than valinomycin. Chem. Eur. J. 1996, 2, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Kumar, N.; Bhalla, V. Ratiometric detection of Hg2+ ions: An allosterically synchronized Hg2+/Li+ switch based on thiacalix[4]crown. Dalton Trans. 2011, 40, 5170–5175. [Google Scholar] [CrossRef]
- Bouhroum, S.; Arnaud-Neu, F.; Asfari, Z.; Vicens, J. Ionic recognition by thiacalixarenes: Binding of alkali and heavy metal ions by thiacalix[4]arene-bis-crown[n]ethers. Russ. Chem. Bull. 2004, 53, 1544–1548. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, S.K.; Bartsch, R.A.; Vicens, J.; Miyano, S.; Kim, J.S. Rapid metal ions shuttling through 1,3-alternate thiacalix[4]crown tubes. J. Org. Chem. 2003, 68, 6720–6725. [Google Scholar] [CrossRef]
- Csokai, V.; Grün, A.; Parlagh, G.; Bitter, I. Synthesis and alkali cation extraction ability of 1,3-alt-thiacalix[4]mono(crown) ethers. Tetrahedron Lett. 2002, 43, 7627–7629. [Google Scholar] [CrossRef]
- Ghidini, E.; Ugozzoli, F.; Ungaro, R.; Harkema, S.; El-Fadl, A.A.; Reinhoudt, D.N. Complexation of alkali metal cations by conformationally rigid, stereoisomeric calix[4]arene crown ethers: A quantitative evaluation of preorganization. J. Am. Chem. Soc. 1990, 112, 6979–6985. [Google Scholar] [CrossRef]
- Park, J.-Y.; Kim, B.-C.; Park, S.-M. Molecular recognition of protonated polyamines at calix[4]crown-5 self-assembled monolayer modified electrodes by impedance measurements. Anal. Chem. 2007, 79, 1890–1896. [Google Scholar] [CrossRef]
- Zhang, S.; Song, F.; Echegoyen, L. Synthesis, self-assembled monolayers and alkaline earth metal ion recognition of p-tert-butylcalix[4]arene derivatives. Eur. J. Org. Chem. 2004, 2004, 2936–2943. [Google Scholar] [CrossRef]
- Muravev, A.A.; Solovieva, S.E.; Kochetkov, E.N.; Mel’nikova, N.B.; Safiullin, R.A.; Kadirov, M.K.; Latypov, S.K.; Antipin, I.S.; Konovalov, A.I. Thiacalix[4]monocrowns substituted by sulfur-containing anchoring groups: New ligands for gold surface modification. Macroheterocycles 2013, 6, 302–307. [Google Scholar] [CrossRef]
- Jágerszki, G.; Grün, A.; Bitter, I.; Tóth, K.; Gyurcsányi, R.E. Ionophore–gold nanoparticle conjugates for Ag+-selective sensors with nanomolar detection limit. Chem. Commun. 2010, 46, 607–609. [Google Scholar] [CrossRef]
- Ahn, J.; Lim, N.Y.; Park, J.S.; Choi, Y.; Jung, J.H. Fabrication of calix[4]arene-attached mesoporous ammonium molybdophosphate–silica hybrid and its application as an adsorbent for cesium ions. New J. Chem. 2017, 41, 3196–3203. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, X.; Feng, N.; Tian, D.; Deng, H.; Li, H. Cation-induced pesticide binding and release by a functionalized calix[4]arene molecular host. Sci. Rep. 2015, 5, 8982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capuzzi, G.; Fratini, E.; Dei, L.; LoNostro, P.; Casnati, A.; Gilles, R.; Baglioni, P. Counterion complexation of calixarene ligands in monolayers and micellar solutions. Colloid Surf. A 2000, 167, 105–113. [Google Scholar] [CrossRef]
- Nag, R.; Vashishtha, M.; Rao, C.P. Switching the ion selectivity from Fe3+ to Al3+ by a triazole-appended calix[4]arene-based amphiphile. ChemistrySelect 2018, 3, 1248–1256. [Google Scholar] [CrossRef]
- Csokai, V.; Grün, A.; Bitter, I. Unprecedented cyclisations of calix[4]arenes with glycols under the Mitsunobu protocol. Part 1: A new perspective for the synthesis of calixcrowns. Tetrahedron Lett. 2003, 44, 4681–4684. [Google Scholar] [CrossRef]
- van Leeuwen, F.W.B.; Beijleveld, H.; Kooijman, H.; Spek, A.L.; Verboom, W.; Reinhoudt, D.N. Synthesis and conformational evaluation of p-tert-butylthiacalix[4]arene-crowns. J. Org. Chem. 2004, 69, 3928–3936. [Google Scholar] [CrossRef]
- Muravev, A.A.; Laishevtsev, A.I.; Galieva, F.B.; Bazanova, O.B.; Rizvanov, I.K.; Korany, A.; Solovieva, S.E.; Antipin, I.S.; Konovalov, A.I. Azide-akyne click approach to the preparation of dendrimer-type multi(thia)calix[4]arenes with triazole linkers. Macroheterocycles 2017, 10, 203–214. [Google Scholar] [CrossRef]
- Latypov, S.K.; Kharlamov, S.V.; Muravev, A.A.; Balandina, A.A.; Solovieva, S.E.; Antipin, I.S.; Konovalov, A.I. Conformational diversity and dynamics of distally disubstituted calix and thiacalix[4]arenes in solution. J. Phys. Org. Chem. 2013, 26, 407–414. [Google Scholar] [CrossRef]
- Muravev, A.A.; Yakupov, A.T.; Solovieva, S.E.; Antipin, I.S. A new approach to the synthesis of thiacrowns on a thiacalix[4]arene scaffold. Dokl. Chem. 2019, 487, 188–191. [Google Scholar] [CrossRef]
- Bitter, I.; Csokai, V. An expedient route to p-tert-butylthiacalix[4]arene 1,3-diethers via Mitsunobu reactions. Tetrahedron Lett. 2003, 44, 2261–2265. [Google Scholar] [CrossRef]
- Muravev, A.A.; Solovieva, S.E.; Latypov, S.K.; Antipin, I.S.; Konovalov, A.I. Synthesis and characterization of thiacalix[4]monocrowns modified by thioether groups on the lower rim. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 499–502. [Google Scholar] [CrossRef]
- Solovieva, S.E.; Muravev, A.A.; Zakirzyanov, R.T.; Latypov, S.K.; Antipin, I.S.; Konovalov, A.I. Synthesis, structure, and extraction ability of tetrasubstituted thiacalix[4]arenes with crown ether fragments on the lower rim. Macroheterocycles 2012, 5, 17–22. [Google Scholar] [CrossRef]
- Lang, J.; Dvořáková, H.; Bartošová, I.; Lhoták, P.; Stibor, I.; Hrabal, R. Conformational flexibility of a novel tetraethylether of thiacalix[4]arene. A comparison with the “classical” methylene-bridged compounds. Tetrahedron Lett. 1999, 40, 373–376. [Google Scholar] [CrossRef]
- Šimánová, M.; Dvořáková, H.; Stibor, I.; Pojarová, M.; Lhoták, P. Synthesis and conformational behaviour of lower-rim tetraacetylated thiacalix[4]arenes. Tetrahedron Lett. 2008, 49, 1026–1029. [Google Scholar] [CrossRef]
- van Leeuwen, F.W.B.; Beijleveld, H.; Kooijman, H.; Spek, A.L.; Verboom, W.; Reinhoudt, D.N. Cation control on the synthesis of p-t-butylthiacalix[4](bis)crown ethers. Tetrahedron Lett. 2002, 43, 9675–9678. [Google Scholar] [CrossRef] [Green Version]
- Danil de Namor, A.F.; Pugliese, A.; Casal, A.R.; Llerena, M.B.; Aymonino, P.J.; Sueros Velarde, F.J. The various factors involved in the extraction of alkali metal picrates by calixarene ester derivatives in the mutually saturated water–dichloromethane solvent system. Phys. Chem. Chem. Phys. 2000, 2, 4355–4360. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Steed, J.W. First-and second-sphere coordination chemistry of alkali metal crown ether complexes. Coord. Chem. Rev. 2001, 215, 171–221. [Google Scholar] [CrossRef]
- Chi, X.; Peters, G.M.; Hammel, F.; Brockman, C.; Sessler, J.L. Molecular recognition under interfacial conditions: calix[4]pyrrole-based cross-linkable micelles for ion pair extraction. J. Am. Chem. Soc. 2017, 139, 9124–9127. [Google Scholar] [CrossRef]
- Ellis, R.J.; Meridiano, Y.; Muller, J.; Berthon, L.; Guilbaud, P.; Zorz, N.; Antonio, M.R.; Demars, T.; Zemb, T. Complexation-induced supramolecular assembly drives metal-ion extraction. Chem. Eur. J. 2014, 20, 12796–12807. [Google Scholar] [CrossRef]
- Nayak, S.; Lovering, K.; Uysal, A. Ion-specific clustering of metal–amphiphile complexes in rare earth separations. Nanoscale 2020, 12, 20202–20210. [Google Scholar] [CrossRef]
- Bourgeois, D.; El Maangar, A.; Dourdain, S. Importance of weak interactions in the formulation of organic phases for efficient liquid/liquid extraction of metals. Curr. Opin. Colloid Interface Sci. 2020, 46, 36–51. [Google Scholar] [CrossRef]
- Smirnov, I.V.; Stepanova, E.S.; Tyupina, M.Y.; Ivenskaya, N.M.; Zaripov, S.R.; Kleshnina, S.R.; Solovieva, S.E.; Antipin, I.S. Americium and cesium extraction from alkaline media by calix[8]arenes with p-tert-butyl and isononyl substituents on the upper rim: Aggregation effect. Macroheterocycles 2017, 10, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Muravev, A.A.; Trushina, E.A.; Yakupov, A.T.; Solovieva, S.E.; Antipin, I.S. Ag-selective nanotubes based on bisthiacalix[4]arene with ethylene sulfide bridges. Dokl. Chem. 2019, 487, 212–214. [Google Scholar] [CrossRef]
- Sieffert, N.; Chaumont, A.; Wippf, G. Importance of the liquid-liquid interface in assisted ion extraction: New molecular dynamics studies of cesium picrate extraction by a calix[4]arene. J. Phys. Chem. C 2009, 113, 10610–10622. [Google Scholar] [CrossRef]
- Muravev, A.; Gerasimova, T.; Fayzullin, R.; Babaeva, O.; Rizvanov, I.; Khamatgalimov, A.; Kadirov, M.; Katsyuba, S.; Litvinov, I.; Latypov, S.; et al. Thermally stable nitrothiacalixarene chromophores: Conformational study and aggregation behavior. Int. J. Mol. Sci. 2020, 21, 6916. [Google Scholar] [CrossRef]
- Gaines, G.L.J. Insoluble Monolayers at Liquid-Gas. Interfaces; Wiley: New York, NY, USA, 1966. [Google Scholar]
- Muravev, A.A.; Agarkov, A.S.; Galieva, F.B.; Yakupov, A.T.; Bazanova, O.B.; Rizvanov, I.K.; Shokurov, A.V.; Zaitseva, A.V.; Selektor, S.L.; Solovieva, S.E.; et al. New terpyridine derivatives of thiacalix[4]arenes in solution and at the water—air interface. Russ. Chem. Bull. 2020, 69, 339–350. [Google Scholar] [CrossRef]
- Moradi, M.; Tulli, L.G.; Nowakowski, J.; Baljozovic, M.; Jung, T.A.; Shahgaldian, P. Two-dimensional calix[4]arene-based metal-organic coordination networks of tunable crystallinity. Angew. Chem. Int. Ed. 2017, 56, 14395–14399. [Google Scholar] [CrossRef]
- Dinadayalane, T.C.; Hassan, A.; Leszczynski, J. A theoretical study of cation–π interactions: Li+, Na+, K+, Be2+, Mg2+ and Ca2+ complexation with mono- and bicyclic ring-fused benzene derivatives. Theor. Chem. Acc. 2012, 131, 1131. [Google Scholar] [CrossRef]
- Lonetti, B.; Lo Nostro, P.; Ninham, B.W.; Baglioni, P. Anion effects on calixarene monolayers: A Hofmeister series study. Langmuir 2005, 21, 2242–2249. [Google Scholar] [CrossRef]
- Selektor, S.L.; Shcherbina, M.A.; Bakirov, A.V.; Batat, P.; Grauby-Heywang, C.; Grigorian, S.; Arslanov, V.V.; Chvalun, S.N. Cation-controlled excimer packing in Langmuir−Blodgett films of hemicyanine amphiphilic chromoionophores. Langmuir 2016, 32, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Shokurov, A.V.; Shcherbina, M.A.; Bakirov, A.V.; Alexandrova, A.V.; Raitman, O.A.; Arslanov, V.V.; Chvalun, S.N.; Selektor, S.L. Rational design of hemicyanine Langmuir monolayers by cation-induced preorganization of their structure for sensory response enhancement. Langmuir 2018, 34, 7690–7697. [Google Scholar] [CrossRef] [PubMed]
- Shokurov, A.V.; Alexandrova, A.V.; Lukovskaya, E.V.; Arslanov, V.V.; Selektor, S.L. The effect of alkyl substituent length on receptor properties of dithiaaza-crown-hemicyanine monolayers. Macroheterocycles 2017, 10, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Leontidis, E. Chaotropic salts interacting with soft matter: Beyond the lyotropic series. Curr. Opin. Colloid Interface Sci. 2016, 23, 100–109. [Google Scholar] [CrossRef]
- Thuéry, P.; Nierlich, M.; Lamare, V.; Dozol, J.-F.; Asfari, Z.; Vicens, J. Bis(crown ether) and azobenzocrown derivatives of calix[4]arene. a review of structural information from crystallographic and modelling studies. J. Incl. Phenom. Macrocycl. Chem. 2000, 36, 375–408. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Armarego, W.L.F. Purification of Laboratory Chemicals; Butterworth-Heinemann: Oxford, UK, 2009. [Google Scholar]
- Iki, N.; Kabuto, C.; Fukushima, T.; Kumagai, H.; Takeya, H.; Miyanari, S.; Miyashi, T.; Miyano, S. Synthesis of p-tert-butylthiacalix[4]arene and its inclusion property. Tetrahedron 2000, 56, 1437–1443. [Google Scholar] [CrossRef]
- Wu, D.; Chen, A.; Johnson, C.S. An improved diffusion-ordered spectroscopy experiment incorporating bipolar-gradient pulses. J. Magn. Reson., Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- Stuchebryukov, S.D.; Selektor, S.L.; Silantieva, D.A.; Shokurov, A.V. Peculiarities of the reflection-absorption and transmission spectra of ultrathin films under normal incidence of light. Prot. Met. Phys. Chem. Surf. 2013, 49, 189–197. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Laikov, D.N. A new class of atomic basis functions for accurate electronic structure calculations of molecules. Chem. Phys. Lett. 2005, 416, 116–120. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford CT, UK, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford CT, UK, 2016. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Bergner, A.; Dolg, M.; Küchle, W.; Stoll, H.; Preuß, H. Ab initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol. Phys. 1993, 80, 1431–1441. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Häser, M.; Treutler, O.; Ahlrichs, R. Calculation of excitation energies within time-dependent density functional theory using auxiliary basis set expansions. Chem. Phys. Lett. 1997, 264, 573–578. [Google Scholar] [CrossRef]
- Furche, F. On the density matrix based approach to time-dependent density functional response theory. J. Chem. Phys. 2001, 114, 5982–5992. [Google Scholar] [CrossRef]
- Rudberg, E.; Sałek, P. Calculations of two-photon charge-transfer excitations using Coulomb-attenuated density-functional theory. J. Chem. Phys. 2005, 123, 184108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, R.; Amos, R.D. The application of CAM-B3LYP to the charge-transfer band problem of the zincbacteriochlorin-bacteriochlorin complex. Chem. Phys. Lett. 2006, 420, 106–109. [Google Scholar] [CrossRef]
- Cai, Z.L.; Crossley, M.J.; Reimers, J.R.; Kobayashi, R.; Amos, R.D. Density functional theory for charge transfer: The nature of the N-Bands of porphyrins and chlorophylls revealed through CAM-B3LYP, CASPT2, and SAC-CI calculations. J. Phys. Chem. B 2006, 110, 15624–15632. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | c, 10−3M | Ds, 10−10 m2/s | Rh, Å | Naggr (from cmin) |
| (5a) 5 | 6.10 | 7.2 | 1.0 | |
| (5a) 30 | 5.67 | 7.8 | 1.2 | |
| (7) 5 | 7.61 | 5.8 | 1.0 | |
| (7) 30 | 6.59 | 6.7 | 1.5 |
| Compound | Analyzed Phase | Metal Salt | Size, nm | Polydispersity Index |
|---|---|---|---|---|
| 2c | CH2Cl2 and Water | –/CsPic | – | – |
| CH2Cl2 | CsNO3 | 313.1 ± 14.5 | 0.95 ± 0.05 | |
| Water | 298.7 ± 36.7 | 0.39 ± 0.08 | ||
| 5c | CH2Cl2 | – | 285.4 ± 32.1 | 1.00 |
| CsPic | 426.4 ± 26.1 | 0.47 ± 0.09 | ||
| CsNO3 | 165.9 ± 34.1 | 0.45 ± 0.18 | ||
| Water | – | – | – | |
| CsPic | 80.9 ± 5.6 | 0.46 ± 0.04 | ||
| CsNO3 | 262.7 ± 21.5 | 0.37 ± 0.06 | ||
| 6c | CH2Cl2 | – | – | – |
| CsPic | 76.3 ± 15.2 | 0.59 ± 0.13 | ||
| CsNO3 | 259.3 ± 58.3 | 0.36 ± 0.23 | ||
| Water | – | 331.3 ± 38.2 | 0.30 ± 0.05 | |
| CsPic/CsNO3 | – | – |
| Ligand | A0, Å2 | ΔV, mV 3 | λmax, nm | ||||
|---|---|---|---|---|---|---|---|
| 2a | 195 ± 6 | 40 | 117 1 | 162 | 508 | 1.35 | 302 |
| 2b | 182 ± 5 | 50 | 104 2 | 171 | 517 | 1.37 | 303 |
| 2c | 176 ± 5 | 37 | 103 2 | 193 | 740 | 1.96 | 304 |
| 5a | 201 ± 6 | 29 | 129 | 240 | 263 | 0.70 | 271 |
| 5b | 185 ± 6 | 27 | 132 | 252 | 295 | 0.78 | 269 |
| 5c | 177 ± 5 | 45 | 127 | 250 | 375 | 0.99 | 271 |
| 6a | 392 ± 12 | 25 | 109 | 122 | 225 | 1.19 | 270 |
| 6b | 467 ± 14 | 38 | 108 | 149 | 281 | 1.49 | 271 |
| 6c | 506 ± 15 | 42 | 103 | 163 | 301 | 1.60 | 270 |
| Compound | Number of Monolayers | Ra, nm | h, nm | TR | πtransfer, mN/m |
|---|---|---|---|---|---|
| quartz | n/a | 0.20 | n/a | n/a | n/a |
| 2a (1 × 10−5 M) | 1 | 0.30 | n/a | 0.76 | 10 |
| 2b (1 × 10−5 M) | 1 | 0.25 | n/a | 0.93 | 12 |
| 5a (1 × 10−5 M) | 1 | 2.3 | n/a | 0.84 | 6 |
| 5b (1 × 10−5 M) | 1 | 2.5 | n/a | 1.15 | 12 |
| 6a (1 × 10−5 M) | 1 | 1.0 | n/a | 0.98 | 10 |
| Ion (r, Å) 1 | A0, Å2 | |||||
|---|---|---|---|---|---|---|
| 2a | 2b | 2c | 5a | 5b | 5c | |
| – | 195 ± 6 | 182 ± 5 | 176 ± 5 | 201 ± 6 | 185 ± 6 | 177 ± 5 |
| Li+ (0.76) | 213 ± 6 | 200 ± 6 | 181 ± 5 | 201 ± 6 | 188 ± 6 | 178 ± 5 |
| Na+ (1.02) | 187 ± 6 | 173 ± 5 | 172 ± 5 | 234 ± 7 | 211 ± 6 | 179 ± 5 |
| K+ (1.38) | 205 ± 6 | 264 ± 8 | 211 ± 6 | 215 ± 6 | 178 ± 6 | 181 ± 5 |
| Rb+ (1.53) | 189 ± 6 | 238 ± 7 | 207 ± 6 | 228 ± 7 | 208 ± 6 | 197 ± 6 |
| Cs+ (1.67) | 195 ± 6 | 208 ± 6 | 211 ± 6 | 211 ± 6 | 192 ± 6 | 212 ± 6 |
| Mg2+ (0.72) | 192 ± 6 | 186 ± 5 | 172 ± 5 | 220 ± 6 | 192 ± 6 | 183 ± 6 |
| Ca2+ (1.00) | 201 ± 6 | 189 ± 5 | 177 ± 5 | 235 ± 7 | 228 ± 8 | 199 ± 6 |
| Sr2+ (1.18) | 241 ± 7 | 200 ± 6 | 177 ± 5 | 213 ± 6 | 197 ± 6 | 181 ± 5 |
| Ba2+ (1.35) | 226 ± 7 | 180 ± 5 | 178 ± 5 | 211 ± 6 | 210 ± 6 | 184 ± 6 |
| Pb2+ (1.19) | 190 ± 6 | 214 ± 6 | 195 ± 6 | 221 ± 6 | 204 ± 6 | 179 ± 5 |
| Ag+ (1.15) | 221 ± 7 | 181 ± 5 | 223 ± 7 | 190 ± 6 | 191 ± 6 | 180 ± 5 |
| Eu3+ (0.95) | 203 ± 6 | 182 ± 5 | 179 ± 5 | 212 ± 6 | 182 ± 6 | 176 ± 5 |
| Tb3+ (0.94) | 203 ± 6 | 206 ± 6 | 180 ± 5 | 205 ± 6 | 193 ± 6 | 178 ± 5 |
| Gd3+ (0.92) | 204 ± 6 | 214 ± 6 | 180 ± 5 | 211 ± 6 | 179 ± 6 | 178 ± 5 |
| Compound | ΔG (metal ion) | ||
|---|---|---|---|
| 2a | 1.87 (Sr2+) | 1.36 (Ba2+) | 1.26 (Ag+) |
| 2b | 6.52 (K+) | 2.83 (Rb+) | 2.46 (Gd3+) |
| 2c | 2.19 (K+) | 3.76 (Ag+) | 2.08 (Pb2+) |
| 5a | 1.95 (Na+) | 1.18 (Rb+) | 1.65 (Ca2+) |
| 5b | 1.27 (Na+) | 5.17 (Ca2+) | 1.13 (Ba2+) |
| 5c | 1.02 (Rb+) | 1.94 (Cs+) | 1.32 (Ca2+) |
| Conditions of Interaction | Ligand | |||||
|---|---|---|---|---|---|---|
| 2a | 2b | 2c | 5a | 5b | 5c | |
| Liquid–liquid interface 1 | Ag+ | Ag+ | Ag+ | Ag+ | Ag+/Rb+ | Cs+ |
| Air–water interface 2 | Sr2+ | K+ | Ag+ | Na+ | Ca2+ | Cs+ |
| Gas phase 3 | Li+ | Na+ | K+ | Li+/Na+ | K+ | Li+/Cs+ |
| Compound | HAr-1 | HAr-2 | CH2–1 | CH2–2 | CH2–3 | CH2–4 | CH2–5 | OMe |
|---|---|---|---|---|---|---|---|---|
| 6c | 0.10 | 0.05 | 0.25 | 0.20 | 0.27 | 0.27 | 0.21 | −0.08 |
| 2c | 0.17 | 0.12 | −0.41 | 0.08 | 0.16 | −0.01 | −0.02 | n/a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muravev, A.; Yakupov, A.; Gerasimova, T.; Nugmanov, R.; Trushina, E.; Babaeva, O.; Nizameeva, G.; Syakaev, V.; Katsyuba, S.; Selektor, S.; et al. Switching Ion Binding Selectivity of Thiacalix[4]arene Monocrowns at Liquid–Liquid and 2D-Confined Interfaces. Int. J. Mol. Sci. 2021, 22, 3535. https://doi.org/10.3390/ijms22073535
Muravev A, Yakupov A, Gerasimova T, Nugmanov R, Trushina E, Babaeva O, Nizameeva G, Syakaev V, Katsyuba S, Selektor S, et al. Switching Ion Binding Selectivity of Thiacalix[4]arene Monocrowns at Liquid–Liquid and 2D-Confined Interfaces. International Journal of Molecular Sciences. 2021; 22(7):3535. https://doi.org/10.3390/ijms22073535
Chicago/Turabian StyleMuravev, Anton, Ayrat Yakupov, Tatiana Gerasimova, Ramil Nugmanov, Ekaterina Trushina, Olga Babaeva, Guliya Nizameeva, Viktor Syakaev, Sergey Katsyuba, Sofiya Selektor, and et al. 2021. "Switching Ion Binding Selectivity of Thiacalix[4]arene Monocrowns at Liquid–Liquid and 2D-Confined Interfaces" International Journal of Molecular Sciences 22, no. 7: 3535. https://doi.org/10.3390/ijms22073535
APA StyleMuravev, A., Yakupov, A., Gerasimova, T., Nugmanov, R., Trushina, E., Babaeva, O., Nizameeva, G., Syakaev, V., Katsyuba, S., Selektor, S., Solovieva, S., & Antipin, I. (2021). Switching Ion Binding Selectivity of Thiacalix[4]arene Monocrowns at Liquid–Liquid and 2D-Confined Interfaces. International Journal of Molecular Sciences, 22(7), 3535. https://doi.org/10.3390/ijms22073535