
Pathway from TDP-43-Related Pathology to Neuronal Dysfunction in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
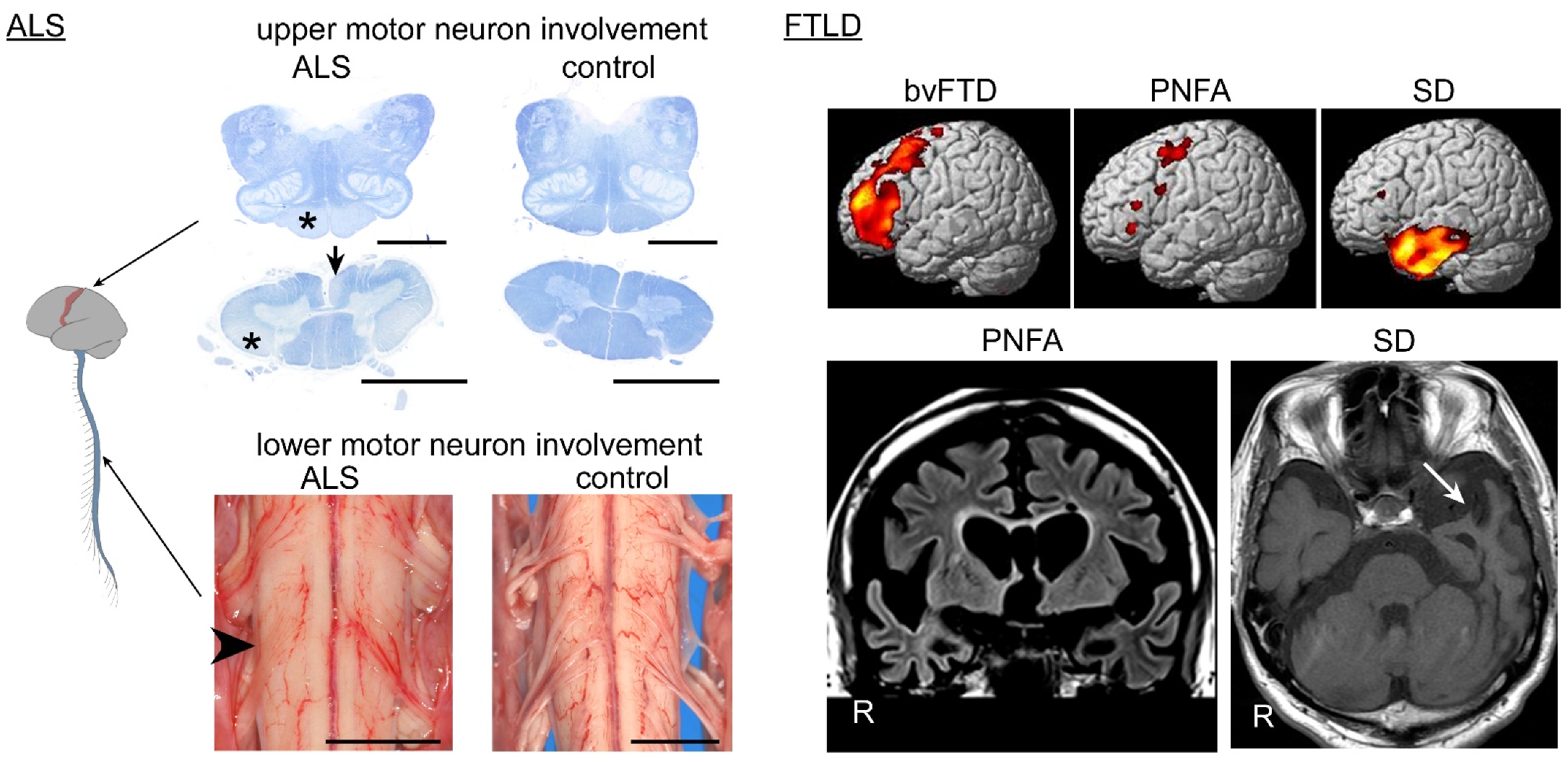
2. Clinical Findings of ALS and FTLD
3. TDP-43 Pathology in ALS and FTLD
4. Gain-of-Neurotoxicity and Loss-of-Function
5. Anatomical Spreading of TDP-43 Pathology
6. Are ALS-TDP and FTLD-TDP Synaptopathies?
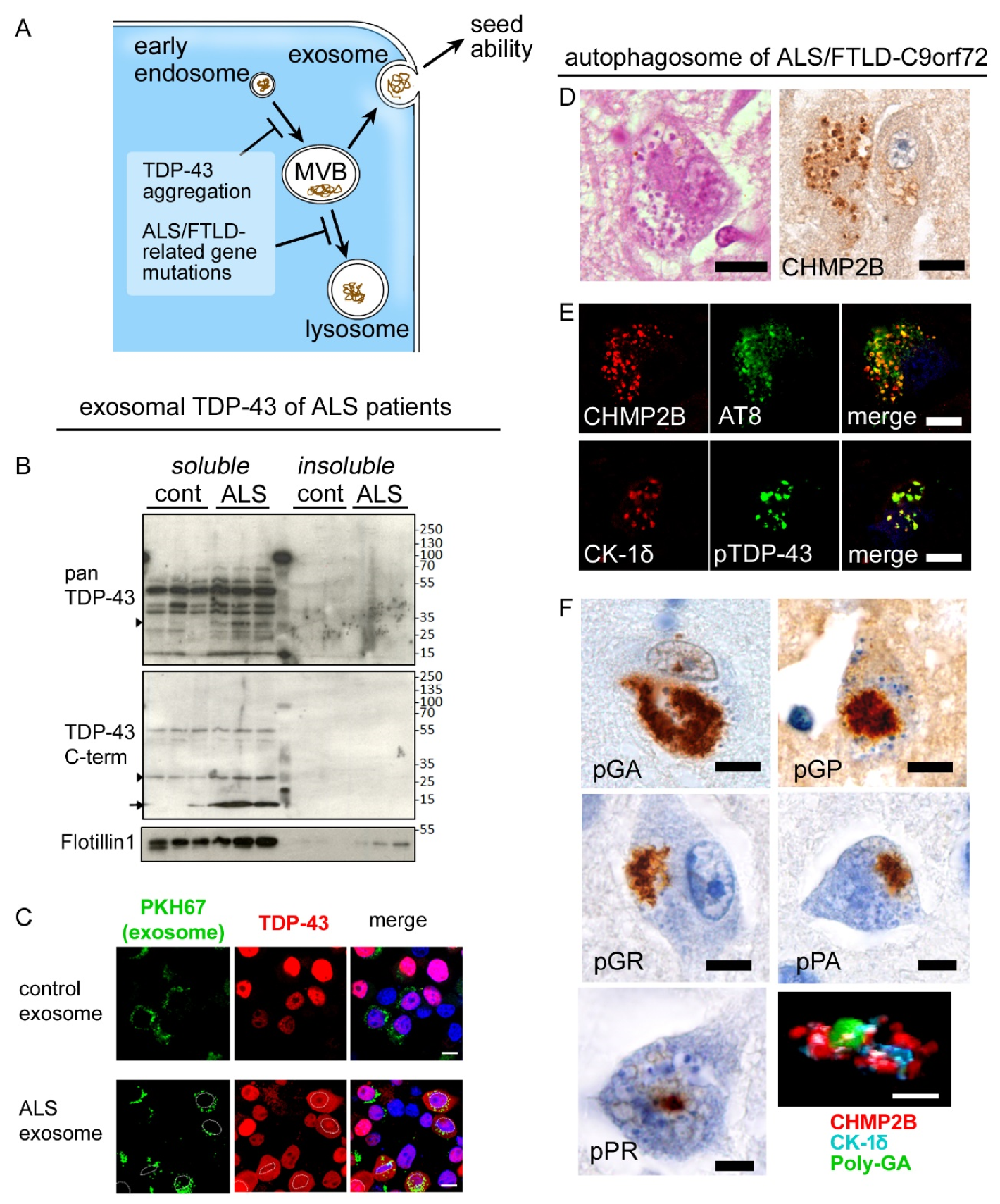
7. Impaired Clearance of TDP-43
8. Linkage between TDP-43 and tau Pathology
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer disease |
| ALS | amyotrophic lateral sclerosis |
| ALS-TDP | TDP-43-related ALS |
| bvFTD | behavioral variant FTD |
| CBD | corticobasal degeneration |
| CHMP2B | charged multivesicular body protein 2B |
| CK1δ | casein kinase 1δ |
| CMA | chaperon-mediated autophagy |
| CSF | cerebrospinal fluid |
| FTD | frontotemporal dementia |
| FTD-MND | frontotemporal dementia with motor neuron disease |
| FTLD | frontotemporal lobar degeneration |
| FTLD-TDP | TDP-43-related FTLD |
| FUS | fused-in-sarcoma |
| GVD | granulovacuolar degeneration |
| HSP-70 | 70 kDa heat shock proteins |
| LATE | limbic-predominant age-related TDP-43 encephalopathy |
| LBD | Lewy body disease |
| MAPT | microtubule-associated protein tau |
| MVB | multivesicular body |
| NF-κB | nuclear factor kappa B |
| PDC | parkinsonism-dementia complex |
| PNFA | progressive non-fluent aphasia |
| PSP | progressive supranuclear palsy |
| SD | semantic dementia |
| SFPQ | splicing factor proline/glutamine-rich proteins |
| TDP-43 | Transactivation response DNA binding protein 43 kDa |
References
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Buratti, E.; Dörk, T.; Zuccato, E.; Pagani, F.; Romano, M.; Baralle, F.E. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001, 20, 1774–1784. [Google Scholar] [CrossRef]
- Brooks, B.; Miller, R.; Swash, M.; Munsat, T. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2016, 138, 225–238. [Google Scholar] [PubMed]
- Logroscino, G.; Piccininni, M. Amyotrophic lateral sclerosis descriptive epidemiology: The origin of geographic difference. Neuroepidemiology 2019, 52, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef] [Green Version]
- Coyle-Gilchrist, I.T.; Dick, K.M.; Patterson, K.; Vázquez Rodríquez, P.; Wehmann, E.; Wilcox, A.; Lansdall, C.J.; Dawson, K.E.; Wiggins, J.; Mead, S.; et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 2016, 86, 1736–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of primary progressive aphasia and its variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95. [Google Scholar] [CrossRef]
- Tan, R.H.; Ke, Y.D.; Ittner, L.M.; Halliday, G.M. ALS/FTLD: Experimental models and reality. Acta Neuropathol. 2017, 133, 177–196. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Arai, K.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Lee, E.B.; Kuwabara, S.; Shibuya, K.; Irwin, D.J.; Fang, L. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 2014, 128, 423–437. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, Y.; Fujita, Y.; Takatama, M.; Okamoto, K. Comparison of phosphorylated TDP-43-positive inclusions in oculomotor neurons in patients with non-ALS and ALS disorders. J. Neurol. Sci. 2012, 315, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Co-localization of Bunina bodies and TDP-43 inclusions in lower motor neurons in amyotrophic lateral sclerosis. Neuropathology 2014, 34, 71–76. [Google Scholar] [CrossRef]
- Okamoto, K.; Mizuno, Y.; Fujita, Y. Bunina bodies in amyotrophic lateral sclerosis. Neuropathology 2008, 28, 109–115. [Google Scholar] [CrossRef]
- Josephs, K.A.; Hodges, J.R.; Snowden, J.S.; Mackenzie, I.R.; Neumann, M.; Mann, D.M.; Dickson, D.W. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011, 122, 137–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.; Lee, V.M. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122, 111–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampathu, D.M.; Neumann, M.; Kwong, L.K.; Chou, T.T.; Micsenyi, M.; Truax, A.; Bruce, J.; Grossman, M.; Trojanowski, J.Q.; Lee, V.M. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am. J. Pathol. 2006, 169, 1343–1352. [Google Scholar] [CrossRef] [Green Version]
- Cairns, N.J.; Neumann, M.; Bigio, E.H.; Holm, I.E.; Troost, D.; Hatanpaa, K.J.; Foong, C.; White, C.L., 3rd; Schneider, J.A.; Kretzschmar, H.A.; et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 2007, 171, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Josephs, K.A.; Stroh, A.; Dugger, B.; Dickson, D.W. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 2009, 118, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Takeda, T.; Seilhean, D.; Le Ber, I.; Millecamps, S.; Sazdovitch, V.; Kitagawa, K.; Uchihara, T.; Duyckaerts, C. Amygdala TDP-43 pathology in frontotemporal lobar degeneration and motor neuron disease. J. Neuropathol. Exp. Neurol. 2017, 76, 800–812. [Google Scholar] [CrossRef]
- Snowden, J.; Neary, D.; Mann, D. Frontotemporal lobar degeneration: Clinical and pathological relationships. Acta Neuropathol. 2007, 114, 31–38. [Google Scholar] [CrossRef]
- Geser, F.; Martinez-Lage, M.; Robinson, J.; Uryu, K.; Neumann, M.; Brandmeir, N.J.; Xie, S.X.; Kwong, L.K.; Elman, L.; McCluskey, L.; et al. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch. Neurol. 2009, 66, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Riku, Y.; Watanabe, H.; Yoshida, M.; Tatsumi, S.; Mimuro, M.; Iwasaki, Y.; Katsuno, M.; Iguchi, Y.; Masuda, M.; Senda, J.; et al. Lower motor neuron involvement in TAR DNA-binding protein of 43 kDa-related frontotemporal lobar degeneration and amyotrophic lateral sclerosis. JAMA Neurol. 2014, 71, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Riku, Y.; Atsuta, N.; Yoshida, M.; Tatsumi, S.; Iwasaki, Y.; Mimuro, M.; Watanabe, H.; Ito, M.; Senda, J.; Nakamura, R.; et al. Differential motor neuron involvement in progressive muscular atrophy: A comparative study with amyotrophic lateral sclerosis. BMJ Open 2014, 4, e005213. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Arai, T.; Kametani, F.; Nonaka, T.; Yamashita, M.; Suzukake, M.; Hosokawa, M.; Yoshida, M.; Hatsuta, H.; Takao, M.; et al. Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 2012, 135, 3380–3391. [Google Scholar] [CrossRef] [Green Version]
- Laferrière, F.; Maniecka, Z.; Pérez-Berlanga, M.; Hruska-Plochan, M.; Gilhespy, L.; Hock, E.M.; Wagner, U.; Afroz, T.; Boersema, P.J.; Barmettler, G.; et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci. 2019, 22, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Wils, H.; Kleinberger, G.; Janssens, J.; Pereson, S.; Joris, G.; Cuijt, I.; Smits, V.; Ceuterick-de Groote, C.; Van Broeckhoven, C.; Kumar-Singh, S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 3858–3863. [Google Scholar] [CrossRef] [Green Version]
- Baskaran, P.; Shaw, C.; Guthrie, S. TDP-43 causes neurotoxicity and cytoskeletal dysfunction in primary cortical neurons. PLoS ONE 2018, 13, e0196528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebstein, S.Y.; Yagudayeva, I.; Shneider, N.A. Mutant TDP-43 causes early-stage dose-dependent motor neuron degeneration in a TARDBP knock in mouse model of ALS. Cell Rep. 2019, 26, 364–373.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest. 2011, 121, 726–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.S.; Cheng, W.C.; Chen, C.Y.; Wu, M.C.; Wang, Y.C.; Tseng, Y.H.; Chuang, T.J.; Shen, C.J. Transcriptomopathies of pre- and post-symptomatic frontotemporal dementia-like mice with TDP-43 depletion in forebrain neurons. Acta Neuropathol. Commun. 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donde, A.; Sun, M.; Ling, J.P.; Braunstein, K.E.; Pang, B.; Wen, X.; Cheng, X.; Chen, L.; Wong, P.C. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol. 2019, 138, 813–826. [Google Scholar] [CrossRef]
- Saldi, T.K.; Ash, P.E.; Wilson, G.; Gonzales, P.; Garrido-Lecca, A.; Roberts, C.M.; Dostal, V.; Gendron, T.F.; Stein, L.D.; Blumenthal, T.; et al. TDP-1, the Caenorhabditis elegans ortholog of TDP-43, limits the accumulation of double-stranded RNA. EMBO J. 2014, 33, 2947–2966. [Google Scholar] [CrossRef]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Irwin, D.J.; Grossman, M.; Robinson, J.L.; Toledo, J.B.; Fang, L.; Van Deerlin, V.M.; Ludolph, A.C.; Lee, V.M.; et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 2014, 127, 423–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riku, Y.; Watanabe, H.; Yoshida, M.; Mimuro, M.; Iwasaki, Y.; Masuda, M.; Ishigaki, S.; Katsuno, M.; Sobue, G. Marked involvement of the striatal efferent system in TAR DNA-binding protein 43 kDa-related frontotemporal lobar degeneration and amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2016, 75, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riku, Y.; Watanabe, H.; Yoshida, M.; Mimuro, M.; Iwasaki, Y.; Masuda, M.; Ishigaki, S.; Katsuno, M.; Sobue, G. Pathologic involvement of glutamatergic striatal inputs from the cortices in TAR DNA-binding protein 43 kDa-related frontotemporal lobar degeneration and amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2017, 76, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Riku, Y. Reappraisal of the anatomical spreading and propagation hypothesis about TDP-43 aggregation in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Neuropathology 2020, 40, 426–435. [Google Scholar] [CrossRef]
- Van den Bos, M.A.J.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and diagnosis of ALS: Insights from advances in neurophysiological techniques. Int. J. Mol. Sci. 2019, 20, 2818. [Google Scholar] [CrossRef] [Green Version]
- Müller, H.P.; Turner, M.R.; Grosskreutz, J.; Abrahams, S.; Bede, P.; Govind, V.; Prudlo, J.; Ludolph, A.C.; Filippi, M.; Kassubek, J.; et al. A large-scale multicentre cerebral diffusion tensor imaging study in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 570–579. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.; de Reus, M.A.; Scholtens, L.H.; van den Berg, L.H.; van den Heuvel, M.P. Simulating disease propagation across white matter connectome reveals anatomical substrate for neuropathology staging in amyotrophic lateral sclerosis. Neuroimage 2016, 124, 762–769. [Google Scholar] [CrossRef] [Green Version]
- Trojsi, F.; Caiazzo, G.; Corbo, D.; Piccirillo, G.; Cristillo, V.; Femiano, C.; Ferrantino, T.; Cirillo, M.; Monsurrò, M.R.; Esposito, F.; et al. Microstructural changes across different clinical milestones of disease in amyotrophic lateral sclerosis. PLoS ONE 2015, 10, e0119045. [Google Scholar] [CrossRef]
- Schulthess, I.; Gorges, M.; Müller, H.P.; Lulé, D.; Del Tredici, K.; Ludolph, A.C.; Kassubek, J. Functional connectivity changes resemble patterns of pTDP-43 pathology in amyotrophic lateral sclerosis. Sci. Rep. 2016, 6, 38391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, T.; Iijima, M.; Uchihara, T.; Ohashi, T.; Seilhean, D.; Duyckaerts, C.; Uchiyama, S. TDP-43 pathology progression along the olfactory pathway as a possible substrate for olfactory impairment in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2015, 74, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, T.; Uchihara, T.; Arai, N.; Mizutani, T.; Iwata, M. Progression of hippocampal degeneration in amyotrophic lateral sclerosis with or without memory impairment: Distinction from Alzheimer disease. Acta Neuropathol. 2009, 117, 35–44. [Google Scholar] [CrossRef]
- Dedeene, L.; Van Schoor, E.; Vandenberghe, R.; Van Damme, P.; Poesen, K.; Thal, D.R. Circadian sleep/wake-associated cells show dipeptide repeat protein aggregates in C9orf72-related ALS and FTLD cases. Acta Neuropathol. Commun. 2019, 7, 189. [Google Scholar] [CrossRef]
- Porta, S.; Xu, Y.; Restrepo, C.R.; Kwong, L.K.; Zhang, B.; Brown, H.J.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 2018, 9, 4220. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Iwata, M. Ultrastructural change of synapses of Betz cells in patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1999, 268, 29–32. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1996, 204, 53–56. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Sideris, D.I.; Carroll, E.; Rotariu, S.; Salomonsson, S.; Tzioras, M.; McKenzie, C.A.; Smith, C.; von Arnim, C.A.F.; Ludolph, A.C.; et al. Synapse loss in the prefrontal cortex is associated with cognitive decline in amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Fallini, C.; Bassell, G.J.; Rossoll, W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 2012, 21, 3703–3718. [Google Scholar] [CrossRef] [Green Version]
- Heyburn, L.; Hebron, M.L.; Smith, J.; Winston, C.; Bechara, J.; Li, Z.; Lonskaya, I.; Burns, M.P.; Harris, B.T.; Moussa, C.E. Tyrosine kinase inhibition reverses TDP-43 effects on synaptic protein expression, astrocytic function and amino acid dis-homeostasis. J. Neurochem. 2016, 139, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Jawaid, A.; Henstridge, C.M.; Valeri, A.; Merlini, M.; Robinson, J.L.; Lee, E.B.; Rose, J.; Appel, S.; Lee, V.M.; et al. TDP-43 depletion in microglia promotes amyloid clearance but also induces synapse loss. Neuron 2017, 95, 297–308.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, B.; Hruscha, A.; Hogl, S.; Banzhaf-Strathmann, J.; Strecker, K.; van der Zee, J.; Teucke, M.; Eimer, S.; Hegermann, J.; Kittelmann, M.; et al. Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc. Natl. Acad. Sci. USA 2013, 110, 4986–4991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klim, J.R.; Williams, L.A.; Limone, F.; Guerra San Juan, I.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R.; et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef]
- Udagawa, T.; Fujioka, Y.; Tanaka, M.; Honda, D.; Yokoi, S.; Riku, Y.; Ibi, D.; Nagai, T.; Yamada, K.; Watanabe, H.; et al. FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun. 2015, 6, 7098. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar] [CrossRef]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Jo, M.; Lee, S.; Kim, K.; Lee, S.; Kim, S.R.; Kim, H.J. Inhibition of MEK5 suppresses TDP-43 toxicity via the mTOR-independent activation of the autophagy-lysosome pathway. Biochem. Biophys. Res. Commun. 2019, 513, 925–932. [Google Scholar] [CrossRef]
- Leibiger, C.; Deisel, J.; Aufschnaiter, A.; Ambros, S.; Tereshchenko, M.; Verheijen, B.M.; Büttner, S.; Braun, R.J. TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum. Mol. Genet. 2018, 27, 1593–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A.; et al. Pathological proteins are transported by extracellular vesicles of sporadic amyotrophic lateral sclerosis patients. Front. Neurosci. 2018, 12, 487. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Wu, D.; Hu, C.J.; Chen, H.Y.; Hsieh, Y.C.; Huang, C.C. Exosomal TAR DNA-binding protein-43 and neurofilaments in plasma of amyotrophic lateral sclerosis patients: A longitudinal follow-up study. J. Neurol. Sci. 2020, 418, 117070. [Google Scholar] [CrossRef]
- Hardy, J.; Rogaeva, E. Motor neuron disease and frontotemporal dementia: Sometimes related, sometimes not. Exp. Neurol. 2014, 262, 75–83. [Google Scholar] [CrossRef]
- Smith, K.R.; Damiano, J.; Franceschetti, S.; Carpenter, S.; Canafoglia, L.; Morbin, M.; Rossi, G.; Pareyson, D.; Mole, S.E.; Staropoli, J.F.; et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet. 2012, 90, 1102–1107. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.H.; Hung, S.T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- Riku, Y.; Duyckaerts, C.; Boluda, S.; Plu, I.; Le Ber, I.; Millecamps, S.; Salachas, F.; Yoshida, M.; Ando, T.; Brainbank NeuroCEB Neuropathology Network. Increased prevalence of granulovacuolar degeneration in C9orf72 mutation. Acta Neuropathol. 2019, 138, 783–793. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef]
- Van Schoor, E.; Koper, M.J.; Ospitalieri, S.; Dedeene, L.; Tomé, S.O.; Vandenberghe, R.; Brenner, D.; Otto, M.; Weishaupt, J.; Ludolph, A.C.; et al. Necrosome-positive granulovacuolar degeneration is associated with TDP-43 pathological lesions in the hippocampus of ALS/FTLD cases. Neuropathol. Appl. Neurobiol. 2021, 47, 328–345. [Google Scholar] [CrossRef]
- Adachi, H.; Katsuno, M.; Minamiyama, M.; Sang, C.; Pagoulatos, G.; Angelidis, C.; Kusakabe, M.; Yoshiki, A.; Kobayashi, Y.; Doyu, M.; et al. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J. Neurosci. 2003, 23, 2203–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, N.; Katsuno, M.; Riku, Y.; Sobue, G. HSF inhibits the progression of age-related neurodegenerative diseases. In Heat Shock Factor; Springer: Tokyo, Japan, 2016; pp. 213–242. [Google Scholar]
- Chen, H.J.; Mitchell, J.C.; Novoselov, S.; Miller, J.; Nishimura, A.L.; Scotter, E.L.; Vance, C.A.; Cheetham, M.E.; Shaw, C.E. The heat shock response plays an important role in TDP-43 clearance: Evidence for dysfunction in amyotrophic lateral sclerosis. Brain 2016, 139, 1417–1432. [Google Scholar] [CrossRef] [Green Version]
- Ormeño, F.; Hormazabal, J.; Moreno, J.; Riquelme, F.; Rios, J.; Criollo, A.; Albornoz, A.; Alfaro, I.E.; Budini, M. Chaperone mediated autophagy degrades TDP-43 protein and is affected by TDP-43 aggregation. Front. Mol. Neurosci. 2020, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lu, S.; Gasior, K.; Singh, D.; Vazquez-Sanchez, S.; Tapia, O.; Toprani, D.; Beccari, M.S.; Yates, J.R., 3rd; Da Cruz, S.; et al. HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 2021, 371, eabb4309. [Google Scholar] [CrossRef]
- Braak, H.; Ludolph, A.C.; Neumann, M.; Ravits, J.; Del Tredici, K. Pathological TDP-43 changes in Betz cells differ from those in bulbar and spinal α-motoneurons in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Giordana, M.T.; Piccinini, M.; Grifoni, S.; De Marco, G.; Vercellino, M.; Magistrello, M.; Pellerino, A.; Buccinnà, B.; Lupino, E.; Rinaudo, M.T. TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol. 2010, 20, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasetto, L.; Pozzi, S.; Castelnovo, M.; Basso, M.; Estevez, A.G.; Fumagalli, S.; De Simoni, M.G.; Castellaneta, V.; Bigini, P.; Restelli, E.; et al. Targeting extracellular cyclophilin A reduces neuroinflammation and extends survival in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2017, 37, 1413–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smethurst, P.; Risse, E.; Tyzack, G.E.; Mitchell, J.S.; Taha, D.M.; Chen, Y.R.; Newcombe, J.; Collinge, J.; Sidle, K.; Patani, R. Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis. Brain 2020, 143, 430–440. [Google Scholar] [CrossRef]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef]
- LaRocca, T.J.; Mariani, A.; Watkins, L.R.; Link, C.D. TDP-43 knockdown causes innate immune activation via protein kinase R in astrocytes. Neurobiol. Dis. 2019, 132, 104514. [Google Scholar] [CrossRef]
- Haidet-Phillips, A.M.; Gross, S.K.; Williams, T.; Tuteja, A.; Sherman, A.; Ko, M.; Jeong, Y.H.; Wong, P.C.; Maragakis, N.J. Altered astrocytic expression of TDP-43 does not influence motor neuron survival. Exp. Neurol. 2013, 250, 250–259. [Google Scholar] [CrossRef]
- Josephs, K.A.; Martin, P.R.; Weigand, S.D.; Tosakulwong, N.; Buciuc, M.; Murray, M.E.; Petrucelli, L.; Senjem, M.L.; Spychalla, A.J.; Knopman, D.S.; et al. Protein contributions to brain atrophy acceleration in Alzheimer’s disease and primary age-related tauopathy. Brain 2020, 143, 3463–3476. [Google Scholar] [CrossRef]
- Tomé, S.O.; Vandenberghe, R.; Ospitalieri, S.; Van Schoor, E.; Tousseyn, T.; Otto, M.; von Arnim, C.A.F.; Thal, D.R. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: Relationship with clinical phenotypes. Acta Neuropathol. Commun. 2020, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Sanchez-Contreras, M.; Josephs, K.A.; Uitti, R.J.; Graff-Radford, N.; van Gerpen, J.A.; Cheshire, W.P.; Wszolek, Z.K.; Rademakers, R.; Dickson, D.W. Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov. Disord. 2017, 32, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, S.; Kouri, N.; Walton, R.L.; Ebbert, M.T.W.; Josephs, K.A.; Litvan, I.; Graff-Radford, N.; Ahlskog, J.E.; Uitti, R.J.; van Gerpen, J.A.; et al. Corticobasal degeneration with TDP-43 pathology presenting with progressive supranuclear palsy syndrome: A distinct clinicopathologic subtype. Acta Neuropathol. 2018, 136, 389–404. [Google Scholar] [CrossRef] [Green Version]
- McAleese, K.E.; Walker, L.; Erskine, D.; Thomas, A.J.; McKeith, I.G.; Attems, J. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol. 2017, 27, 472–479. [Google Scholar] [CrossRef]
- Aoki, N.; Murray, M.E.; Ogaki, K.; Fujioka, S.; Rutherford, N.J.; Rademakers, R.; Ross, O.A.; Dickson, D.W. Hippocampal sclerosis in Lewy body disease is a TDP-43 proteinopathy similar to FTLD-TDP Type A. Acta Neuropathol. 2015, 129, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima-Yasuda, H.; Uryu, K.; Robinson, J.; Xie, S.X.; Hurtig, H.; Duda, J.E.; Arnold, S.E.; Siderowf, A.; Grossman, M.; Leverenz, J.B.; et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007, 114, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef]
- Amador-Ortiz, C.; Lin, W.L.; Ahmed, Z.; Personett, D.; Davies, P.; Duara, R.; Graff-Radford, N.R.; Hutton, M.L.; Dickson, D.W. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 2007, 61, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Kara, E.; Bandopadhyay, R.; Hardy, J.; Holton, J.; Xiromerisiou, G.; Lees, A.; Houlden, H.; Revesz, T. TDP-43 pathology in a patient carrying G2019S LRRK2 mutation and a novel p.Q124E MAPT. Neurobiol Aging. 2013, 34, 2889.e5–2889.e9. [Google Scholar] [CrossRef] [Green Version]
- Kenney, K.; Iacono, D.; Edlow, B.L.; Katz, D.I.; Diaz-Arrastia, R.; Dams-O’Connor, K.; Daneshvar, D.H.; Stevens, A.; Moreau, A.L.; Tirrell, L.S.; et al. Dementia after moderate-severe traumatic brain injury: Coexistence of multiple proteinopathies. J. Neuropathol. Exp. Neurol. 2018, 77, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.; Yan, N.; Caswell, C.; Xie, S.X.; Suh, E.; Van Deerlin, V.M.; Gibbons, G.; Irwin, D.J.; Grossman, M.; Lee, E.B.; et al. Primary tau pathology, not copathology, correlates with clinical symptoms in PSP and CBD. J. Neuropathol. Exp. Neurol. 2020, 79, 296–304. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Braak, H.; Brion, J.P.; Buée, L.; Del Tredici, K.; Goedert, M.; Halliday, G.; Neumann, M.; Spillantini, M.G.; Tolnay, M.; et al. PART is part of Alzheimer disease. Acta Neuropathol. 2015, 129, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.H.; Guthrie, N.J.; van Roijen, M.; Halliday, G.M.; Ooi, L. Increased tau phosphorylation in motor neurons from clinically pure sporadic amyotrophic lateral sclerosis patients. J. Neuropathol. Exp. Neurol. 2019, 78, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Behrouzi, R.; Liu, X.; Wu, D.; Robinson, A.C.; Tanaguchi-Watanabe, S.; Rollinson, S.; Shi, J.; Tian, J.; Hamdalla, H.H.; Ealing, J.; et al. Pathological tau deposition in motor neurone disease and frontotemporal lobar degeneration associated with TDP-43 proteinopathy. Acta Neuropathol. Commun. 2016, 4, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieniek, K.F.; Murray, M.E.; Rutherford, N.J.; Castanedes-Casey, M.; DeJesus-Hernandez, M.; Liesinger, A.M.; Baker, M.C.; Boylan, K.B.; Rademakers, R.; Dickson, D.W. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol. 2013, 125, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.C.; Thompson, J.C.; Weedon, L.; Rollinson, S.; Pickering-Brown, S.; Snowden, J.S.; Davidson, Y.S.; Mann, D.M. No interaction between tau and TDP-43 pathologies in either frontotemporal lobar degeneration or motor neurone disease. Neuropathol. Appl. Neurobiol. 2014, 40, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Mimuro, M.; Yoshida, M.; Kuzuhara, S.; Kokubo, Y. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: A multiple proteinopathy? Neuropathology 2018, 38, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheijen, B.M.; Morimoto, S.; Sasaki, R.; Oyanagi, K.; Kokubo, Y.; Kuzuhara, S.; van Leeuwen, F.W. Expression of mutant ubiquitin and proteostasis impairment in Kii amyotrophic lateral sclerosis/parkinsonism-dementia complex brains. J. Neuropathol. Exp. Neurol. 2020, 79, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Riku, Y.; Fujioka, Y.; Endo, K.; Iwade, N.; Kawai, K.; Ishibashi, M.; Yokoi, S.; Katsuno, M.; Watanabe, H.; et al. Aberrant interaction between FUS and SFPQ in neurons in a wide range of FTLD spectrum diseases. Brain 2020, 143, 2398–2405. [Google Scholar] [CrossRef]
- Ishigaki, S.; Fujioka, Y.; Okada, Y.; Riku, Y.; Udagawa, T.; Honda, D.; Yokoi, S.; Endo, K.; Ikenaka, K.; Takagi, S.; et al. Altered tau isoform ratio caused by loss of FUS and SFPQ function leads to FTLD-like phenotypes. Cell Rep. 2017, 18, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Kim, E.; Duffy, A.; Adalbert, R.; Phillips, B.U.; Peters, O.M.; Stephenson, J.; Yang, S.; Massenzio, F.; Lin, Z.; et al. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat. Neurosci. 2018, 21, 552–563. [Google Scholar] [CrossRef]
- Yokoyama, J.S.; Karch, C.M.; Fan, C.C.; Bonham, L.W.; Kouri, N.; Ross, O.A.; Rademakers, R.; Kim, J.; Wang, Y.; Höglinger, G.U.; et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol. 2017, 133, 825–837. [Google Scholar] [CrossRef]
- Karch, C.M.; Wen, N.; Fan, C.C.; Yokoyama, J.S.; Kouri, N.; Ross, O.A.; Höglinger, G.; Müller, U.; Ferrari, R.; Hardy, J.; et al. Selective genetic overlap between amyotrophic lateral sclerosis and diseases of the frontotemporal dementia spectrum. JAMA Neurol. 2018, 75, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Cali, C.P.; Patino, M.; Tai, Y.K.; Ho, W.Y.; McLean, C.A.; Morris, C.M.; Seeley, W.W.; Miller, B.L.; Gaig, C.; Vonsattel, J.P.G.; et al. C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathol. 2019, 138, 795–811. [Google Scholar] [CrossRef]
- Uryu, K.; Nakashima-Yasuda, H.; Forman, M.S.; Kwong, L.K.; Clark, C.M.; Grossman, M.; Miller, B.L.; Kretzschmar, H.A.; Lee, V.M.; Trojanowski, J.Q.; et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 2008, 67, 555–564. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riku, Y.; Seilhean, D.; Duyckaerts, C.; Boluda, S.; Iguchi, Y.; Ishigaki, S.; Iwasaki, Y.; Yoshida, M.; Sobue, G.; Katsuno, M. Pathway from TDP-43-Related Pathology to Neuronal Dysfunction in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. Int. J. Mol. Sci. 2021, 22, 3843. https://doi.org/10.3390/ijms22083843
Riku Y, Seilhean D, Duyckaerts C, Boluda S, Iguchi Y, Ishigaki S, Iwasaki Y, Yoshida M, Sobue G, Katsuno M. Pathway from TDP-43-Related Pathology to Neuronal Dysfunction in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. International Journal of Molecular Sciences. 2021; 22(8):3843. https://doi.org/10.3390/ijms22083843
Chicago/Turabian StyleRiku, Yuichi, Danielle Seilhean, Charles Duyckaerts, Susana Boluda, Yohei Iguchi, Shinsuke Ishigaki, Yasushi Iwasaki, Mari Yoshida, Gen Sobue, and Masahisa Katsuno. 2021. "Pathway from TDP-43-Related Pathology to Neuronal Dysfunction in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration" International Journal of Molecular Sciences 22, no. 8: 3843. https://doi.org/10.3390/ijms22083843
APA StyleRiku, Y., Seilhean, D., Duyckaerts, C., Boluda, S., Iguchi, Y., Ishigaki, S., Iwasaki, Y., Yoshida, M., Sobue, G., & Katsuno, M. (2021). Pathway from TDP-43-Related Pathology to Neuronal Dysfunction in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. International Journal of Molecular Sciences, 22(8), 3843. https://doi.org/10.3390/ijms22083843