
Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation


Abstract
:1. Introduction
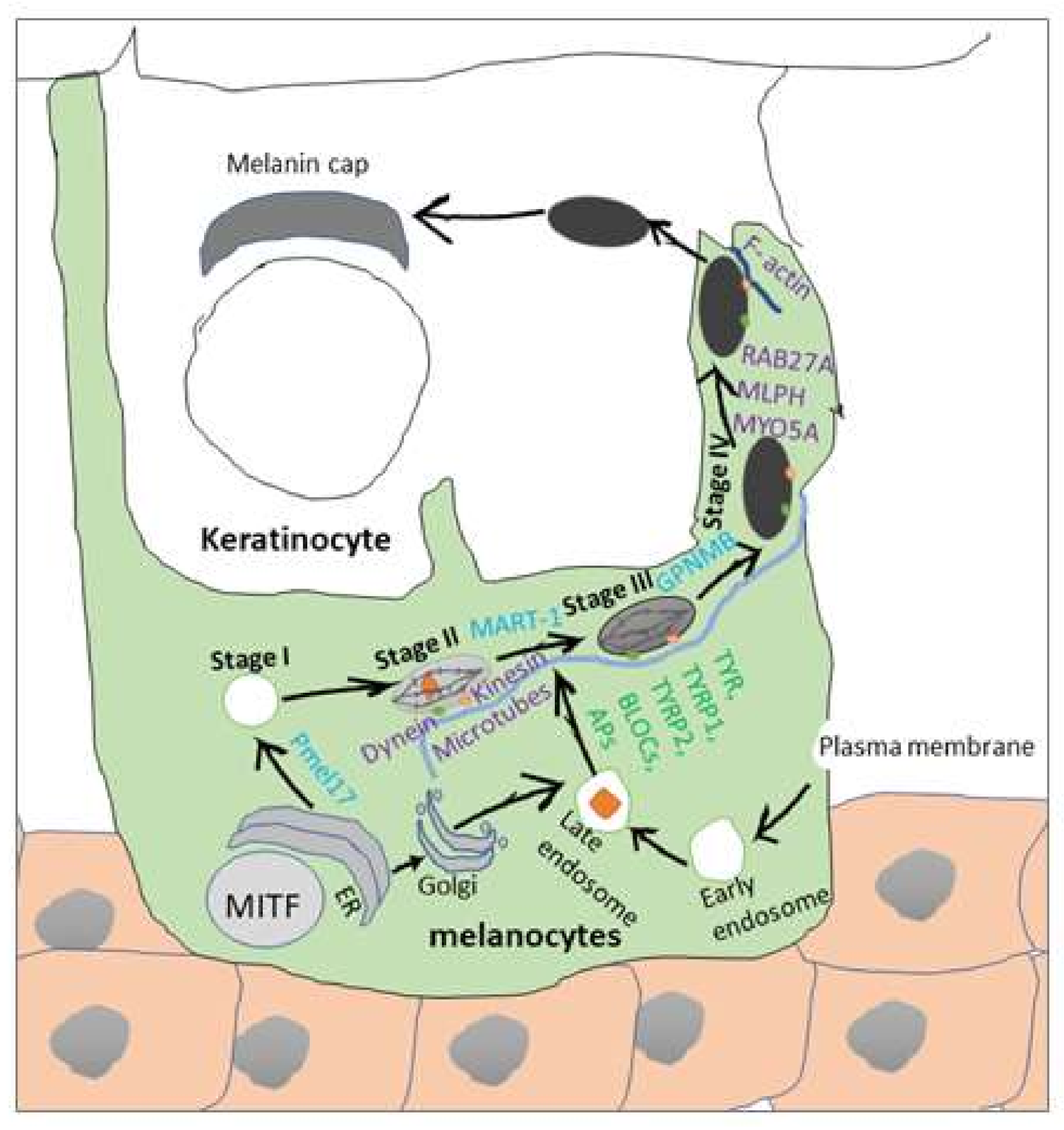
2. Melanogenesis Process
3. Melanogenesis Regulation
Involvement of Signaling Pathways in Melanogenesis Regulation
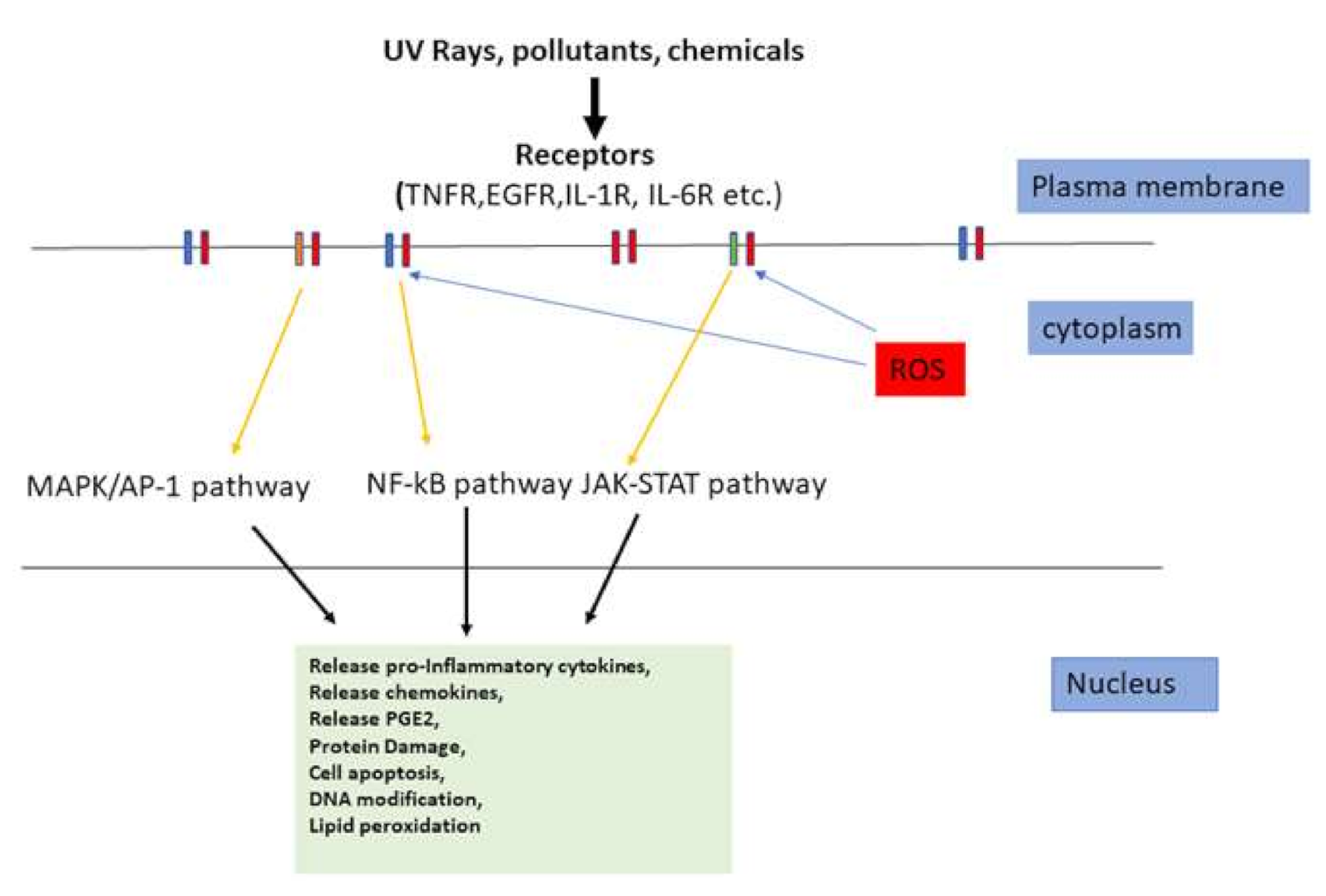
4. Skin Inflammation
5. Inflammatory Cytokines
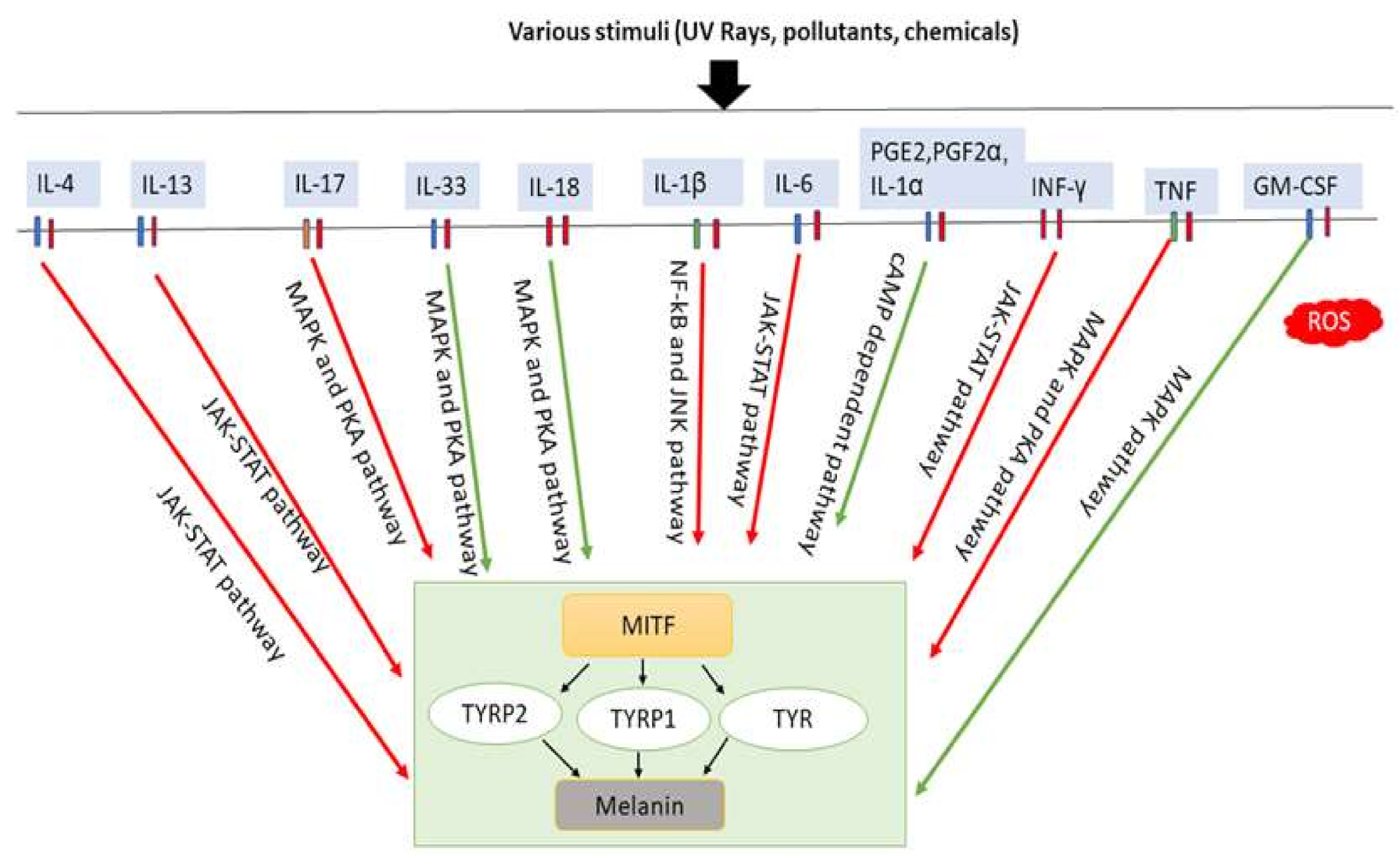
6. Inflammatory Cytokines Induce Melanogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokines | Main Source | Cells Used for Experiment | Impact on Melanogenesis | Mechanisms of Melanogenesis | Refs. |
|---|---|---|---|---|---|
| IL-4 | Th-cells | Melanocytes | Inhibition | JAK2-STAT6 signaling pathway is used for downregulating the expression of MITF, TYRP1, TYRP2 | [16] |
| IL-13 | Th2 | γδ T-cell | Inhibition | Suppress tyrosinase and DCT through the JAK2–STAT6 | [107] |
| IL-17 | Epithelial cells, Endothelial cells, Fibroblasts | Melanocytes, Primary pooled human keratinocytes | Inhibition | TNF and IL-17 simultaneously inhibit melanin formation through PKA and MAPK signaling pathways | [93] |
| IL-33 | Keratinocytes, Fibroblasts | Keratinocytes | Promotion | Activate p38 and PKA pathways and thus promote MITF, TYR, TYRP1, TYRP2 expression | [111,112] |
| IL-18 | Monocytes/Macrophages, Keratinocytes | Melanocytes | Promotion | Increase the activity of tyrosinase and upregulate the expression Of TYRP1 and TYRP2 | [113] |
| IL-1α | Langerhans cells, Melanocytes, keratinocytes | Primary melanocytes and swine skin | Promotion | Melanin deposition is increased when IL-1α is combined with KGF | [114] |
| IL-1β | Monocytes/Macrophages, Keratinocytes | Melanoma cell lines (LB2259-MEL and CP50-MEL) | Inhibition | Through the NF-κB and JNK pathways, it downregulates MITF-M expression | [115] |
| IL-6 | Macrophages, T-cells, Adipocyte | Melanocytes | Inhibition | Tyrosinase activity is declined | [15] |
| PGE2 and PGF2α | Fibroblasts, Keratinocytes | Keratinocytes | Promotion | Activate cAMP-dependent pathway and stimulate melanocyte dendrite formation | [116] |
| INF-γ | T-cells, NK cells, NKT cells | B16F10 | Inhibition | Block melanosomal maturation and upregulate STAT1 phosphorylation | [117,118] |
| TNF | Monocytes, Macrophages, Keratinocytes, Dendritic cells, Th1, Th17 and Th22 | Melanocytes, Primary pooled human keratinocytes | Promotion | Stimulate PKA and MAPK signaling pathways with combination of IL17, and thus inhibit melanin formation | [93] |
| GM-CSF | Macrophages, Keratinocytes and Th cells | Melanocytes | Promotion | Stimulate MAPK pathway and promote melanocyte proliferation, melanin and synthesis | [119] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ER | Endoplasmic reticulum |
| PMEL17 | Melanocytes lineage-specific antigen gp100 |
| MART-1 | Melanoma antigen recognized by T cell-1 |
| GPNMB | Glycoprotein nonmetastatic melanoma protein b |
| TYR | Tyrosinase |
| TYRP1 | Tyrosinase-related protein-1 |
| TYRP2 | Tyrosinase-related protein-2 |
| BLOCs | Biogenesis of lysosome-related organelles complexes |
| AP-1 | Activator protein 1 |
| MLPH | Melanophilin |
| MYO5A | Myosin-Va |
| RAB27A | Ras-related protein Rab27A |
| MITF | Microphthalmia-associated transcription factor |
| DAMPs | Damage-associated molecular patterns |
| PAMPs | Pathogen-associated molecular patterns |
| PRRs | Pattern-recognition receptors |
| TLRs | Toll-like receptors; |
| NLRs | NOD-like receptors |
| RLRs | Retinoic acid-inducible gene-I-like receptors |
| MyD88 | Myeloid differentiation primary response protein-88 |
| TAM | Tyro3, Axl, and Mer receptors |
| NF-κβ | Nuclear factor kappa-B |
| IRF | Interferon regulatory factor |
| IL | Interleukin |
| TNF | Tumor necrosis factor |
| IKK | IκB kinase |
| JAK-STAT | Janus kinase-signal transducer and activator of transcription |
| MAPKs | Mitogen-activated protein kinases |
| MKK | MAPK kinase |
| MKKK | MAPK kinase kinase |
| Erk1/2 | Etxracellular signal-regulated kinase ½ |
| JNK | c-Jun N-terminal kinases |
| UVR | Ultraviolet radiation |
| TNFR | Tumor necrosis factor receptor |
| EGFR | Epidermal growth factor receptor |
| PGE2 | Prostaglandin E2 |
| PGF2α | Prostaglandin F2α |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| IFN-γ | Interferon-γ |
| PKA | Protein kinase A |
| NK cell | Natural killer cell |
| NKT cell | Natural killer T cell |
| Th | T helper cell |
| cAMP | Cyclic adenosine monophosphate |
| PAX3 | Paired box protein Pax-3 |
| SOX10 | Sex-determining region Y-box 10 |
| KIT | Stem cell growth factor receptor (CD117) |
| SCF | Stem cell growth factor |
| EDN | Endothelin |
| NGF | Nerve growth factor |
| HGF | Hepatocyte growth factor |
| α-MSH | α-Melanocyte-stimulating hormone |
| ACTH | Adrenocorticotropic hormone |
| MLPH | melanophilin |
| CREB | cAMP responsive-element binding protein |
| SIK | Salt-inducible kinase |
| CRTC | CREB-regulated transcription coactivator |
| MITF-M | Melanocytes-specific MITF isoform |
References
- Chuong, C.M.; Nickoloff, B.J.; Elias, P.M.; Goldsmith, L.A.; Macher, E.; Maderson, P.A.; Sundberg, J.P.; Tagami, H.; Plonka, P.M.; Thestrup-Pederson, K.; et al. What is the ‘true’ function of skin? Exp. Dermatol. 2002, 11, 159–187. [Google Scholar] [CrossRef]
- Brenner, M.; Hearing, V.J. The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 2008, 84, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Bonaventure, J.; Domingues, M.J.; Larue, L. Cellular and molecular mechanisms controlling the migration of melanocytes and melanoma cells. Pigment. Cell Melanoma Res. 2013, 26, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Jan Borovansky, P.A.R. Melanins and Melanosomes Biosynthesis, Biogenesis, Physiological, and Pathological Functions; Jan Borovansky, P.A.R., Ed.; Wiley-Blackwell: Weinheim, Germany, 2011; p. 429. [Google Scholar]
- Lei, T.C.; Virador, V.; Yasumoto, K.; Vieira, W.D.; Toyofuku, K.; Hearing, V.J. Stimulation of melanoblast pigmentation by 8-methoxypsoralen:the involvement of microphthalmia-associated transcription factor, the protein kinase a signal pathway, and proteasome-mediated degradation. J. Investig. Dermatol. 2002, 119, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Sviderskaya, E.V.; Hill, S.P.; Balachandar, D.; Barsh, G.S.; Bennett, D.C. Agouti signaling protein and other factors modulating differentiation and proliferation of immortal melanoblasts. Dev. Dyn. 2001, 221, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Tolleson, W.H. Human melanocyte biology, toxicology, and pathology. J. Environ. Sci. Health C Environ. Carcinog Ecotoxicol. Rev. 2005, 23, 105–161. [Google Scholar] [CrossRef]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef]
- Martin, S.F. Contact dermatitis: From pathomechanisms to immunotoxicology. Exp. Dermatol. 2012, 21, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.S.; Cho, J.S. Immunity against Staphylococcus aureus cutaneous infections. Nat. Rev. Immunol. 2011, 11, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Behrends, U.; Peter, R.U.; Hintermeier-Knabe, R.; Eissner, G.; Holler, E.; Bornkamm, G.W.; Caughman, S.W.; Degitz, K. Ionizing radiation induces human intercellular adhesion molecule-1 in vitro. J. Investig. Dermatol. 1994, 103, 726–730. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, J.; Kern, H. Modulation of UV-light-induced skin inflammation by D-alpha-tocopherol and L-ascorbic acid: A clinical study using solar simulated radiation. Free Radic. Biol. Med. 1998, 25, 1006–1012. [Google Scholar] [CrossRef]
- Bäsler, K.; Brandner, J.M. Tight junctions in skin inflammation. Pflugers Arch. 2017, 469, 3–14. [Google Scholar] [CrossRef]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Swope, V.B.; Abdel-Malek, Z.; Kassem, L.M.; Nordlund, J.J. Interleukins 1 alpha and 6 and tumor necrosis factor-alpha are paracrine inhibitors of human melanocyte proliferation and melanogenesis. J. Investig. Dermatol. 1991, 96, 180–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Choi, H.; Han, J.; Jin, S.H.; Park, J.Y.; Shin, D.W.; Lee, T.R.; Kim, K.; Lee, A.Y.; Noh, M. IL-4 inhibits the melanogenesis of normal human melanocytes through the JAK2-STAT6 signaling pathway. J. Investig. Dermatol. 2013, 133, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Park, H.Y.; Kosmadaki, M.; Yaar, M.; Gilchrest, B.A. Cellular mechanisms regulating human melanogenesis. Cell Mol. Life Sci. 2009, 66, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Fistarol, S.K.; Itin, P.H. Disorders of pigmentation. J. Dtsch. Dermatol. Ges. 2010, 8, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Tsatmali, M.; Ancans, J.; Thody, A.J. Melanocyte function and its control by melanocortin peptides. J. Histochem. Cytochem. 2002, 50, 125–133. [Google Scholar] [CrossRef]
- Costin, G.E.; Hearing, V.J. Human skin pigmentation: Melanocytes modulate skin color in response to stress. FASEB J. 2007, 21, 976–994. [Google Scholar] [CrossRef]
- Dessinioti, C.; Stratigos, A.J.; Rigopoulos, D.; Katsambas, A.D. A review of genetic disorders of hypopigmentation: Lessons learned from the biology of melanocytes. Exp. Dermatol. 2009, 18, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Imokawa, G. Melanocyte Activation Mechanisms and Rational Therapeutic Treatments of Solar Lentigos. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imokawa, G.; Ishida, K. Inhibitors of intracellular signaling pathways that lead to stimulated epidermal pigmentation: Perspective of anti-pigmenting agents. Int. J. Mol. Sci. 2014, 15, 8293–8315. [Google Scholar] [CrossRef] [Green Version]
- Seiberg, M. Keratinocyte-melanocyte interactions during melanosome transfer. Pigment. Cell Res. 2001, 14, 236–242. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K. Chemistry of mixed melanogenesis--pivotal roles of dopaquinone. Photochem. Photobiol. 2008, 84, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Yaar, M.; Byers, H.R.; Goukassian, D.; Fine, R.E.; Gonsalves, J.; Gilchrest, B.A. Kinesin participates in melanosomal movement along melanocyte dendrites. J. Investig. Dermatol. 2000, 114, 438–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, H.R.; Yaar, M.; Eller, M.S.; Jalbert, N.L.; Gilchrest, B.A. Role of cytoplasmic dynein in melanosome transport in human melanocytes. J. Investig. Dermatol. 2000, 114, 990–997. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, M.V. Signaling pathways in melanosome biogenesis and pathology. Int. J. Biochem. Cell Biol. 2010, 42, 1094–1104. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Hearing, V.J. Physiological factors that regulate skin pigmentation. Biofactors 2009, 35, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Hoashi, T.; Watabe, H.; Muller, J.; Yamaguchi, Y.; Vieira, W.D.; Hearing, V.J. MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J. Biol. Chem. 2005, 280, 14006–14016. [Google Scholar] [CrossRef] [Green Version]
- Hoashi, T.; Sato, S.; Yamaguchi, Y.; Passeron, T.; Tamaki, K.; Hearing, V.J. Glycoprotein nonmetastatic melanoma protein b, a melanocytic cell marker, is a melanosome-specific and proteolytically released protein. FASEB J. 2010, 24, 1616–1629. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Liu, W.; Zhu, C.; Yuan, X.; Li, D.; Gu, W.; Ma, H.; Xie, X.; Gao, T. Silencing of GPNMB by siRNA inhibits the formation of melanosomes in melanocytes in a MITF-independent fashion. PLoS ONE 2012, 7, e42955. [Google Scholar] [CrossRef] [Green Version]
- Schallreuter, K.U.; Kothari, S.; Chavan, B.; Spencer, J.D. Regulation of melanogenesis--controversies and new concepts. Exp. Dermatol. 2008, 17, 395–404. [Google Scholar] [CrossRef]
- Park, H.Y.; Perez, J.M.; Laursen, R.; Hara, M.; Gilchrest, B.A. Protein kinase C-beta activates tyrosinase by phosphorylating serine residues in its cytoplasmic domain. J. Biol. Chem. 1999, 274, 16470–16478. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Hearing, V.J. Update on the regulation of mammalian melanocyte function and skin pigmentation. Expert Rev. Dermatol. 2011, 6, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, T.S.; Ariga, H.; Fukuda, M. The actin-binding domain of Slac2-a/melanophilin is required for melanosome distribution in melanocytes. Mol. Cell Biol. 2003, 23, 5245–5255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Bossche, K.; Naeyaert, J.M.; Lambert, J. The quest for the mechanism of melanin transfer. Traffic 2006, 7, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Vachtenheim, J.; Borovanský, J. “Transcription physiology” of pigment formation in melanocytes: Central role of MITF. Exp. Dermatol. 2010, 19, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Fisher, D.E. Lighting a path to pigmentation: Mechanisms of MITF induction by UV. Pigment. Cell Melanoma Res. 2010, 23, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y. Biology of melanocytes. In Fitzpatrick’s Dermatology in General Medicine; McGraw-Hill: New York, NY, USA, 2008; pp. 591–608. [Google Scholar]
- Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef]
- Mujahid, N.; Liang, Y.; Murakami, R.; Choi, H.G.; Dobry, A.S.; Wang, J.; Suita, Y.; Weng, Q.Y.; Allouche, J.; Kemeny, L.V.; et al. A UV-Independent Topical Small-Molecule Approach for Melanin Production in Human Skin. Cell Rep. 2017, 19, 2177–2184. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Brenner, M.; Hearing, V.J. The regulation of skin pigmentation. J. Biol. Chem. 2007, 282, 27557–27561. [Google Scholar] [CrossRef] [Green Version]
- Bennett, D.C.; Lamoreux, M.L. The color loci of mice—a genetic century. Pigment. Cell Res. 2003, 16, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.H.; Jin, Z.H. Paracrine regulation of melanogenesis. Br. J. Dermatol. 2018, 178, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Swope, V.B.; Abdel-Malek, Z.A. MC1R: Front and Center in the Bright Side of Dark Eumelanin and DNA Repair. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grando, S.A.; Pittelkow, M.R.; Schallreuter, K.U. Adrenergic and cholinergic control in the biology of epidermis: Physiological and clinical significance. J. Investig. Dermatol. 2006, 126, 1948–1965. [Google Scholar] [CrossRef] [Green Version]
- Besmer, P.; Murphy, J.E.; George, P.C.; Qiu, F.H.; Bergold, P.J.; Lederman, L.; Snyder, H.W., Jr.; Brodeur, D.; Zuckerman, E.E.; Hardy, W.D. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature 1986, 320, 415–421. [Google Scholar] [CrossRef]
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. Embo J. 1987, 6, 3341–3351. [Google Scholar] [CrossRef]
- Lim, X.; Nusse, R. Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Dorsky, R.I.; Moon, R.T.; Raible, D.W. Control of neural crest cell fate by the Wnt signalling pathway. Nature 1998, 396, 370–373. [Google Scholar] [CrossRef]
- Takeda, K.; Yasumoto, K.; Takada, R.; Takada, S.; Watanabe, K.; Udono, T.; Saito, H.; Takahashi, K.; Shibahara, S. Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. J. Biol. Chem. 2000, 275, 14013–14016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, E.J.; Erickson, C.A.; Takada, S.; Burrus, L.W. Wnt and BMP signaling govern lineage segregation of melanocytes in the avian embryo. Dev. Biol. 2001, 233, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Dunn, K.J.; Brady, M.; Ochsenbauer-Jambor, C.; Snyder, S.; Incao, A.; Pavan, W.J. WNT1 and WNT3a promote expansion of melanocytes through distinct modes of action. Pigment. Cell Res. 2005, 18, 167–180. [Google Scholar] [CrossRef]
- Dorsky, R.I.; Raible, D.W.; Moon, R.T. Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev. 2000, 14, 158–162. [Google Scholar] [PubMed]
- Flaherty, K.T.; Hodi, F.S.; Fisher, D.E. From genes to drugs: Targeted strategies for melanoma. Nat. Rev. Cancer 2012, 12, 349–361. [Google Scholar] [CrossRef]
- Widlund, H.R.; Horstmann, M.A.; Price, E.R.; Cui, J.; Lessnick, S.L.; Wu, M.; He, X.; Fisher, D.E. Beta-catenin-induced melanoma growth requires the downstream target Microphthalmia-associated transcription factor. J. Cell Biol. 2002, 158, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Benson, H.A.E.; Grice, J.E.; Mohammed, Y.; Namjoshi, S.; Roberts, M.S. Topical and Transdermal Drug Delivery: From Simple Potions to Smart Technologies. Curr. Drug Deliv. 2019, 16, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.S.; Chang, H.; Hamm, F.; Elsner, P.; Thiele, J.J. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int. J. Dermatol. 2004, 43, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Kohen, R. Skin antioxidants: Their role in aging and in oxidative stress—New approaches for their evaluation. Biomed Pharm. 1999, 53, 181–192. [Google Scholar] [CrossRef]
- Trouba, K.J.; Hamadeh, H.K.; Amin, R.P.; Germolec, D.R. Oxidative stress and its role in skin disease. Antioxid. Redox Signal. 2002, 4, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Komarova, E.A. p53 and the Carcinogenicity of Chronic Inflammation. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Brusselle, G.; Bracke, K. Targeting immune pathways for therapy in asthma and chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. 5), S322–S328. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adib-Conquy, M.; Cavaillon, J.M. Stress molecules in sepsis and systemic inflammatory response syndrome. FEBS Lett. 2007, 581, 3723–3733. [Google Scholar] [CrossRef] [Green Version]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Hendrayani, S.F.; Al-Harbi, B.; Al-Ansari, M.M.; Silva, G.; Aboussekhra, A. The inflammatory/cancer-related IL-6/STAT3/NF-κB positive feedback loop includes AUF1 and maintains the active state of breast myofibroblasts. Oncotarget 2016, 7, 41974–41985. [Google Scholar] [CrossRef] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [Green Version]
- Henríquez-Olguín, C.; Altamirano, F.; Valladares, D.; López, J.R.; Allen, P.D.; Jaimovich, E. Altered ROS production, NF-κB activation and interleukin-6 gene expression induced by electrical stimulation in dystrophic mdx skeletal muscle cells. Biochim. Biophys. Acta 2015, 1852, 1410–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.L.; Labasi, J.M.; Zhu, Y.; Tang, X.; McClure, K.; Gabel, C.A.; Athar, M.; Bickers, D.R. Role of p38 MAPK in UVB-induced inflammatory responses in the skin of SKH-1 hairless mice. J. Investig. Dermatol. 2005, 124, 1318–1325. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Lee, E.; Lowes, M.A.; Haider, A.S.; Fuentes-Duculan, J.; Abello, M.V.; Chamian, F.; Cardinale, I.; Krueger, J.G. Prominent production of IL-20 by CD68+/CD11c+ myeloid-derived cells in psoriasis: Gene regulation and cellular effects. J. Investig. Dermatol. 2006, 126, 1590–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 increases the innate immunity of tissues. Immunity 2004, 21, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Dhar, A.; Young, M.R.; Colburn, N.H. The role of AP-1, NF-kappaB and ROS/NOS in skin carcinogenesis: The JB6 model is predictive. Mol. Cell Biochem. 2002, 234–235, 185–193. [Google Scholar] [CrossRef]
- Endoh, I.; Di Girolamo, N.; Hampartzoumian, T.; Cameron, B.; Geczy, C.L.; Tedla, N. Ultraviolet B irradiation selectively increases the production of interleukin-8 in human cord blood-derived mast cells. Clin. Exp. Immunol. 2007, 148, 161–167. [Google Scholar] [CrossRef]
- Valencia, A.; Kochevar, I.E. Nox1-based NADPH oxidase is the major source of UVA-induced reactive oxygen species in human keratinocytes. J. Investig. Dermatol. 2008, 128, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beak, S.M.; Lee, Y.S.; Kim, J.A. NADPH oxidase and cyclooxygenase mediate the ultraviolet B-induced generation of reactive oxygen species and activation of nuclear factor-kappaB in HaCaT human keratinocytes. Biochimie 2004, 86, 425–429. [Google Scholar] [CrossRef]
- Glady, A.; Tanaka, M.; Moniaga, C.S.; Yasui, M.; Hara-Chikuma, M. Involvement of NADPH oxidase 1 in UVB-induced cell signaling and cytotoxicity in human keratinocytes. Biochem. Biophys. Rep. 2018, 14, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shao, Y.; Zhou, J.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation-induces epidermal growth factor receptor (EGFR) nuclear translocation in human keratinocytes. J. Cell Biochem. 2009, 107, 873–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.P.; Wu, J.X.; Fan, Y.; Adamson, E.D. UV activates growth factor receptors via reactive oxygen intermediates. J. Cell Biol. 1996, 133, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tober, K.L.; Thomas-Ahner, J.M.; Kusewitt, D.F.; Oberyszyn, T.M. Effects of UVB on E prostanoid receptor expression in murine skin. J. Investig. Dermatol. 2007, 127, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Soontrapa, K.; Honda, T.; Sakata, D.; Yao, C.; Hirata, T.; Hori, S.; Matsuoka, T.; Kita, Y.; Shimizu, T.; Kabashima, K.; et al. Prostaglandin E2-prostaglandin E receptor subtype 4 (EP4) signaling mediates UV irradiation-induced systemic immunosuppression. Proc. Natl. Acad. Sci. USA 2011, 108, 6668–6673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthusamy, V.; Piva, T.J. The UV response of the skin: A review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Arch. Dermatol. Res. 2010, 302, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.S.; Wang, Z.Q.; Voorhees, J.; Fisher, G. EGF receptor crosstalks with cytokine receptors leading to the activation of c-Jun kinase in response to UV irradiation in human keratinocytes. Cell Signal. 2001, 13, 139–144. [Google Scholar] [CrossRef]
- Blanton, R.A.; Kupper, T.S.; McDougall, J.K.; Dower, S. Regulation of interleukin 1 and its receptor in human keratinocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 1273–1277. [Google Scholar] [CrossRef] [Green Version]
- López-Camarillo, C.; Ocampo, E.A.; Casamichana, M.L.; Pérez-Plasencia, C.; Alvarez-Sánchez, E.; Marchat, L.A. Protein kinases and transcription factors activation in response to UV-radiation of skin: Implications for carcinogenesis. Int. J. Mol. Sci. 2012, 13, 142–172. [Google Scholar] [CrossRef]
- Rosette, C.; Karin, M. Ultraviolet light and osmotic stress: Activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 1996, 274, 1194–1197. [Google Scholar] [CrossRef]
- Peus, D.; Meves, A.; Vasa, R.A.; Beyerle, A.; O’Brien, T.; Pittelkow, M.R. H2O2 is required for UVB-induced EGF receptor and downstream signaling pathway activation. Free Radic. Biol. Med. 1999, 27, 1197–1202. [Google Scholar] [CrossRef]
- Madson, J.G.; Hansen, L.A. Multiple mechanisms of Erbb2 action after ultraviolet irradiation of the skin. Mol. Carcinog. 2007, 46, 624–628. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Finkel, T. Signal. transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. An. inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015, 26, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Q.F.; Akalu, Y.T.; Suarez-Farinas, M.; Gonzalez, J.; Mitsui, H.; Lowes, M.A.; Orlow, S.J.; Manga, P.; Krueger, J.G. IL-17 and TNF synergistically modulate cytokine expression while suppressing melanogenesis: Potential relevance to psoriasis. J. Investig. Dermatol. 2013, 133, 2741–2752. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T.R.; Sad, S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 1996, 17, 138–146. [Google Scholar] [CrossRef]
- O’Garra, A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 1998, 8, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Reiner, S.L.; Seder, R.A. Dealing from the evolutionary pawnshop: How lymphocytes make decisions. Immunity 1999, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Barata, L.T.; Ying, S.; Meng, Q.; Barkans, J.; Rajakulasingam, K.; Durham, S.R.; Kay, A.B. IL-4- and IL-5-positive T lymphocytes, eosinophils, and mast cells in allergen-induced late-phase cutaneous reactions in atopic subjects. J. Allergy Clin. Immunol. 1998, 101, 222–230. [Google Scholar] [CrossRef]
- Min, B.; Prout, M.; Hu-Li, J.; Zhu, J.; Jankovic, D.; Morgan, E.S.; Urban, J.F., Jr.; Dvorak, A.M.; Finkelman, F.D.; LeGros, G.; et al. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J. Exp. Med. 2004, 200, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, M.; Laddha, N.C.; Dwivedi, M.; Mansuri, M.S.; Singh, J.; Rani, R.; Gokhale, R.S.; Sharma, V.K.; Marfatia, Y.S.; Begum, R. Interleukin-4 genetic variants correlate with its transcript and protein levels in patients with vitiligo. Br. J. Dermatol. 2012, 167, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Salgame, P.; Abrams, J.S.; Clayberger, C.; Goldstein, H.; Convit, J.; Modlin, R.L.; Bloom, B.R. Differing lymphokine profiles of functional subsets of human CD4 and CD8 T cell clones. Science 1991, 254, 279–282. [Google Scholar] [CrossRef]
- Basak, P.Y.; Adiloglu, A.K.; Ceyhan, A.M.; Tas, T.; Akkaya, V.B. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J. Am. Acad. Dermatol. 2009, 60, 256–260. [Google Scholar] [CrossRef]
- Nouri-Koupaee, A.; Mansouri, P.; Jahanbini, H.; Sanati, M.H.; Jadali, Z. Differential expression of mRNA for T-bet and GATA-3 transcription factors in peripheral blood mononuclear cells of patients with vitiligo. Clin. Exp. Dermatol. 2015, 40, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Rael, E.L.; Lockey, R.F. Interleukin-13 signaling and its role in asthma. World Allergy Organ J. 2011, 4, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, Y.; Nitto, T.; Inoue, T.; Node, K. IL-13 attenuates vascular tube formation via JAK2-STAT6 pathway. Circ. J. 2008, 72, 469–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renauld, J.C. New insights into the role of cytokines in asthma. J. Clin. Pathol. 2001, 54, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Han, J.; Lee, T.R.; Shin, D.W.; Chang, H.; Cho, A.R.; Choi, S.J.; Jo, S.J.; Kwon, O. Comparative Analysis of Human Epidermal and Peripheral Blood γδ T Cell Cytokine Profiles. Ann. Dermatol. 2014, 26, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Lee, E.; Kim, E.; Yeom, M.H.; Kwon, O.; Yoon, T.H.; Lee, T.R.; Kim, K. Role of epidermal γδ T-cell-derived interleukin 13 in the skin-whitening effect of Ginsenoside F1. Exp. Dermatol. 2014, 23, 860–862. [Google Scholar] [CrossRef]
- Speeckaert, R.; Lambert, J.; Grine, L.; Van Gele, M.; De Schepper, S.; van Geel, N. The many faces of interleukin-17 in inflammatory skin diseases. Br. J. Dermatol. 2016, 175, 892–901. [Google Scholar] [CrossRef]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupé, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Meephansan, J.; Komine, M.; Tsuda, H.; Tominaga, S.; Ohtsuki, M. Ultraviolet B irradiation induces the expression of IL-33 mRNA and protein in normal human epidermal keratinocytes. J. Dermatol. Sci. 2012, 65, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Song, J.; Ping, F.; Shang, J. Enhancement of the p38 MAPK and PKA signaling pathways is associated with the pro-melanogenic activity of Interleukin 33 in primary melanocytes. J. Dermatol. Sci. 2014, 73, 110–116. [Google Scholar] [CrossRef]
- Zhou, J.; Shang, J.; Song, J.; Ping, F. Interleukin-18 augments growth ability of primary human melanocytes by PTEN inactivation through the AKT/NF-κB pathway. Int. J. Biochem. Cell Biol. 2013, 45, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Hu, Y.; Li, W.H.; Eisinger, M.; Seiberg, M.; Lin, C.B. The role of keratinocyte growth factor in melanogenesis: A possible mechanism for the initiation of solar lentigines. Exp. Dermatol. 2010, 19, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Kholmanskikh, O.; van Baren, N.; Brasseur, F.; Ottaviani, S.; Vanacker, J.; Arts, N.; van der Bruggen, P.; Coulie, P.; De Plaen, E. Interleukins 1alpha and 1beta secreted by some melanoma cell lines strongly reduce expression of MITF-M and melanocyte differentiation antigens. Int. J. Cancer 2010, 127, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.; Leopardi, S.; Printup, S.; Malhi, N.; Seiberg, M.; Lapoint, R. Proteinase-activated receptor-2 stimulates prostaglandin production in keratinocytes: Analysis of prostaglandin receptors on human melanocytes and effects of PGE2 and PGF2alpha on melanocyte dendricity. J. Investig. Dermatol. 2004, 122, 1214–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, V.T.; Ganju, P.; Singh, A.; Vijayan, V.; Kirty, K.; Yadav, S.; Puntambekar, S.; Bajaj, S.; Dani, P.P.; Kar, H.K.; et al. IFN-γ signaling maintains skin pigmentation homeostasis through regulation of melanosome maturation. Proc. Natl. Acad. Sci. USA 2014, 111, 2301–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Ling, J.; Wang, Y.; Shang, J.; Ping, F. Cross-talk between interferon-gamma and interleukin-18 in melanogenesis. J. Photochem. Photobiol. B 2016, 163, 133–143. [Google Scholar] [CrossRef]
- Videira, I.F.; Moura, D.F.; Magina, S. Mechanisms regulating melanogenesis. An. Bras. Dermatol. 2013, 88, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 2008, 223, 20–38. [Google Scholar] [CrossRef]
- Byrne, S.N.; Beaugie, C.; O’Sullivan, C.; Leighton, S.; Halliday, G.M. The immune-modulating cytokine and endogenous Alarmin interleukin-33 is upregulated in skin exposed to inflammatory UVB radiation. Am. J. Pathol. 2011, 179, 211–222. [Google Scholar] [CrossRef]
- Ali, S.; Huber, M.; Kollewe, C.; Bischoff, S.C.; Falk, W.; Martin, M.U. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc. Natl. Acad. Sci. USA 2007, 104, 18660–18665. [Google Scholar] [CrossRef] [Green Version]
- Allakhverdi, Z.; Smith, D.E.; Comeau, M.R.; Delespesse, G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J. Immunol. 2007, 179, 2051–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulin, D.; Donzé, O.; Talabot-Ayer, D.; Mézin, F.; Palmer, G.; Gabay, C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine 2007, 40, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theoharides, T.C.; Zhang, B.; Kempuraj, D.; Tagen, M.; Vasiadi, M.; Angelidou, A.; Alysandratos, K.D.; Kalogeromitros, D.; Asadi, S.; Stavrianeas, N.; et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc. Natl. Acad. Sci. USA 2010, 107, 4448–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushparaj, P.N.; Tay, H.K.; H’Ng, S.C.; Pitman, N.; Xu, D.; McKenzie, A.; Liew, F.Y.; Melendez, A.J. The cytokine interleukin-33 mediates anaphylactic shock. Proc. Natl. Acad. Sci. USA 2009, 106, 9773–9778. [Google Scholar] [CrossRef] [Green Version]
- Kurowska-Stolarska, M.; Stolarski, B.; Kewin, P.; Murphy, G.; Corrigan, C.J.; Ying, S.; Pitman, N.; Mirchandani, A.; Rana, B.; van Rooijen, N.; et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 2009, 183, 6469–6477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, T.; Oboki, K.; Kajiwara, N.; Morii, E.; Aozasa, K.; Flavell, R.A.; Okumura, K.; Saito, H.; Nakae, S. Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. J. Immunol. 2009, 183, 7890–7897. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hueber, A.J.; Alves-Filho, J.C.; Asquith, D.L.; Michels, C.; Millar, N.L.; Reilly, J.H.; Graham, G.J.; Liew, F.Y.; Miller, A.M.; McInnes, I.B. IL-33 induces skin inflammation with mast cell and neutrophil activation. Eur. J. Immunol. 2011, 41, 2229–2237. [Google Scholar] [CrossRef]
- Suzukawa, M.; Iikura, M.; Koketsu, R.; Nagase, H.; Tamura, C.; Komiya, A.; Nakae, S.; Matsushima, K.; Ohta, K.; Yamamoto, K.; et al. An.IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J. Immunol. 2008, 181, 5981–5989. [Google Scholar] [CrossRef]
- Rank, M.A.; Kobayashi, T.; Kozaki, H.; Bartemes, K.R.; Squillace, D.L.; Kita, H. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 2009, 123, 1047–1054. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.L.; Chhiba, K.D.; Krier-Burris, R.; Hosakoppal, S.; Berdnikovs, S.; Miller, M.L.; Bryce, P.J. Allergic inflammation is initiated by IL-33-dependent crosstalk between mast cells and basophils. PLoS ONE 2020, 15, e0226701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cevikbas, F.; Steinhoff, M. IL-33: A novel danger signal system in atopic dermatitis. J. Investig. Dermatol. 2012, 132, 1326–1329. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Interleukin-1. Rev. Infect. Dis. 1984, 6, 51–95. [Google Scholar] [CrossRef]
- Kampschmidt, R.F. The numerous postulated biological manifestations of interleukin-1. J. Leukoc. Biol. 1984, 36, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupper, T.S. Immune and inflammatory processes in cutaneous tissues. Mechanisms and speculations. J. Clin. Investig. 1990, 86, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Auron, P.E.; Webb, A.C.; Rosenwasser, L.J.; Mucci, S.F.; Rich, A.; Wolff, S.M.; Dinarello, C.A. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc. Natl. Acad. Sci. USA 1984, 81, 7907–7911. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.E.; March, C.J.; Cosman, D.; Widmer, M.B.; MacDonald, H.R.; McMahan, C.J.; Grubin, C.E.; Wignall, J.M.; Jackson, J.L.; Call, S.M.; et al. cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science 1988, 241, 585–589. [Google Scholar] [CrossRef]
- March, C.J.; Mosley, B.; Larsen, A.; Cerretti, D.P.; Braedt, G.; Price, V.; Gillis, S.; Henney, C.S.; Kronheim, S.R.; Grabstein, K.; et al. Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature 1985, 315, 641–647. [Google Scholar] [CrossRef]
- Groves, R.W.; Sherman, L.; Mizutani, H.; Dower, S.K.; Kupper, T.S. Detection of interleukin-1 receptors in human epidermis. Induction of the type II receptor after organ culture and in psoriasis. Am. J. Pathol. 1994, 145, 1048–1056. [Google Scholar]
- Martin, M.U.; Wesche, H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim. Biophys. Acta 2002, 1592, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.; Gilchrest, B.A. Regulation of keratinocyte growth factor gene expression in human skin fibroblasts. J. Dermatol. Sci. 1996, 11, 41–50. [Google Scholar] [CrossRef]
- Groves, R.W.; Ross, E.; Barker, J.N.; Ross, J.S.; Camp, R.D.; MacDonald, D.M. Effect of in vivo interleukin-1 on adhesion molecule expression in normal human skin. J. Investig. Dermatol. 1992, 98, 384–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, S.; Sauder, D.N.; Kono, T.; Galley, K.A.; McKenzie, R.C. Differential modulation of interleukin-1 alpha (IL-1 alpha) and interleukin-1 beta (IL-1 beta) in human epidermal keratinocytes by UVB. Exp. Dermatol. 1994, 3, 29–39. [Google Scholar] [CrossRef]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Pentland, A.P.; Mahoney, M.; Jacobs, S.C.; Holtzman, M.J. Enhanced prostaglandin synthesis after ultraviolet injury is mediated by endogenous histamine stimulation. A mechanism for irradiation erythema. J. Clin. Investig. 1990, 86, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Pentland, A.P.; Mahoney, M.G. Keratinocyte prostaglandin synthesis is enhanced by IL-1. J. Investig. Dermatol. 1990, 94, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Fosslien, E. Molecular pathology of cyclooxygenase-2 in neoplasia. Ann. Clin. Lab. Sci. 2000, 30, 3–21. [Google Scholar]
- Pentland, A.P.; Needleman, P. Modulation of keratinocyte proliferation in vitro by endogenous prostaglandin synthesis. J. Clin. Investig. 1986, 77, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Scott, G.; Jacobs, S.; Leopardi, S.; Anthony, F.A.; Learn, D.; Malaviya, R.; Pentland, A. Effects of PGF2alpha on human melanocytes and regulation of the FP receptor by ultraviolet radiation. Exp. Cell Res. 2005, 304, 407–416. [Google Scholar] [CrossRef]
- Holtzman, M.J.; Zhang, V.; Hussain, H.; Roswit, W.T.; Wilson, J.D. Prostaglandin H synthase and lipoxygenase gene families in the epithelial cell barrier. Ann. N. Y. Acad. Sci. 1994, 744, 58–77. [Google Scholar] [CrossRef]
- Bach, E.A.; Aguet, M.; Schreiber, R.D. The IFN gamma receptor: A paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [Google Scholar] [CrossRef]
- Carnaud, C.; Lee, D.; Donnars, O.; Park, S.H.; Beavis, A.; Koezuka, Y.; Bendelac, A. Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J. Immunol. 1999, 163, 4647–4650. [Google Scholar]
- Frucht, D.M.; Fukao, T.; Bogdan, C.; Schindler, H.; O’Shea, J.J.; Koyasu, S. IFN-gamma production by antigen-presenting cells: Mechanisms emerge. Trends Immunol. 2001, 22, 556–560. [Google Scholar] [CrossRef]
- Flaishon, L.; Hershkoviz, R.; Lantner, F.; Lider, O.; Alon, R.; Levo, Y.; Flavell, R.A.; Shachar, I. Autocrine secretion of interferon gamma negatively regulates homing of immature B cells. J. Exp. Med. 2000, 192, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, E.K.; Suzuki, M.; Igras, V.; Du, J.; Lonning, S.; Miyachi, Y.; Roes, J.; Beermann, F.; Fisher, D.E. Key roles for transforming growth factor beta in melanocyte stem cell maintenance. Cell Stem Cell 2010, 6, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Malek, Z.; Scott, M.C.; Suzuki, I.; Tada, A.; Im, S.; Lamoreux, L.; Ito, S.; Barsh, G.; Hearing, V.J. The melanocortin-1 receptor is a key regulator of human cutaneous pigmentation. Pigment. Cell Res. 2000, 13 (Suppl. 8) (Suppl. 8), 156–162. [Google Scholar] [CrossRef]
- Harris, J.E.; Harris, T.H.; Weninger, W.; Wherry, E.J.; Hunter, C.A.; Turka, L.A. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-γ for autoreactive CD8+ T-cell accumulation in the skin. J. Investig. Dermatol. 2012, 132, 1869–1876. [Google Scholar] [CrossRef] [Green Version]
- Gregg, R.K.; Nichols, L.; Chen, Y.; Lu, B.; Engelhard, V.H. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J. Immunol. 2010, 184, 1909–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wei, Y.; Sun, Y.; Shi, W.; Yang, J.; Zhu, L.; Li, M. Interferon-gamma Inhibits Melanogenesis and Induces Apoptosis in Melanocytes: A Pivotal Role of CD8+ Cytotoxic T Lymphocytes in Vitiligo. Acta Derm. Venereol. 2015, 95, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, M.; Chu, C.Q.; Eedy, D.J.; Feldmann, M.; Brennan, F.M.; Breathnach, S.M. Localization of tumour necrosis factor-alpha (TNF-alpha) and its receptors in normal and psoriatic skin: Epidermal cells express the 55-kD but not the 75-kD TNF receptor. Clin. Exp. Immunol. 1993, 94, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Meephansan, J.; Tsuda, H.; Komine, M.; Tominaga, S.; Ohtsuki, M. Regulation of IL-33 expression by IFN-γ and tumor necrosis factor-α in normal human epidermal keratinocytes. J. Investig. Dermatol. 2012, 132, 2593–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meephansan, J.; Komine, M.; Tsuda, H.; Karakawa, M.; Tominaga, S.; Ohtsuki, M. Expression of IL-33 in the epidermis: The mechanism of induction by IL-17. J. Dermatol. Sci. 2013, 71, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Kim, D.S.; Kim, H.J.; Seo, K.I.; Kim, K.H.; Chung, J.H.; Eun, H.C.; Jung, H.C. Growth related secretion and production of GM-CSF by epithelial cell line. J. Dermatol. Sci. 2001, 25, 53–58. [Google Scholar] [CrossRef]
- Wu, X.G.; Hong, W.S.; Xu, A. GM-CSF: A possible prognostic serum biomarker of vitiligo patients’ considered for transplantation treatment with cultured autologous melanocytes: A pilot study. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1409–1411. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, M.R.; Ansary, T.M.; Komine, M.; Ohtsuki, M. Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation. Int. J. Mol. Sci. 2021, 22, 3970. https://doi.org/10.3390/ijms22083970
Hossain MR, Ansary TM, Komine M, Ohtsuki M. Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation. International Journal of Molecular Sciences. 2021; 22(8):3970. https://doi.org/10.3390/ijms22083970
Chicago/Turabian StyleHossain, Md Razib, Tuba M. Ansary, Mayumi Komine, and Mamitaro Ohtsuki. 2021. "Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation" International Journal of Molecular Sciences 22, no. 8: 3970. https://doi.org/10.3390/ijms22083970
APA StyleHossain, M. R., Ansary, T. M., Komine, M., & Ohtsuki, M. (2021). Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation. International Journal of Molecular Sciences, 22(8), 3970. https://doi.org/10.3390/ijms22083970