
The Role of GPR109a Signaling in Niacin Induced Effects on Fed and Fasted Hepatic Metabolism


Abstract
:1. Introduction
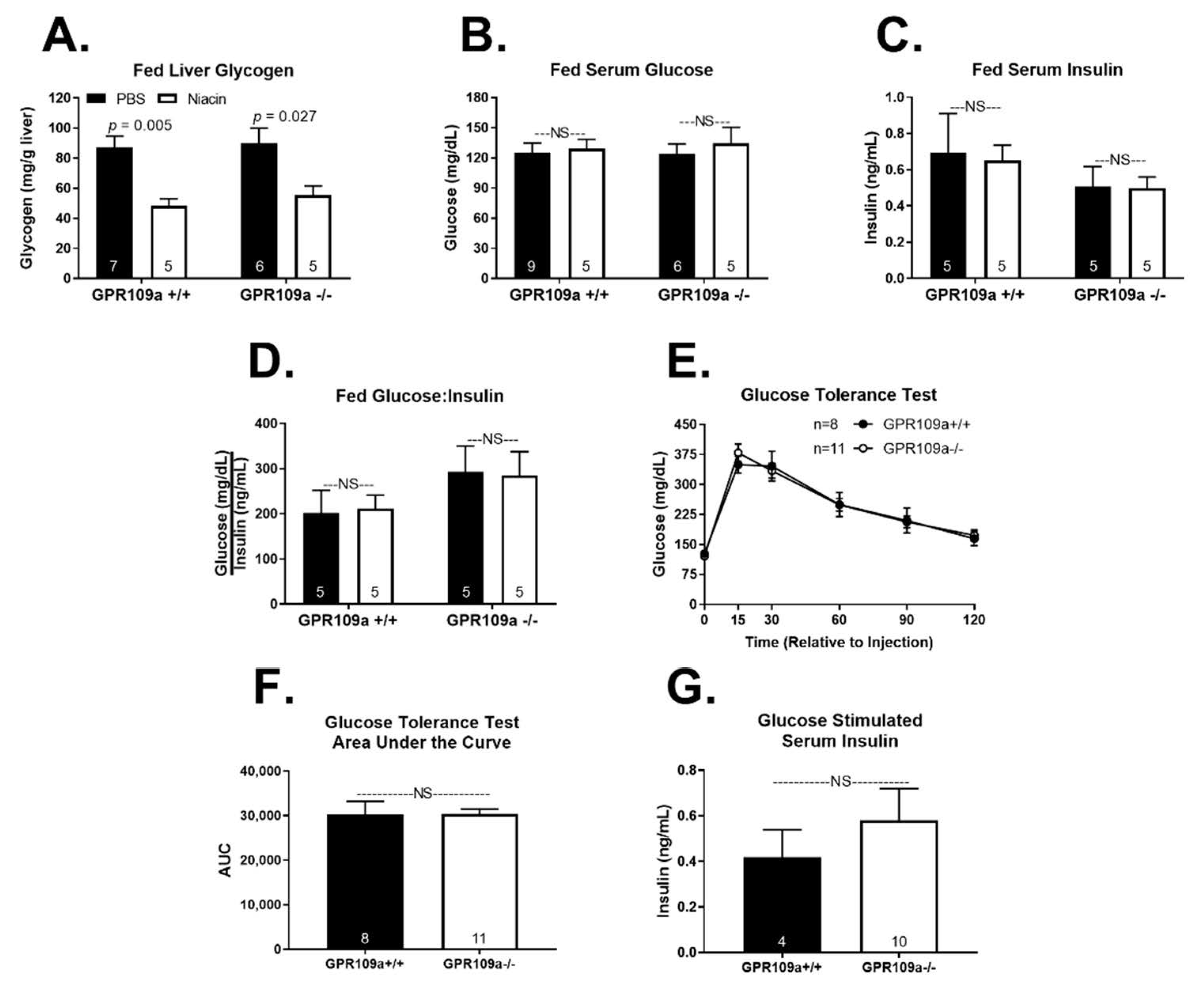
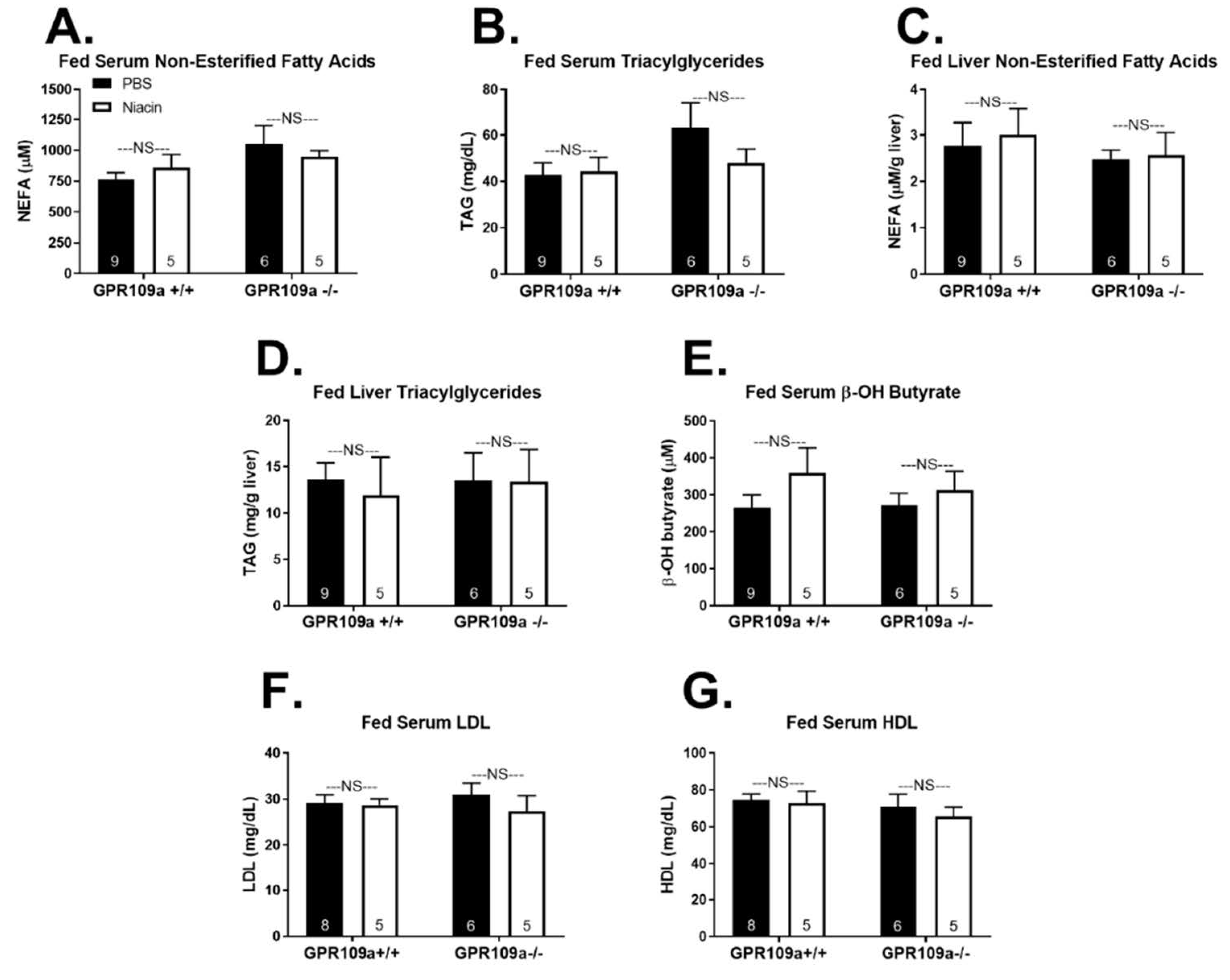
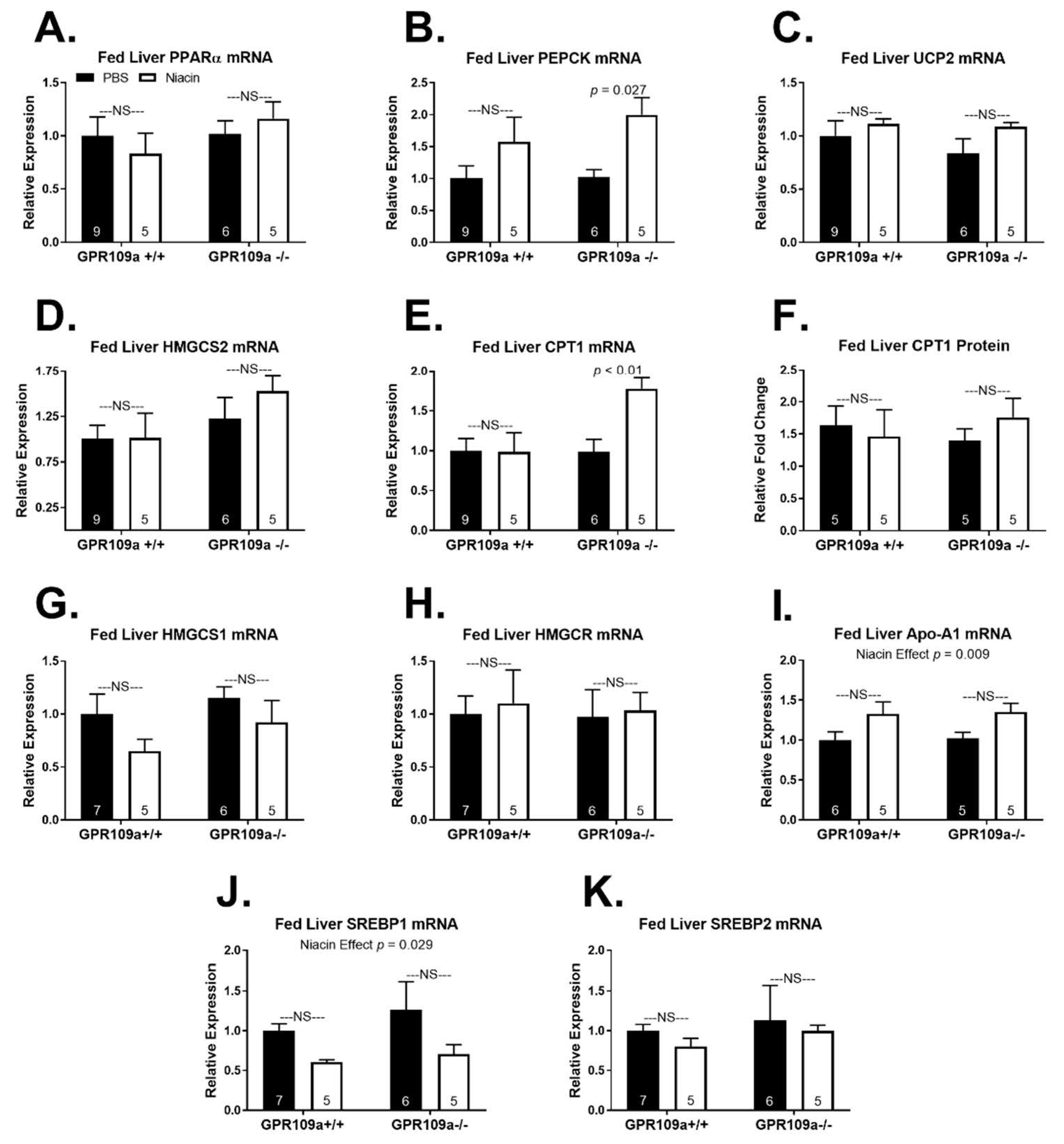
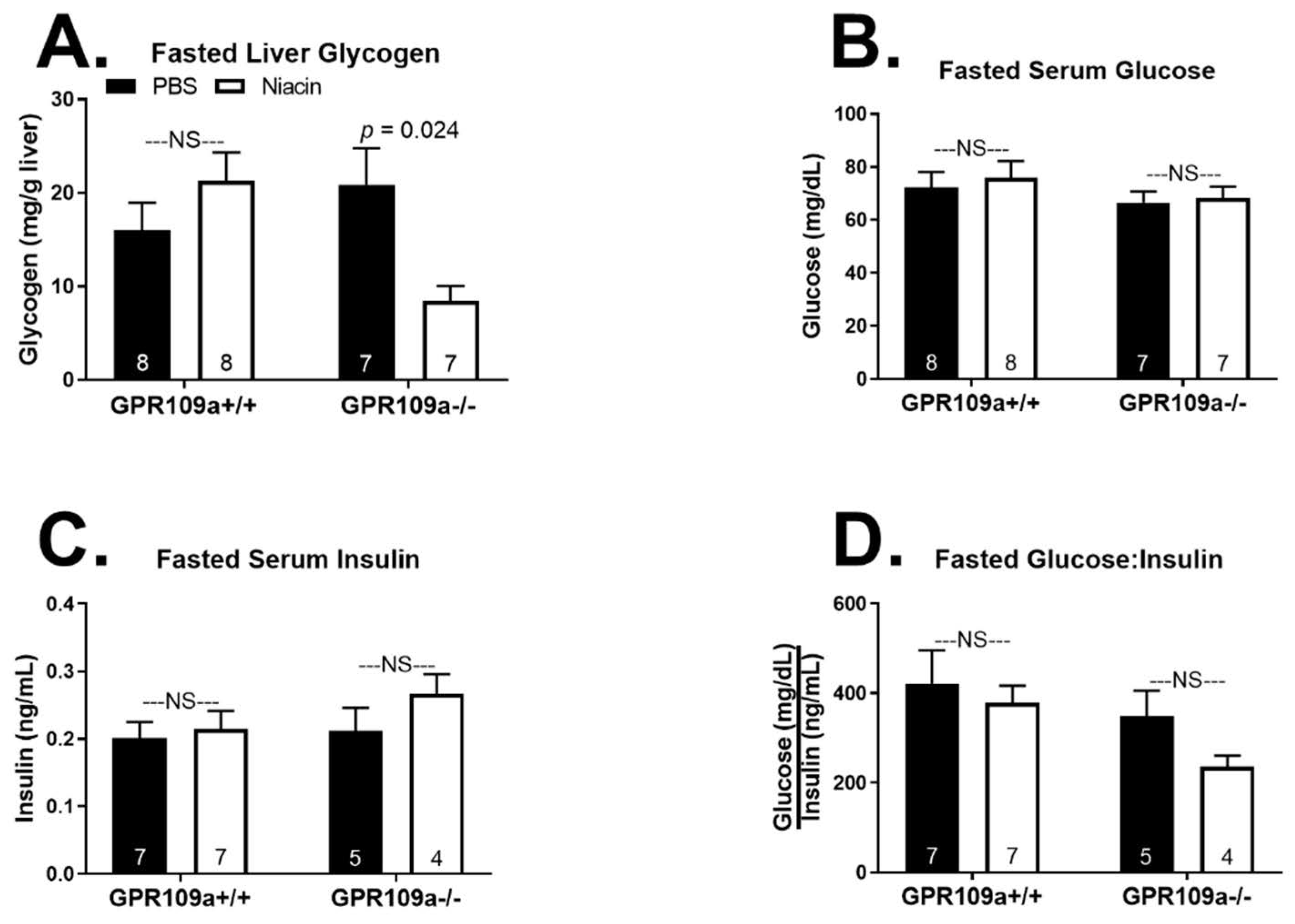
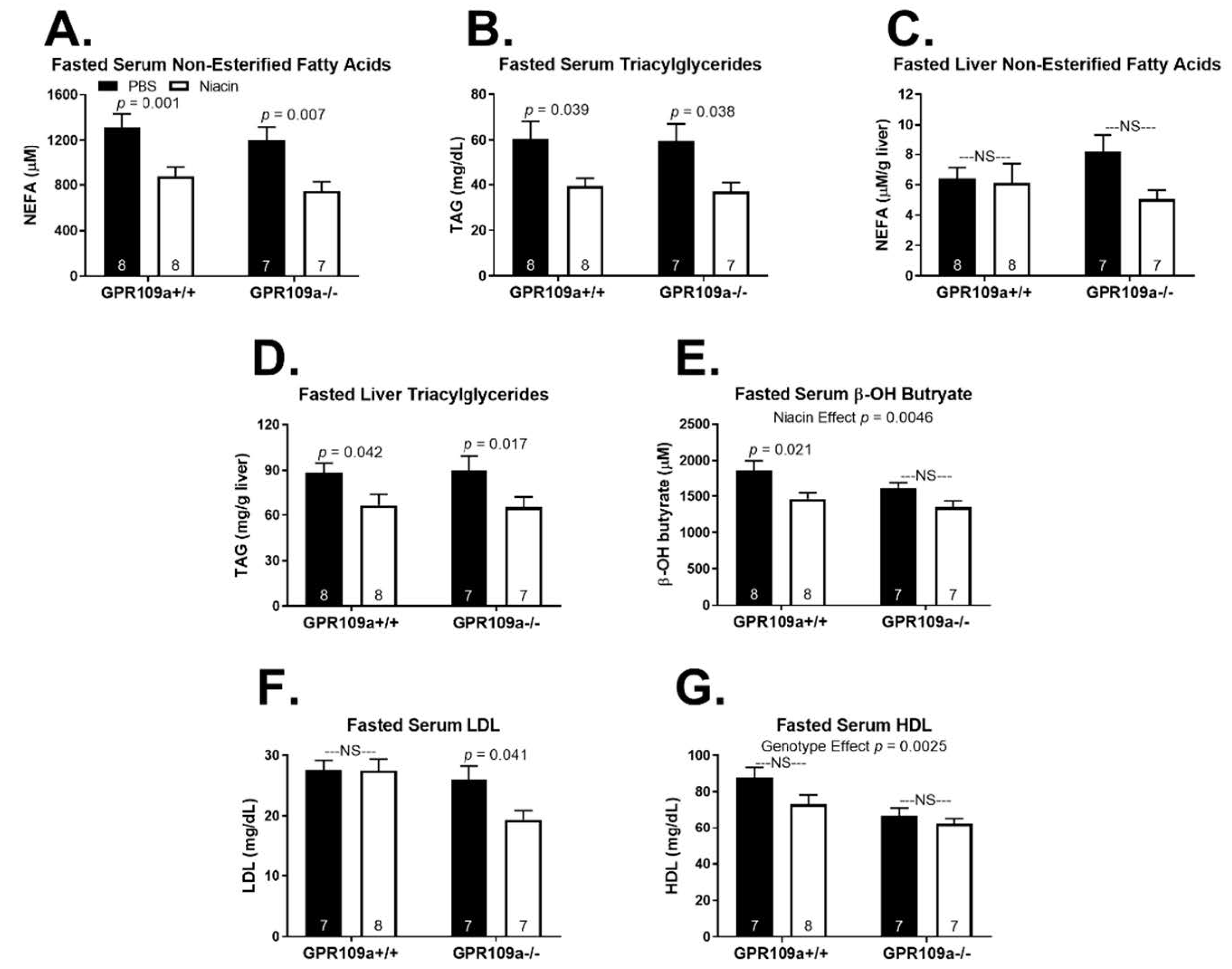
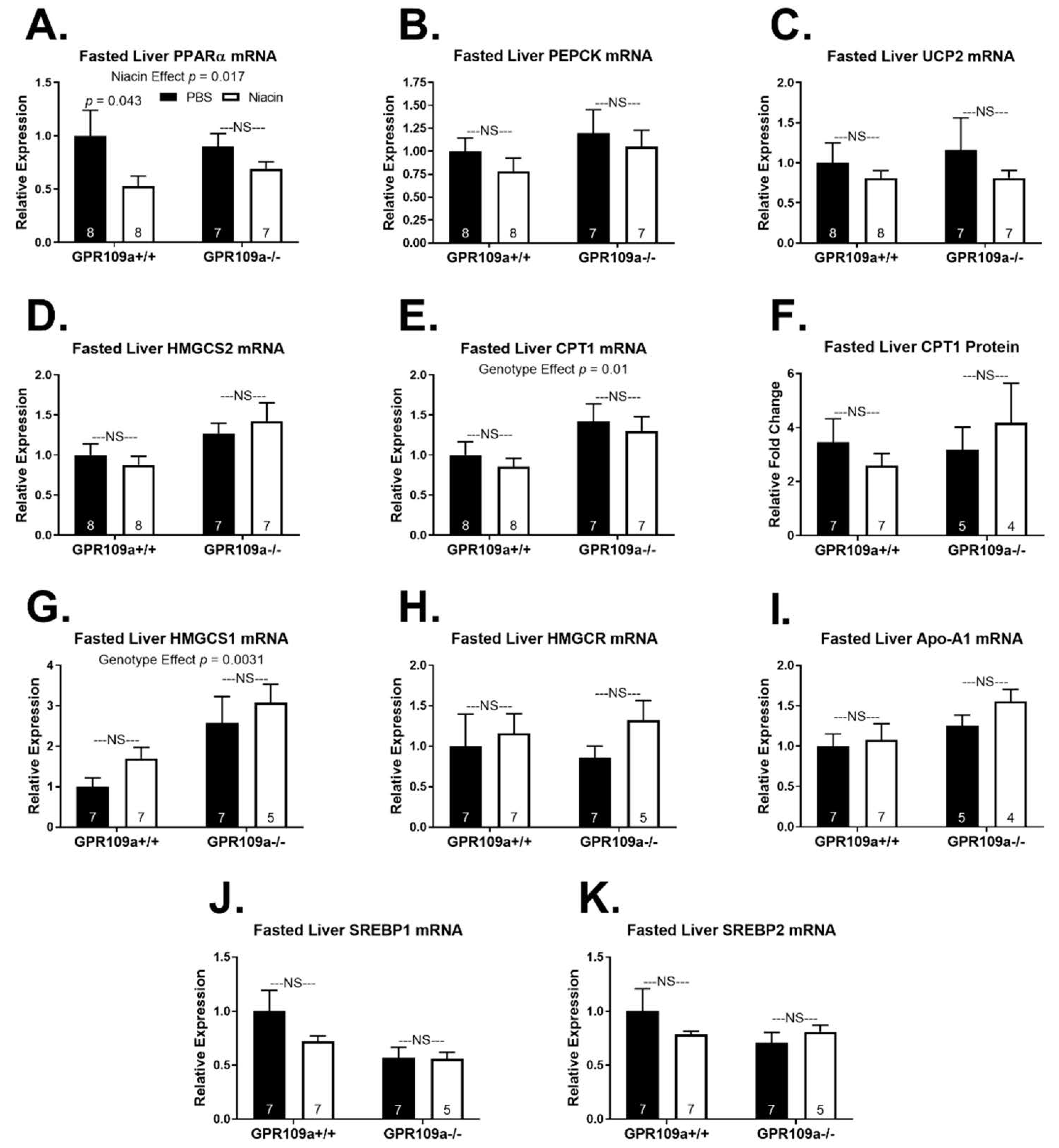
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Injection Studies
4.3. Glucose Tolerance Test
4.4. Tissue Collection
4.5. Serum Assays
4.6. Liver Analyses
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CPT1 | Carnitine Palmitoyltransferase 1 |
| HMGCS2/HMG-CoA synthase II | 3-hydroxy-3-methyl glutaryl CoenzymeA Synthase II |
| HMGCS1/HMG-CoA synthase I | 3-hydroxy-3-methyl glutaryl CoenzymeA Synthase I |
| ACTβ | β-Actin |
| PPARα | Peroxisome Proliferator Activated Receptor α |
| UCP2 | Uncoupling Protein 2 |
| PEPCK | Phosphoenolypyruvate Carboxykinase |
| NEFA | Non-Esterified Fatty Acid |
| TAG | Triacylglyceride |
| LDL | Low Density Lipoprotein |
| HDL | High Density Lipoprotein |
| SREBP1 | Sterol Regulatory Element Binding Protein I |
| SREBP2 | Sterol Regulatory Element Binding Protein II |
| HMGCR | 3-hydroxy-3-methyl glutaryl CoenzymeA Reductase |
References
- Tunaru, S.; Kero, J.; Schaub, A.; Wufka, C.; Blaukat, A.; Pfeffer, K.; Offermanns, S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat. Med. 2003, 9, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Taggart, A.K.P.; Kero, J.; Gan, X.; Cai, T.-Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.-J.; et al. (d)-β-Hydroxybutyrate Inhibits Adipocyte Lipolysis via the Nicotinic Acid Receptor PUMA-G. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, C.E.; Hepler, C.; Higgins, M.R.; Renquist, B.J. Hepatic adaptations to maintain metabolic homeostasis in response to fasting and refeeding in mice. Nutr. Metab. 2016, 13, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganji, S.H.; Kukes, G.D.; Lambrecht, N.; Kashyap, M.L.; Kamanna, V.S. Therapeutic role of niacin in the prevention and regression of hepatic steatosis in rat model of nonalcoholic fatty liver disease. Am. J. Physiol. Liver Physiol. 2014, 306, G320–G327. [Google Scholar] [CrossRef] [Green Version]
- Knowles, H.J.; Poole, R.T.; Workman, P.; Harris, A.L. Niacin induces PPARγ expression and transcriptional activation in macrophages via HM74 and HM74a-mediated induction of prostaglandin synthesis pathways. Biochem. Pharmacol. 2006, 71, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Ringseis, R.; Rosenbaum, S.; Gessner, D.K.; Herges, L.; Kubens, J.F.; Mooren, F.-C.; Krüger, K.; Eder, K. Supplementing Obese Zucker Rats with Niacin Induces the Transition of Glycolytic to Oxidative Skeletal Muscle Fibers. J. Nutr. 2013, 143, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.-H.; Zhao, S.-P. Niacin Promotes Cholesterol Efflux through Stimulation of the PPARγ-LXRα-ABCA1 Pathway in 3T3-L1 Adipocytes. Pharmacology 2009, 84, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Altschul, R.; Hoffer, A.; Stephen, J. Influence of nicotinic acid on serum cholesterol in man. Arch. Biochem. Biophys. 1955, 54, 558–559. [Google Scholar] [CrossRef]
- Grundy, S.M.; Mok, H.Y.; Zech, L.; Berman, M. Influence of nicotinic acid on metabolism of cholesterol and triglycerides in man. J. Lipid Res. 1981, 22, 24–36. [Google Scholar] [CrossRef]
- Wahlberg, G.; Walldius, G.; Olsson, A.G.; Kirstein, P. Effects of nicotinic acid on serum cholesterol concentrations of high density lipoprotein subfractions HDL2, and HDL3, in hyperlipoproteinaemia. J. Intern. Med. 1990, 228, 151–158. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, E.; Hey, S.P.; Ramirez, C.L.; Kesselheim, A.S. Assessment of the Role of Niacin in Managing Cardiovascular Disease Outcomes: A Systematic Review and Meta-analysis. JAMA Netw. Open 2019, 2, e192224. [Google Scholar] [CrossRef] [Green Version]
- The AIM-HIGH Investigators. Niacin in Patients with Low HDL Cholesterol Levels Receiving Intensive Statin Therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Madan, N. Role of niacin in current clinical practice. Minerva Med. 2019, 110, 79–83. [Google Scholar] [CrossRef]
- Lauring, B.; Taggart, A.K.P.; Tata, J.R.; Dunbar, R.; Caro, L.; Cheng, K.; Chin, J.; Colletti, S.L.; Cote, J.; Khalilieh, S.; et al. Niacin Lipid Efficacy Is Independent of Both the Niacin Receptor GPR109A and Free Fatty Acid Suppression. Sci. Transl. Med. 2012, 4, 148ra115. [Google Scholar] [CrossRef]
- Benyó, Z.; Gille, A.; Kero, J.; Csiky, M.; Suchánková, M.C.; Nüsing, R.M.; Moers, A.; Pfeffer, K.; Offermanns, S. GPR109A (PUMA-G/HM74A) mediates nicotinic acid–induced flushing. J. Clin. Investig. 2005, 115, 3634–3640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, T.A. A “Hot” Topic in Dyslipidemia Management—“How to Beat a Flush”: Optimizing Niacin Tolerability to Promote Long-term Treatment Adherence and Coronary Disease Prevention. Mayo Clin. Proc. 2010, 85, 365–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecqueur, C.; Bui, T.; Gelly, C.; Hauchard, J.; Barbot, C.; Bouillaud, F.; Ricquier, D.; Miroux, B.; Thompson, C.B. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J. 2007, 22, 9–18. [Google Scholar] [CrossRef]
- Sheets, A.R.; Fülöp, P.; Derdák, Z.; Kassai, A.; Sabo, E.; Mark, N.M.; Paragh, G.; Wands, J.R.; Baffy, G. Uncoupling protein-2 modulates the lipid metabolic response to fasting in mice. Am. J. Physiol. Liver Physiol. 2008, 294, G1017–G1024. [Google Scholar] [CrossRef] [Green Version]
- Van Der Vorst, E.P.C. High-Density Lipoproteins and Apolipoprotein A1. Subcell. Biochem. 2020, 94, 399–420. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liang, G.; Ou, J.; Goldstein, J.L.; Brown, M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 11245–11250. [Google Scholar] [CrossRef] [Green Version]
- Amemiya-Kudo, M.; Shimano, H.; Hasty, A.H.; Yahagi, N.; Yoshikawa, T.; Matsuzaka, T.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; et al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J. Lipid Res. 2002, 43, 1220–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. A critical role for the peroxisome proliferator-activated receptor (PPAR) in the cellular fasting response: The PPAR -null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 1999, 96, 7473–7478. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator–activated receptor α mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.C.; Gil-Gomez, G.; Hegardt, F.G.; Haro, D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J. Biol. Chem. 1994, 269, 18767–18772. [Google Scholar] [CrossRef]
- Aoyama, T.; Peters, J.M.; Iritani, N.; Nakajima, T.; Furihata, K.; Hashimoto, T.; Gonzalez, F.J. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem. 1998, 273, 5678–5684. [Google Scholar] [CrossRef] [Green Version]
- Yabuuchi, H.; Niijima, S.; Takematsu, H.; Ida, T.; Hirokawa, T.; Hara, T.; Ogawa, T.; Minowa, Y.; Tsujimoto, G.; Okuno, Y. Analysis of multiple compound–protein interactions reveals novel bioactive molecules. Mol. Syst. Biol. 2011, 7, 472. [Google Scholar] [CrossRef]
- Blond, E.; Rieusset, J.; Alligier, M.; Lambert-Porcheron, S.; Bendridi, N.; Gabert, L.; Chétiveaux, M.; Debard, C.; Chauvin, M.-A.; Normand, S.; et al. Nicotinic Acid Effects on Insulin Sensitivity and Hepatic Lipid Metabolism: An In Vivo to In Vitro Study. Horm. Metab. Res. 2014, 46, 390–396. [Google Scholar] [CrossRef]
- Fraterrigo, G.; Fabbrini, E.; Mittendorfer, B.; Rahilly, S.O.; Scherer, P.E.; Patterson, B.W.; Klein, S. Relationship between Changes in Plasma Adiponectin Concentration and Insulin Sensitivity after Niacin Therapy. Cardiorenal Med. 2012, 2, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.J.; Lawson, J.A.; Campbell, L.V.; Storlien, L.H.; Jenkins, A.B.; Whitworth, J.A.; O’Sullivan, A.J. Effects of nicotinic acid on insulin sensitivity and blood pressure in healthy subjects. J. Hum. Hypertens. 2000, 14, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Ringseis, R.; Mooren, F.-C.; Krüger, K.; Most, E.; Eder, K. Niacin supplementation increases the number of oxidative type I fibers in skeletal muscle of growing pigs. BMC Veter-Res. 2013, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Poynten, A.M.; Gan, S.K.; Kriketos, A.D.; O’Sullivan, A.; Kelly, J.J.; Ellis, B.A.; Chisholm, D.J.; Campbell, L.V. Nicotinic acid-induced insulin resistance is related to increased circulating fatty acids and fat oxidation but not muscle lipid content. Metabolism 2003, 52, 699–704. [Google Scholar] [CrossRef]
- Pereira, J.N. The plasma free fatty acid rebound induced by nicotinic acid. J. Lipid Res. 1967, 8, 239–244. [Google Scholar] [CrossRef]
- Heemskerk, M.M.; Berg, S.A.A.V.D.; Pronk, A.C.M.; Van Klinken, J.-B.; Boon, M.R.; Havekes, L.M.; Rensen, P.C.N.; Van Dijk, K.W.; Van Harmelen, V. Long-term niacin treatment induces insulin resistance and adrenergic responsiveness in adipocytes by adaptive downregulation of phosphodiesterase 3B. Am. J. Physiol. Metab. 2014, 306, E808–E813. [Google Scholar] [CrossRef] [Green Version]
- Kroon, T.; Kjellstedt, A.; Thalén, P.; Gabrielsson, J.; Oakes, N.D. Dosing profile profoundly influences nicotinic acid’s ability to improve metabolic control in rats. J. Lipid Res. 2015, 56, 1679–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.T.; Oh, K.-S.; Choi, Y.M.; Jokiaho, A.; Donovan, C.; Choi, S.; Kang, I.; Youn, J.H. Continuous 24-h nicotinic acid infusion in rats causes FFA rebound and insulin resistance by altering gene expression and basal lipolysis in adipose tissue. Am. J. Physiol. Metab. 2011, 300, E1012–E1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; So, W.Y.; Li, S.Y.; Cheng, Q.; Boucher, B.J.; Leung, P.S. Niacin-induced hyperglycemia is partially mediated via niacin receptor GPR109a in pancreatic islets. Mol. Cell. Endocrinol. 2015, 404, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-M.; Zhang, M.; Xu, S.-T.; Li, D.-Z.; Zhu, L.-Y.; Peng, S.-W.; Chen, G.-Q.; Martin, P.M.; Ganapathy, V.; Wei, C.-J. Nicotinic Acid Inhibits Glucose-Stimulated Insulin Secretion Via the G Protein-Coupled Receptor PUMA-G in Murine Islet β Cells. Pancreas 2011, 40, 615–621. [Google Scholar] [CrossRef]
- Li, D.; Luo, N.; Ma, Q.; Li, S.-Z.; Shi, Q.; Cao, Y.; Zhou, S.-S. Excessive nicotinic acid increases methyl consumption and hydrogen peroxide generation in rats. Pharm. Biol. 2012, 51, 8–12. [Google Scholar] [CrossRef]
- Rosebrough, R.; Steele, N. Effect of Supplemental Dietary Chromium or Nicotinic Acid on Carbohydrate Metabolism During Basal, Starvation, and Refeeding Periods in Poults. Poult. Sci. 1981, 60, 407–417. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, S.; Lin, Y.; Xu, W.; Ye, D.; Xiong, Y.; Zhao, S.; Guan, K.-L. Acetylation Negatively Regulates Glycogen Phosphorylase by Recruiting Protein Phosphatase 1. Cell Metab. 2012, 15, 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of Cellular NAD Levels by Nicotinic Acid and Involvement of Nicotinic Acid Phosphoribosyltransferase in Human Cells*. J. Biol. Chem. 2007, 282, 24574–24582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, T.M.; Rawling, J.M.; Roebuck, B.D.; Kirkland, J.B. Large supplements of nicotinic acid and nicotinamide increase tissue NAD+ and poly(ADP-ribose) levels but do not affect diethylnitrosamine-induced altered hepatic foci in Fischer-344 rats. J. Nutr. 1995, 125, 1455–1461. [Google Scholar] [PubMed]
- Romani, M.; Hofer, D.C.; Katsyuba, E.; Auwerx, J. Niacin: An old lipid drug in a new NAD+ dress. J. Lipid Res. 2019, 60, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, F.-Y.; Kamanna, V.S.; Kashyap, M.L. Niacin accelerates intracellular ApoB degradation by inhibiting triacylglycerol synthesis in human hepatoblastoma (HepG2) cells. Arter. Thromb. Vasc. Biol. 1999, 19, 1051–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, L.A. Studies on the incorporation of injected palmitic acid-I-C into liver and plasma lipids in man. Acta Soc. Med. Ups. 1960, 65, 85–90. [Google Scholar]
- Carlson, L.A. Nicotinic acid: The broad-spectrum lipid drug. A 50th anniversary review. J. Intern. Med. 2005, 258, 94–114. [Google Scholar] [CrossRef]
- Chennamsetty, I.; Kostner, K.M.; Claudel, T.; Vinod, M.; Frank, S.; Weiss, T.S.; Trauner, M.; Kostner, G.M. Nicotinic acid inhibits hepatic APOA gene expression: Studies in humans and in transgenic mice. J. Lipid Res. 2012, 53, 2405–2412. [Google Scholar] [CrossRef] [Green Version]
- Ganji, S.H.; Tavintharan, S.; Zhu, D.; Xing, Y.; Kamanna, V.S.; Kashyap, M.L. Niacin noncompetitively inhibits DGAT2 but not DGAT1 activity in HepG2 cells. J. Lipid Res. 2004, 45, 1835–1845. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Millar, J.S.; Brownell, N.; Briand, F.; Rader, D.J. Modulation of HDL metabolism by the niacin receptor GPR109A in mouse hepatocytes. Biochem. Pharmacol. 2010, 80, 1450–1457. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, T.; Zhao, S.; Zhang, S. Niacin regulates apolipoprotein M expression via liver X receptor‑α. Mol. Med. Rep. 2019, 20, 3285–3291. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-H.; Kamanna, V.S.; Zhang, M.C.; Kashyap, M.L. Niacin inhibits surface expression of ATP synthase β chain in HepG2 cells: Implications for raising HDL. J. Lipid Res. 2008, 49, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-H.; Kamanna, V.S.; Ganji, S.H.; Xiong, X.-M.; Kashyap, M.L. Niacin increases HDL biogenesis by enhancing DR4-dependent transcription of ABCA1 and lipidation of apolipoprotein A-I in HepG2 cells. J. Lipid Res. 2012, 53, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, M.J.; Alamir, A.-R.; Sultan, S.; Chehade, J.M.; Wong, N.C.; Mooradian, A.D. Nicotinic acid induces apolipoprotein A-I gene expression in HepG2 and Caco-2 cell lines. Metabolism 2011, 60, 1790–1796. [Google Scholar] [CrossRef] [PubMed]
- Lamon-Fava, S.; Diffenderfer, M.R.; Barrett, P.H.R.; Buchsbaum, A.; Nyaku, M.; Horvath, K.V.; Asztalos, B.F.; Otokozawa, S.; Ai, M.; Matthan, N.R.; et al. Extended-Release Niacin Alters the Metabolism of Plasma Apolipoprotein (Apo) A-I and ApoB-Containing Lipoproteins. Arter. Thromb. Vasc. Biol. 2008, 28, 1672–1678. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cao, Y.; Fu, S.; Li, W.; Ge, Y.; Cheng, J.; Liu, J. Niacin inhibits the synthesis of milk fat in BMECs through the GPR109A-mediated downstream signalling pathway. Life Sci. 2020, 260, 118415. [Google Scholar] [CrossRef]
- Zimmer, M.; Bista, P.; Benson, E.L.; Lee, D.Y.; Liu, F.; Picarella, D.; Vega, R.B.; Vu, C.B.; Yeager, M.; Ding, M.; et al. CAT-2003: A novel sterol regulatory element-binding protein inhibitor that reduces steatohepatitis, plasma lipids, and atherosclerosis in apolipoprotein E*3-Leiden mice. Hepatol. Commun. 2017, 1, 311–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidarov, I.; Chen, X.; Anthony, T.; Maciejewski-Lenoir, D.; Liaw, C.; Unett, D.J. Differential tissue and ligand-dependent signaling of GPR109A receptor: Implications for anti-atherosclerotic therapeutic potential. Cell. Signal. 2013, 25, 2003–2016. [Google Scholar] [CrossRef]
- Rubic, T.; Trottmann, M.; Lorenz, R.L. Stimulation of CD36 and the key effector of reverse cholesterol transport ATP-binding cassette A1 in monocytoid cells by niacin. Biochem. Pharmacol. 2004, 67, 411–419. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nat. Cell Biol. 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Song, S.; Zhang, Y.; Ma, K.; Jackson-Hayes, L.; Lavrentyev, E.N.; Cook, G.A.; Elam, M.B.; Park, E.A. Peroxisomal proliferator activated receptor gamma coactivator (PGC-1α) stimulates carnitine palmitoyltransferase I (CPT-Iα) through the first intron. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 2004, 1679, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Gerhart-Hines, Z.; Puigserver, P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 2007, 582, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Torra, I.P.; Jamshidi, Y.; Flavell, D.M.; Fruchart, J.-C.; Staels, B. Characterization of the Human PPARα Promoter: Identification of a Functional Nuclear Receptor Response Element. Mol. Endocrinol. 2002, 16, 1013–1028. [Google Scholar] [CrossRef] [Green Version]
- Richman, J.G.; Kanemitsu-Parks, M.; Gaidarov, I.; Cameron, J.S.; Griffin, P.; Zheng, H.; Guerra, N.C.; Cham, L.; Maciejewski-Lenoir, D.; Behan, D.P.; et al. Nicotinic Acid Receptor Agonists Differentially Activate Downstream Effectors. J. Biol. Chem. 2007, 282, 18028–18036. [Google Scholar] [CrossRef] [Green Version]
- Krebs, S.; Fischaleck, M.; Blum, H. A simple and loss-free method to remove TRIzol contaminations from minute RNA samples. Anal. Biochem. 2009, 387, 136–138. [Google Scholar] [CrossRef]
- Ramakersab, C.; Ruijter, J.M.; Deprez, R.H.; Moorman, A.F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 2003, 339, 62–66. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.; Russell, J.C.; Taylor, A.W. Determination of glycogen in small tissue samples. J. Appl. Physiol. 1970, 28, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| HMG-CoA Reductase | 5′-CCTGTGGAATGCCTTGTGATTG-3′ | 5′-AGCCGAAGCAGCACATGAT-3′ |
| HMG-CoA Synthase | 5′-TGGCACAGTACTCACCTC-3′ | 5′-CCTTCATCCAAACTGTGG-3′ |
| SREBP-1 | 5′-GCAGCCACCATCTAGCCTG-3′ | 5′-CAGCAGTGAGTCTGCCTTGAT-3′ |
| SREBP-2 | 5′-GCAGCAACGGGACCATTCT-3′ | 5′-CCCCATGACTAAGTCCTTCAACT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geisler, C.E.; Miller, K.E.; Ghimire, S.; Renquist, B.J. The Role of GPR109a Signaling in Niacin Induced Effects on Fed and Fasted Hepatic Metabolism. Int. J. Mol. Sci. 2021, 22, 4001. https://doi.org/10.3390/ijms22084001
Geisler CE, Miller KE, Ghimire S, Renquist BJ. The Role of GPR109a Signaling in Niacin Induced Effects on Fed and Fasted Hepatic Metabolism. International Journal of Molecular Sciences. 2021; 22(8):4001. https://doi.org/10.3390/ijms22084001
Chicago/Turabian StyleGeisler, Caroline E., Kendra E. Miller, Susma Ghimire, and Benjamin J. Renquist. 2021. "The Role of GPR109a Signaling in Niacin Induced Effects on Fed and Fasted Hepatic Metabolism" International Journal of Molecular Sciences 22, no. 8: 4001. https://doi.org/10.3390/ijms22084001
APA StyleGeisler, C. E., Miller, K. E., Ghimire, S., & Renquist, B. J. (2021). The Role of GPR109a Signaling in Niacin Induced Effects on Fed and Fasted Hepatic Metabolism. International Journal of Molecular Sciences, 22(8), 4001. https://doi.org/10.3390/ijms22084001