
Structural Biology for the Molecular Insight between Aptamers and Target Proteins

 , ,
, , 
Abstract
:1. Introduction
2. Progress in Structure Determination for Aptamer–Protein Complexes by NMR Spectroscopy, X-ray Crystallography and Cryo-EM
2.1. Structural Determination by NMR
2.2. Structural Determination by X-ray
2.3. Structural Determination by Cryo-Electron Microscopy (Cryo-EM)
2.4. Other Methods for Structural Determination
3. Interfaces between Aptamer–Protein Complexes
3.1. Structures of Aptamer–Protein Complex in Database
3.2. The Features of Aptamers That Contribute to Binding
3.3. The Features of Proteins That Contribute to Binding
3.4. Conformational Changes upon Binding
3.5. Factors Contribute to the Stability of Aptamer Structure in Structure Determination
4. Challenges in Structural Determination
4.1. Low Affinity of Aptamers to Proteins
4.2. Methods for Determination of the Binding Affinity between Aptamers and Proteins
4.2.1. Isothermal Titration Calorimetry (ITC)
4.2.2. Surface Plasmon Resonance (SPR)
4.2.3. Atomic Force Microscopy (AFM)
4.2.4. Flow Cytometry
4.2.5. Enzyme-Linked Oligonucleotide Assay (ELONA)
4.2.6. Surface-Enhanced Raman Spectroscopy (SERS)
4.2.7. Microscale Thermophoresis (MST)
4.2.8. Bio-Layer Interferometry (BLI)
4.3. Modifications of Aptamers to Enhance the Stability and Binding Affinity of Aptamers
4.3.1. Chemical Modification Strategies
4.3.2. Structural Modification Strategies
4.3.3. Modified Aptamers in Clinical Trials
4.4. Heterogeneity of the Aptamers
5. Discussion
5.1. Structural Determination for Aptamer–Protein Complexes
5.2. In-Silico Analysis for Aptamer–Protein Binding
5.3. Different Application of Proteins Specific Aptamers
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Haßel, S.; Mayer, G. Aptamers as therapeutic agents: Has the initial euphoria subsided? Mol. Diagn. Ther. 2019, 23, 301–309. [Google Scholar] [CrossRef]
- Sakamoto, T. NMR study of aptamers. Aptamers 2017, 1, 13–18. [Google Scholar]
- Davlieva, M.; Donarski, J.; Wang, J.; Shamoo, Y.; Nikonowicz, E.P. Structure analysis of free and bound states of an RNA aptamer against ribosomal protein S8 from Bacillus anthracis. Nucleic Acids Res. 2014, 42, 10795–10808. [Google Scholar] [CrossRef] [Green Version]
- Hong, M. Resonance assignment of 13C/15N labeled solid proteins by two-and three-dimensional magic-angle-spinning NMR. J. Biomol. NMR 1999, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Barnwal, R.P.; Yang, F.; Varani, G. Applications of NMR to structure determination of RNAs large and small. Arch. Biochem. Biophys. 2017, 628, 42–56. [Google Scholar] [CrossRef]
- Dieckmann, T.; Suzuki, E.; Nakamura, G.K.; Feigon, J. Solution structure of an ATP-binding RNA aptamer reveals a novel fold. RNA 1996, 2, 628–640. [Google Scholar]
- Jiang, F.; Kumar, R.A.; Jones, R.A.; Patel, D.J. Structural basis of RNA folding and recognition in an AMP–RNA aptamer complex. Nature 1996, 382, 183–186. [Google Scholar] [CrossRef]
- Fan, P.; Suri, A.K.; Fiala, R.; Live, D.; Patel, D.J. Molecular recognition in the FMN–RNA aptamer complex. J. Mol. Biol. 1996, 258, 480–500. [Google Scholar] [CrossRef]
- Yang, Y.; Kochoyan, M.; Burgstaller, P.; Westhof, E.; Famulok, M. Structural basis of ligand discrimination by two related RNA aptamers resolved by NMR spectroscopy. Science 1996, 272, 1343–1347. [Google Scholar] [CrossRef]
- Jiang, L.; Suri, A.K.; Fiala, R.; Patel, D.J. Saccharide-RNA recognition in an aminoglycoside antibiotic-RNA aptamer complex. Chem. Biol. 1997, 4, 35–50. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.B.; Yildiz, F.Z.; Lo, J.A.; Wang, B.; D’Souza, V.M. A structure-based mechanism for tRNA and retroviral RNA remodelling during primer annealing. Nature 2014, 515, 591–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayie, T.K.; Thakur, C.S. Site-specific labeling of nucleotides for making RNA for high resolution NMR studies using an E. coli strain disabled in the oxidative pentose phosphate pathway. J. Biomol. NMR 2010, 47, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Longhini, A.P.; LeBlanc, R.M.; Becette, O.; Salguero, C.; Wunderlich, C.H.; Johnson, B.A.; D’Souza, V.M.; Kreutz, C.; Dayie, T.K. Chemo-enzymatic synthesis of site-specific isotopically labeled nucleotides for use in NMR resonance assignment, dynamics and structural characterizations. Nucleic Acids Res. 2016, 44, e52. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M. Practical aspects of paramagnetic relaxation enhancement in biological macromolecules. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 564, pp. 485–497. [Google Scholar]
- Lieberman, R.L.; Peek, M.E.; Watkins, J.D. Determination of soluble and membrane protein structures by X-ray crystallography. In Electron Crystallography of Soluble and Membrane Proteins; Springer: Berlin/Heidelberg, Germany, 2013; pp. 475–493. [Google Scholar]
- McPherson, A. Protein crystallization. In Protein Crystallography; Springer: Berlin/Heidelberg, Germany, 2017; pp. 17–50. [Google Scholar]
- Schmidt, C.; Perbandt, M.; Klussmann, S.; Betzel, C. Molecular characterization of a ghrelin-l-aptamer complex. J. Mol. Struct. 2020, 1204, 127510. [Google Scholar] [CrossRef]
- Renaud, J.-P.; Chari, A.; Ciferri, C.; Liu, W.-t.; Rémigy, H.-W.; Stark, H.; Wiesmann, C. Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug Discov. 2018, 17, 471–492. [Google Scholar] [CrossRef] [PubMed]
- Klein Douwel, D.; Hoogenboom, W.S.; Boonen, R.A.; Knipscheer, P. Recruitment and positioning determine the specific role of the XPF-ERCC 1 endonuclease in interstrand crosslink repair. EMBO J. 2017, 36, 2034–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, H.D.; Laister, R.C.; Martin, S.R.; Arrowsmith, C.H.; West, S.C. The SMX DNA repair tri-nuclease. Mol. Cell 2017, 65, 848–860.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.; Beuron, F.; Borg, A.; Nans, A.; Earl, C.P.; Briggs, D.C.; Snijders, A.P.; Bowles, M.; Morris, E.P.; Linch, M.; et al. Cryo-EM structures of the XPF-ERCC1 endonuclease reveal how DNA-junction engagement disrupts an auto-inhibited conformation. Nat. Commun. 2020, 11, 1120. [Google Scholar] [CrossRef] [Green Version]
- Del Villar-Guerra, R.; Trent, J.O.; Chaires, J.B. G-quadruplex secondary structure obtained from circular dichroism spectroscopy. Angew. Chem. 2018, 130, 7289–7293. [Google Scholar] [CrossRef]
- Jaumot, J.; Eritja, R.; Navea, S.; Gargallo, R. Classification of nucleic acids structures by means of the chemometric analysis of circular dichroism spectra. Anal. Chim. Acta 2009, 642, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Loo, R.R.O.; Loo, J.A. Structural characterization of a thrombin-aptamer complex by high resolution native top-down mass spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Spirig, T.; Malmirchegini, G.R.; Zhang, J.; Robson, S.A.; Sjodt, M.; Liu, M.; Kumar, K.K.; Dickson, C.F.; Gell, D.A.; Lei, B. Staphylococcus aureus uses a novel multidomain receptor to break apart human hemoglobin and steal its heme. J. Biol. Chem. 2013, 288, 1065–1078. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Zhang, H.; Gross, M.L.; Blankenship, R.E. Native electrospray mass spectrometry reveals the nature and stoichiometry of pigments in the FMO photosynthetic antenna protein. Biochemistry 2011, 50, 3502–3511. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Malmirchegini, G.R.; Clubb, R.T.; Loo, J.A. Native top-down mass spectrometry for the structural characterization of human hemoglobin. Eur. J. Mass Spectrom. 2015, 21, 221–231. [Google Scholar] [CrossRef] [Green Version]
- AhYoung, A.P.; Jiang, J.; Zhang, J.; Dang, X.K.; Loo, J.A.; Zhou, Z.H.; Egea, P.F. Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E3179–E3188. [Google Scholar] [CrossRef] [Green Version]
- Novoseltseva, A.; Zavyalova, E.; Golovin, A.; Kopylov, A. An insight into aptamer–protein complexes. Aptamers 2018, 2, 1–19. [Google Scholar]
- Jančaříková, G.; Houser, J.; Dobeš, P.; Demo, G.; Hyršl, P.; Wimmerová, M. Characterization of novel bangle lectin from Photorhabdus asymbiotica with dual sugar-binding specificity and its effect on host immunity. PLoS Pathog. 2017, 13, e1006564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, W.T.; Convery, M.A.; Stonehouse, N.J.; Adams, C.J.; Liljas, L.; Phillips, S.E.; Stockley, P.G. The crystal structure of a high affinity RNA stem-loop complexed with the bacteriophage MS2 capsid: Further challenges in the modeling of ligand-RNA interactions. RNA 2004, 10, 1776–1782. [Google Scholar] [CrossRef] [Green Version]
- Bullock, T.L.; Sherlin, L.D.; Perona, J.J. Tertiary core rearrangements in a tight binding transfer RNA aptamer. Nat. Struct. Biol. 2000, 7, 497–504. [Google Scholar] [CrossRef]
- Huang, D.-B.; Vu, D.; Cassiday, L.A.; Zimmerman, J.M.; Maher, L.J.; Ghosh, G. Crystal structure of NF-κB (p50) 2 complexed to a high-affinity RNA aptamer. Proc. Natl. Acad. Sci. USA 2003, 100, 9268–9273. [Google Scholar] [CrossRef] [Green Version]
- Kettenberger, H.; Eisenführ, A.; Brueckner, F.; Theis, M.; Famulok, M.; Cramer, P. Structure of an RNA polymerase II–RNA inhibitor complex elucidates transcription regulation by noncoding RNAs. Nat. Struct. Mol. Biol. 2006, 13, 44–48. [Google Scholar] [CrossRef]
- Russo Krauss, I.; Pica, A.; Merlino, A.; Mazzarella, L.; Sica, F. Duplex-quadruplex motifs in a peculiar structural organization cooperatively contribute to thrombin binding of a DNA aptamer. Acta Cryst. D Biol. Cryst. 2013, 69, 2403–2411. [Google Scholar] [CrossRef]
- Pica, A.; Russo Krauss, I.; Parente, V.; Tateishi-Karimata, H.; Nagatoishi, S.; Tsumoto, K.; Sugimoto, N.; Sica, F. Through-bond effects in the ternary complexes of thrombin sandwiched by two DNA aptamers. Nucleic Acids Res. 2016, 45, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, J.; Wulffen, B.; Pötzsch, B.; Mayer, G. Multidomain targeting generates a high-affinity thrombin-inhibiting bivalent aptamer. ChemBioChem 2007, 8, 2223–2226. [Google Scholar] [CrossRef] [PubMed]
- Russo Krauss, I.; Merlino, A.; Randazzo, A.; Novellino, E.; Mazzarella, L.; Sica, F. High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucleic Acids Res. 2012, 40, 8119–8128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, B.; Martino, L.; Randazzo, A.; Giancola, C. Stability and binding properties of a modified thrombin binding aptamer. Biophys. J. 2008, 94, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyakawa, S.; Nomura, Y.; Sakamoto, T.; Yamaguchi, Y.; Kato, K.; Yamazaki, S.; Nakamura, Y. Structural and molecular basis for hyperspecificity of RNA aptamer to human immunoglobulin G. RNA 2008, 14, 1154–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, Y.; Sugiyama, S.; Sakamoto, T.; Miyakawa, S.; Adachi, H.; Takano, K.; Murakami, S.; Inoue, T.; Mori, Y.; Nakamura, Y.; et al. Conformational plasticity of RNA for target recognition as revealed by the 2.15 A crystal structure of a human IgG-aptamer complex. Nucleic Acids Res. 2010, 38, 7822–7829. [Google Scholar] [CrossRef] [Green Version]
- Russo Krauss, I.; Merlino, A.; Giancola, C.; Randazzo, A.; Mazzarella, L.; Sica, F. Thrombin-aptamer recognition: A revealed ambiguity. Nucleic Acids Res. 2011, 39, 7858–7867. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.H.; Fremont, D.H.; Diener, J.L.; Schaub, R.G.; Sadler, J.E. A structural explanation for the antithrombotic activity of ARC1172, a DNA aptamer that binds von Willebrand factor domain A1. Structure 2009, 17, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Diener, J.; Daniel Lagasse, H.; Duerschmied, D.; Merhi, Y.; Tanguay, J.F.; Hutabarat, R.; Gilbert, J.; Wagner, D.; Schaub, R. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J. Thromb. Haemost. 2009, 7, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, V.M.; Lennarz, S.; Mayer, G.; Tesmer, J.J. Molecular mechanism for inhibition of g protein-coupled receptor kinase 2 by a selective RNA aptamer. Structure 2012, 20, 1300–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, D.R.; Gelinas, A.D.; Zhang, C.; Rohloff, J.C.; Carter, J.D.; O’Connell, D.; Waugh, S.M.; Wolk, S.K.; Mayfield, W.S.; Burgin, A.B.; et al. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc. Natl. Acad. Sci. USA 2012, 109, 19971–19976. [Google Scholar] [CrossRef] [Green Version]
- Pica, A.; Russo Krauss, I.; Merlino, A.; Nagatoishi, S.; Sugimoto, N.; Sica, F. Dissecting the contribution of thrombin exosite I in the recognition of thrombin binding aptamer. FEBS J. 2013, 280, 6581–6588. [Google Scholar] [CrossRef]
- Choi, S.J.; Ban, C. Crystal structure of a DNA aptamer bound to PvLDH elucidates novel single-stranded DNA structural elements for folding and recognition. Sci. Rep. 2016, 6, 34998. [Google Scholar] [CrossRef] [Green Version]
- Yatime, L.; Maasch, C.; Hoehlig, K.; Klussmann, S.; Andersen, G.R.; Vater, A. Structural basis for the targeting of complement anaphylatoxin C5a using a mixed L-RNA/L-DNA aptamer. Nat. Commun. 2015, 6, 6481. [Google Scholar] [CrossRef]
- Cheung, Y.W.; Kwok, J.; Law, A.W.; Watt, R.M.; Kotaka, M.; Tanner, J.A. Structural basis for discriminatory recognition of Plasmodium lactate dehydrogenase by a DNA aptamer. Proc. Natl. Acad. Sci. USA 2013, 110, 15967–15972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelinas, A.D.; Davies, D.R.; Edwards, T.E.; Rohloff, J.C.; Carter, J.D.; Zhang, C.; Gupta, S.; Ishikawa, Y.; Hirota, M.; Nakaishi, Y.; et al. Crystal structure of interleukin-6 in complex with a modified nucleic acid ligand. J. Biol. Chem. 2014, 289, 8720–8734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberthür, D.; Achenbach, J.; Gabdulkhakov, A.; Buchner, K.; Maasch, C.; Falke, S.; Rehders, D.; Klussmann, S.; Betzel, C. Crystal structure of a mirror-image L-RNA aptamer (Spiegelmer) in complex with the natural L-protein target CCL2. Nat. Commun. 2015, 6, 6923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, T.C.; Davies, D.R.; Hisaminato, A.; Resnicow, D.I.; Gupta, S.; Waugh, S.M.; Nagabukuro, A.; Wadatsu, T.; Hishigaki, H.; Gawande, B.; et al. Non-helical DNA Triplex Forms a Unique Aptamer Scaffold for High Affinity Recognition of Nerve Growth Factor. Structure 2015, 23, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo Krauss, I.; Spiridonova, V.; Pica, A.; Napolitano, V.; Sica, F. Different duplex/quadruplex junctions determine the properties of anti-thrombin aptamers with mixed folding. Nucleic Acids Res. 2016, 44, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Ikeda, H.; Miyakawa, S.; Futakawa, S.; Nonaka, Y.; Fujiwara, M.; Okudaira, S.; Kano, K.; Aoki, J.; Morita, J.; et al. Structural basis for specific inhibition of Autotaxin by a DNA aptamer. Nat. Struct. Mol. Biol. 2016, 23, 395–401. [Google Scholar] [CrossRef]
- Abeydeera, N.D.; Egli, M.; Cox, N.; Mercier, K.; Conde, J.N.; Pallan, P.S.; Mizurini, D.M.; Sierant, M.; Hibti, F.E.; Hassell, T.; et al. Evoking picomolar binding in RNA by a single phosphorodithioate linkage. Nucleic Acids Res. 2016, 44, 8052–8064. [Google Scholar] [CrossRef]
- Ren, X.; Gelinas, A.D.; von Carlowitz, I.; Janjic, N.; Pyle, A.M. Structural basis for IL-1α recognition by a modified DNA aptamer that specifically inhibits IL-1α signaling. Nat. Commun. 2017, 8, 810. [Google Scholar] [CrossRef] [Green Version]
- Dolot, R.; Lam, C.H.; Sierant, M.; Zhao, Q.; Liu, F.W.; Nawrot, B.; Egli, M.; Yang, X. Crystal structures of thrombin in complex with chemically modified thrombin DNA aptamers reveal the origins of enhanced affinity. Nucleic Acids Res. 2018, 46, 4819–4830. [Google Scholar] [CrossRef]
- Grau, F.C.; Jaeger, J.; Groher, F.; Suess, B.; Muller, Y.A. The complex formed between a synthetic RNA aptamer and the transcription repressor TetR is a structural and functional twin of the operator DNA-TetR regulator complex. Nucleic Acids Res. 2020, 48, 3366–3378. [Google Scholar] [CrossRef]
- Ptacek, J.; Zhang, D.; Qiu, L.; Kruspe, S.; Motlova, L.; Kolenko, P.; Novakova, Z.; Shubham, S.; Havlinova, B.; Baranova, P.; et al. Structural basis of prostate-specific membrane antigen recognition by the A9g RNA aptamer. Nucleic Acids Res. 2020, 48, 11130–11145. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, I.; Kolganova, N.; Troisi, R.; Sica, F.; Timofeev, E. Expanding the recognition interface of the thrombin-binding aptamer HD1 through modification of residues T3 and T12. Mol. Nucleic Acids 2021, 23, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, C.; Denton, D.T.; O’Connell, D.; Drolet, D.W.; Geisbrecht, B.V. Inhibition of the Complement Alternative Pathway by Chemically Modified DNA Aptamers That Bind with Picomolar Affinity to Factor B. J. Immunol. 2021, 206, 861–873. [Google Scholar] [CrossRef]
- Padlan, C.S.; Malashkevich, V.N.; Almo, S.C.; Levy, M.; Brenowitz, M.; Girvin, M.E. An RNA aptamer possessing a novel monovalent cation-mediated fold inhibits lysozyme catalysis by inhibiting the binding of long natural substrates. RNA 2014, 20, 447–461. [Google Scholar] [CrossRef] [Green Version]
- Oguro, A.; Yanagida, A.; Fujieda, Y.; Amano, R.; Otsu, M.; Sakamoto, T.; Kawai, G.; Matsufuji, S. Two stems with different characteristics and an internal loop in an RNA aptamer contribute to spermine-binding. J. Biochem. 2017, 161, 197–206. [Google Scholar] [CrossRef]
- Yamaoki, Y.; Nagata, T.; Sakamoto, T.; Katahira, M. Recent progress of in-cell NMR of nucleic acids in living human cells. Biophys. Rev. 2020, 12, 411–417. [Google Scholar] [CrossRef]
- Mao, L.; Wang, Y.; Liu, Y.; Hu, X. Multiple Intermolecular Interaction Modes of Positively Charged Residues with Adenine in ATP-Binding Proteins. J. Am. Chem. Soc. 2003, 125, 14216–14217. [Google Scholar] [CrossRef]
- Rooman, M.; Liévin, J.; Buisine, E.; Wintjens, R. Cation-pi/H-bond stair motifs at protein-DNA interfaces. J. Mol. Biol. 2002, 319, 67–76. [Google Scholar] [CrossRef]
- Wintjens, R.; Liévin, J.; Rooman, M.; Buisine, E. Contribution of cation-pi interactions to the stability of protein-DNA complexes. J. Mol. Biol. 2000, 302, 395–410. [Google Scholar] [CrossRef]
- Gunaratne, R.; Kumar, S.; Frederiksen, J.W.; Stayrook, S.; Lohrmann, J.L.; Perry, K.; Bompiani, K.M.; Chabata, C.V.; Thalji, N.K.; Ho, M.D.; et al. Combination of aptamer and drug for reversible anticoagulation in cardiopulmonary bypass. Nat. Biotechnol. 2018, 36, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Dearborn, A.D.; Eren, E.; Watts, N.R.; Palmer, I.W.; Kaufman, J.D.; Steven, A.C.; Wingfield, P.T. Structure of an RNA Aptamer that Can Inhibit HIV-1 by Blocking Rev-Cognate RNA (RRE) Binding and Rev-Rev. Association. Structure 2018, 26, 1187–1195.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelinas, A.D.; Davies, D.R.; Janjic, N. Embracing proteins: Structural themes in aptamer–protein complexes. Curr. Opin. Struct. Biol. 2016, 36, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashima, T.; Matsugami, A.; Nishikawa, F.; Nishikawa, S.; Katahira, M. Unique quadruplex structure and interaction of an RNA aptamer against bovine prion protein. Nucleic Acids Res. 2009, 37, 6249–6258. [Google Scholar] [CrossRef] [PubMed]
- Tuske, S.; Zheng, J.; Olson, E.D.; Ruiz, F.X.; Pascal, B.D.; Hoang, A.; Bauman, J.D.; Das, K.; DeStefano, J.J.; Musier-Forsyth, K.; et al. Integrative structural biology studies of HIV-1 reverse transcriptase binding to a high-affinity DNA aptamer. Curr. Res. Struct. Biol. 2020, 2, 116–129. [Google Scholar] [CrossRef]
- Reiter, N.J.; Maher, L.J., III; Butcher, S.E. DNA mimicry by a high-affinity anti-NF-κB RNA aptamer. Nucleic Acids Res. 2007, 36, 1227–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Jucker, F.; Pardi, A. Imino proton exchange rates imply an induced-fit binding mechanism for the VEGF165-targeting aptamer, Macugen. FEBS Lett. 2008, 582, 1835–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, T.; Patel, D.J. Adaptive recognition by nucleic acid aptamers. Science 2000, 287, 820–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavery, R.; Sklenar, H. The Definition of Generalized Helicoidal Parameters and of Axis Curvature for Irregular Nucleic Acids. J. Biomol. Struct. Dyn. 1988, 6, 63–91. [Google Scholar] [CrossRef]
- Koldobskaya, Y.; Duguid, E.M.; Shechner, D.M.; Suslov, N.B.; Ye, J.; Sidhu, S.S.; Bartel, D.P.; Koide, S.; Kossiakoff, A.A.; Piccirilli, J.A. A portable RNA sequence whose recognition by a synthetic antibody facilitates structural determination. Nat. Struct. Mol. Biol. 2011, 18, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Suslov, N.B.; Li, N.-S.; Shelke, S.A.; Evans, M.E.; Koldobskaya, Y.; Rice, P.A.; Piccirilli, J.A. A G-quadruplex–containing RNA activates fluorescence in a GFP-like fluorophore. Nat. Chem. Biol. 2014, 10, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Elskens, J.P.; Elskens, J.M.; Madder, A. Chemical Modification of Aptamers for Increased Binding Affinity in Diagnostic Applications: Current Status and Future Prospects. Int. J. Mol. Sci. 2020, 21, 4522. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Freire, E.; Mayorga, O.L.; Straume, M. Isothermal titration calorimetry. Anal. Chem. 1990, 62, 950A–959A. [Google Scholar] [CrossRef]
- Schasfoort, R.B. Handbook of Surface Plasmon Resonance; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- Halpern, A.R.; Chen, Y.; Corn, R.M.; Kim, D. Surface plasmon resonance phase imaging measurements of patterned monolayers and DNA adsorption onto microarrays. Anal. Chem. 2011, 83, 2801–2806. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lee, H.J.; Corn, R.M. Fabrication and characterization of RNA aptamer microarrays for the study of protein-aptamer interactions with SPR imaging. Nucleic Acids Res. 2006, 34, 6416–6424. [Google Scholar] [CrossRef] [Green Version]
- Wegner, G.J.; Lee, H.J.; Corn, R.M. Characterization and optimization of peptide arrays for the study of epitope−Antibody interactions using surface plasmon resonance imaging. Anal. Chem. 2002, 74, 5161–5168. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.A.; Corn, R.M. Surface plasmon resonance imaging as a tool to monitor biomolecular interactions in an array based format. Appl. Spectrosc. 2003, 57, 320A–332A. [Google Scholar] [CrossRef]
- Shumaker-Parry, J.S.; Campbell, C.T. Quantitative methods for spatially resolved adsorption/desorption measurements in real time by surface plasmon resonance microscopy. Anal. Chem. 2004, 76, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, C.P.; Scardino, E.; Layzer, J.; Pitoc, G.A.; Ortel, T.L.; Monroe, D.; Sullenger, B.A. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 2002, 419, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhu, C.; Ling, L.; Wan, L.; Fang, X.; Bai, C. Specific Aptamer−Protein Interaction Studied by Atomic Force Microscopy. Anal. Chem. 2003, 75, 2112–2116. [Google Scholar] [CrossRef]
- Zlatanova, J.; Lindsay, S.M.; Leuba, S.H. Single molecule force spectroscopy in biology using the atomic force microscope. Prog. Biophys. Mol. Biol. 2000, 74, 37–61. [Google Scholar] [CrossRef]
- Willemsen, O.H.; Snel, M.M.; Cambi, A.; Greve, J.; De Grooth, B.G.; Figdor, C.G. Biomolecular interactions measured by atomic force microscopy. Biophys. J. 2000, 79, 3267–3281. [Google Scholar] [CrossRef] [Green Version]
- Yip, C.M. Atomic force microscopy of macromolecular interactions. Curr. Opin. Struct. Biol. 2001, 11, 567–572. [Google Scholar] [CrossRef]
- McKinnon, K.M. Flow cytometry: An overview. Curr. Protoc. Immunol. 2018, 120, 5.1.1–5.1.11. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W. Development of Novel Far-Red/Near-Infrared Dye-hCRBPII Based Imaging Tags for Background-Free Live Cell Imaging; Michigan State University: East Lansing, MI, USA, 2019. [Google Scholar]
- Liang, C.; Guo, B.; Wu, H.; Shao, N.; Li, D.; Liu, J.; Dang, L.; Wang, C.; Li, H.; Li, S. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference–based bone anabolic strategy. Nat. Med. 2015, 21, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Bhagwat, S.R.; Sharma, T.K.; Kumar, A. Analytical techniques for characterization of biological molecules–proteins and aptamers/oligonucleotides. Bioanalysis 2019, 11, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Krafčiková, P.; Víglaský, V.; Strehlitz, B. G-quadruplex aptamer targeting Protein A and its capability to detect Staphylococcus aureus demonstrated by ELONA. Sci. Rep. 2016, 6, 33812. [Google Scholar] [CrossRef]
- Sypabekova, M.; Bekmurzayeva, A.; Wang, R.; Li, Y.; Nogues, C.; Kanayeva, D. Selection, characterization, and application of DNA aptamers for detection of Mycobacterium tuberculosis secreted protein MPT64. Tuberculosis 2017, 104, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, H.; Hasan, D.; Ruoff, R.S.; Wang, A.X.; Fan, D. Near-field enhanced plasmonic-magnetic bifunctional nanotubes for single cell bioanalysis. Adv. Funct. Mater. 2013, 23, 4332–4338. [Google Scholar] [CrossRef] [Green Version]
- Henry, A.I.; Sharma, B.; Cardinal, M.F.; Kurouski, D.; Van Duyne, R.P. Surface-Enhanced Raman Spectroscopy Biosensing: In Vivo Diagnostics and Multimodal Imaging. Anal. Chem. 2016, 88, 6638–6647. [Google Scholar] [CrossRef] [PubMed]
- Cialla-May, D.; Zheng, X.-S.; Weber, K.; Popp, J. Recent progress in surface-enhanced Raman spectroscopy for biological and biomedical applications: From cells to clinics. Chem. Soc. Rev. 2017, 46, 3945–3961. [Google Scholar] [CrossRef]
- Ochsenkühn, M.A.; Campbell, C.J. Probing biomolecular interactions using surface enhanced Raman spectroscopy: Label-free protein detection using a G-quadruplex DNA aptamer. Chem. Commun. 2010, 46, 2799–2801. [Google Scholar] [CrossRef] [Green Version]
- Baaske, P.; Wienken, C.J.; Reineck, P.; Duhr, S.; Braun, D. Optical thermophoresis for quantifying the buffer dependence of aptamer binding. Angew. Chem. Int. Ed. 2010, 49, 2238–2241. [Google Scholar] [CrossRef]
- Entzian, C.; Schubert, T. Mapping the binding site of an aptamer on ATP using microscale thermophoresis. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.M.; Breitsprecher, D.; Duhr, S.; Baaske, P.; Schubert, T.; Längst, G. Microscale thermophoresis: A rapid and precise method to quantify protein–nucleic acid interactions in solution. In Functional Genomics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 151–164. [Google Scholar]
- Plach, M.; Schubert, T. Biophysical Characterization of Aptamer-Target Interactions. Aptamers Biotechnol. 2019, 1–15. [Google Scholar] [CrossRef]
- Skouridou, V.; Jauset-Rubio, M.; Ballester, P.; Bashammakh, A.S.; El-Shahawi, M.S.; Alyoubi, A.O.; O’Sullivan, C.K. Selection and characterization of DNA aptamers against the steroid testosterone. Microchim. Acta 2017, 184, 1631–1639. [Google Scholar] [CrossRef]
- Asmari, M.; Ratih, R.; Alhazmi, H.A.; El Deeb, S. Thermophoresis for characterizing biomolecular interaction. Methods 2018, 146, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Park, C.; Kwon, S.G.; Bang, W.Y.; Kim, S.W.; Kim, C.W.; Lee, K.W. Structural importance of the C-terminal region in pig aldo-keto reductase family 1 member C1 and their effects on enzymatic activity. BMC Struct. Biol. 2015, 15, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjuler, C.T.; Maolanon, N.N.; Sauer, J.; Stougaard, J.; Thygesen, M.B.; Jensen, K.J. Preparation of glycoconjugates from unprotected carbohydrates for protein-binding studies. Nat. Protoc. 2017, 12, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Pioszak, A.A.; Parker, N.R.; Gardella, T.J.; Xu, H.E. Structural Basis for Parathyroid Hormone-related Protein Binding to the Parathyroid Hormone Receptor and Design of Conformation-selective Peptides. J. Biol. Chem. 2009, 284, 28382–28391. [Google Scholar] [CrossRef] [Green Version]
- Grauschopf, U.; Lilie, H.; Honold, K.; Wozny, M.; Reusch, D.; Esswein, A.; Schäfer, W.; Rücknagel, K.P.; Rudolph, R. The N-terminal fragment of human parathyroid hormone receptor 1 constitutes a hormone binding domain and reveals a distinct disulfide pattern. Biochemistry 2000, 39, 8878–8887. [Google Scholar] [CrossRef]
- Wojciech, J.; Daniel, L.H.; Chang, W.C.; McGill, J.; Jankowska, K.I.; Gelinas, A.D.; Nebojsa, J. Modified aptamers as reagents to characterize recombinant human erythropoietin products. Sci. Rep. 2020, 10, 18593. [Google Scholar]
- Zhang, Z.; Guo, L.; Guo, A.; Xu, H.; Tang, J.; Guo, X.; Xie, J. In vitro lectin-mediated selection and characterization of rHuEPO-α-binding ssDNA aptamers. Bioorganic Med. Chem. 2010, 18, 8016–8025. [Google Scholar] [CrossRef]
- Macaya, R.F.; Schultze, P.; Smith, F.W.; Roe, J.A.; Feigon, J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. USA 1993, 90, 3745–3749. [Google Scholar] [CrossRef] [Green Version]
- Bonifacio, L.; Church, F.C.; Jarstfer, M.B. Effect of locked-nucleic acid on a biologically active g-quadruplex. A structure-activity relationship of the thrombin aptamer. Int. J. Mol. Sci. 2008, 9, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Zaitseva, M.; Kaluzhny, D.; Shchyolkina, A.; Borisova, O.; Smirnov, I.; Pozmogova, G. Conformation and thermostability of oligonucleotide d(GGTTGGTGTGGTTGG) containing thiophosphoryl internucleotide bonds at different positions. Biophys. Chem. 2010, 146, 1–6. [Google Scholar] [CrossRef]
- Varizhuk, A.M.; Tsvetkov, V.B.; Tatarinova, O.N.; Kaluzhny, D.N.; Florentiev, V.L.; Timofeev, E.N.; Shchyolkina, A.K.; Borisova, O.F.; Smirnov, I.P.; Grokhovsky, S.L.; et al. Synthesis, characterization and in vitro activity of thrombin-binding DNA aptamers with triazole internucleotide linkages. Eur. J. Med. Chem. 2013, 67, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Pozmogova, G.E.; Zaitseva, M.A.; Smirnov, I.P.; Shvachko, A.G.; Murina, M.A.; Sergeenko, V.I. Anticoagulant Effects of Thioanalogs of Thrombin-Binding DNA-Aptamer and Their Stability in the Plasma. Bull. Exp. Biol. Med. 2010, 150, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Varizhuk, A.M.; Kaluzhny, D.N.; Novikov, R.A.; Chizhov, A.O.; Smirnov, I.P.; Chuvilin, A.N.; Tatarinova, O.N.; Fisunov, G.Y.; Pozmogova, G.E.; Florentiev, V.L. Synthesis of Triazole-Linked Oligonucleotides with High Affinity to DNA Complements and an Analysis of Their Compatibility with Biosystems. J. Org. Chem. 2013, 78, 5964–5969. [Google Scholar] [CrossRef] [PubMed]
- Morvan, F.; Debart, F.; Vasseur, J.J. From anionic to cationic alpha-anomeric oligodeoxynucleotides. Chem. Biodivers 2010, 7, 494–535. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, S.; Yu, Z.; Konik, R.; Cui, Y.; Koirala, D.; Mao, H. G-quadruplex and i-motif are mutually exclusive in ILPR double-stranded DNA. Biophys. J. 2012, 102, 2575–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odeh, F.; Nsairat, H.; Alshaer, W.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Bawab, A.A.; Ismail, S.I. Aptamers chemistry: Chemical modifications and conjugation strategies. Molecules 2020, 25, 3. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhang, Z.K.; Yu, Y.; Zhuo, Z.; Zhang, G.; Zhang, B.T. Pros and Cons of Denosumab Treatment for Osteoporosis and Implication for RANKL Aptamer Therapy. Front. Cell Dev. Biol. 2020, 8, 325. [Google Scholar] [CrossRef] [PubMed]
- Troisi, R.; Napolitano, V.; Spiridonova, V.; Russo Krauss, I.; Sica, F. Several structural motifs cooperate in determining the highly effective anti-thrombin activity of NU172 aptamer. Nucleic Acids Res. 2018, 46, 12177–12185. [Google Scholar] [CrossRef] [Green Version]
- Yadav, A.; Saini, V.; Arora, S. MCP-1: Chemoattractant with a role beyond immunity: A review. Clin. Chim. Acta 2010, 411, 1570–1579. [Google Scholar] [CrossRef]
- Landgraf, G. Pharmacokinetics, pharmacodynamics, safety and tolerability of the CCL2 antagonist NOX-E36, a novel agent being investigated for treatment of diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 960A. [Google Scholar]
- Ng, E.W.M.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef]
- Nikonowicz, E.P.; Sirr, A.; Legault, P.; Jucker, F.M.; Baer, L.M.; Pardi, A. Preparation of 13 C and 15 N labelled RNAs for heteronuclear multi-dimensional NMR studies. Nucleic Acids Res. 1992, 20, 4507–4513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulbachinskiy, A. Methods for selection of aptamers to protein targets. Biochemistry 2007, 72, 1505–1518. [Google Scholar] [CrossRef]
- Rowsell, S.; Stonehouse, N.J.; Convery, M.A.; Adams, C.J.; Ellington, A.D.; Hirao, I.; Peabody, D.S.; Stockley, P.G.; Phillips, S.E. Crystal structures of a series of RNA aptamers complexed to the same protein target. Nat. Struct. Biol. 1998, 5, 970–975. [Google Scholar] [CrossRef]
- Ruigrok, V.J.; Levisson, M.; Hekelaar, J.; Smidt, H.; Dijkstra, B.W.; Van der Oost, J. Characterization of aptamer-protein complexes by X-ray crystallography and alternative approaches. Int. J. Mol. Sci. 2012, 13, 10537–10552. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.; Yan, J.; Xiong, H.; Liu, Y.; Peng, D.; Liu, Z. Investigations on the interface of nucleic acid aptamers and binding targets. Analyst 2018, 143, 5317–5338. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.-C.; McMullan, G.; Scheres, S.H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, L.; Zhang, B.-T.; Lu, A.; Wang, Y.; Yu, Y.; Zhang, G. Artificial Intelligence in aptamer-target binding prediction. Int. J. Mol. Sci. 2021, 22, 3605. [Google Scholar] [CrossRef] [PubMed]
- Tabarzad, M.; Jafari, M. Trends in the Design and Development of Specific Aptamers Against Peptides and Proteins. Protein J. 2016, 35, 81–99. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, F.; Yang, X.; Wang, K.; Wang, H.; Deng, X. Sensitive point-of-care monitoring of cardiac biomarker myoglobin using aptamer and ubiquitous personal glucose meter. Biosens. Bioelectron. 2015, 64, 161–164. [Google Scholar] [CrossRef]
- Mustafa, M.G.; Petersen, J.R.; Ju, H.; Cicalese, L.; Snyder, N.; Haidacher, S.J.; Denner, L.; Elferink, C. Biomarker discovery for early detection of hepatocellular carcinoma in hepatitis C–infected patients. Mol. Cell. Proteom. 2013, 12, 3640–3652. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.M.; Chakraborty, B.; Sen, D.; Yu, H.-Z. Analyte-driven switching of DNA charge transport: De novo creation of electronic sensors for an early lung cancer biomarker. J. Am. Chem. Soc. 2012, 134, 13823–13833. [Google Scholar] [CrossRef]
- Lee, S.J.; Park, J.-W.; Kim, I.-A.; Youn, B.-S.; Gu, M.B. Sensitive detection of adipokines for early diagnosis of type 2 diabetes using enzyme-linked antibody-aptamer sandwich (ELAAS) assays. Sens. Actuators B Chem. 2012, 168, 243–248. [Google Scholar] [CrossRef]
- Lee, S.J.; Youn, B.-S.; Park, J.W.; Niazi, J.H.; Kim, Y.S.; Gu, M.B. ssDNA aptamer-based surface plasmon resonance biosensor for the detection of retinol binding protein 4 for the early diagnosis of type 2 diabetes. Anal. Chem. 2008, 80, 2867–2873. [Google Scholar] [CrossRef] [PubMed]
- Song, K.-M.; Lee, S.; Ban, C. Aptamers and their biological applications. Sensors 2012, 12, 612–631. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Kang, H.; Ryu, S.H.; Lee, D.S.; Lee, J.H.; Kim, S. Bioimaging of nucleolin aptamer-containing 5-(N-benzylcarboxyamide)-2’-deoxyuridine more capable of specific binding to targets in cancer cells. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]





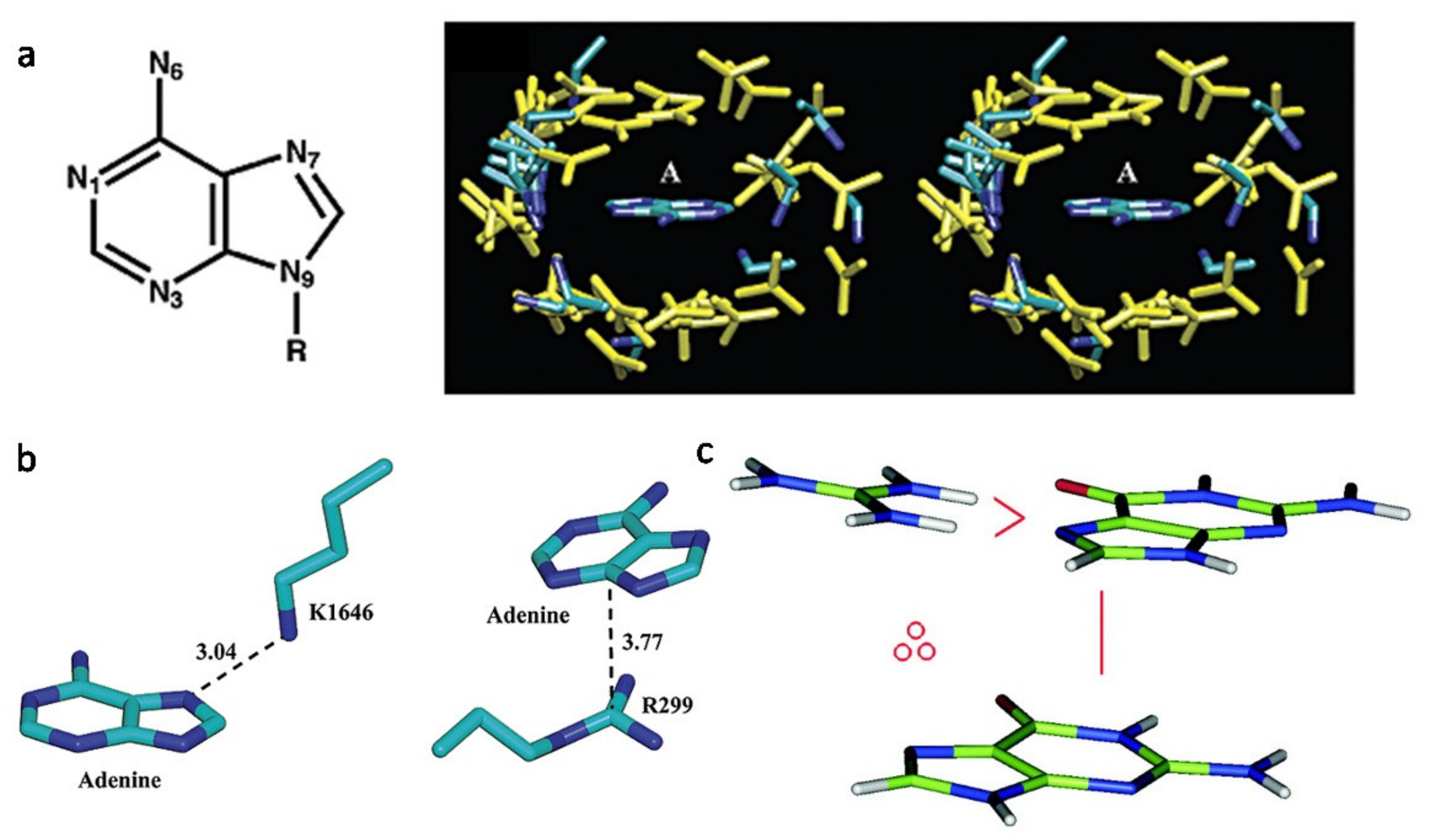{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Sample Requirement | Advantages | Limitations |
|---|---|---|---|
| NMR | Samples must be in solution | (1) Moelcules are studied in solution (2) Protein folding studies can be done by monitoring NMR spectra (3) Efficient in mapping interactions with other molecules | (1) The upper weight limit for NMR structure determination is ~30 kDa (2) Requires a protein sample be soluble in a high-concentrated solution (3) Overlapped spectrum |
| X-ray | Samples must be crystallized in a lattice structure | (1) Provides high-resolution information (2) Does not requie a protein be soluble in a high-concentrated solution (3) Applied to proteins or macromolecules in a wide molecular weight range | (1) Requires a protein crystal (2) Crystal contacts can distort protein structure (3) Not suitable with fairly flexible molecules |
| Cryo-EM | Sample is frozen in its native state | (1) High enough resolution (2) Does not require a protein crystal (3) Does not requie a protein be soluble in a high-concentrated solution | (1) Complex measurements and data analysis (2) Difficult to use for proteins with MW below 300 kDa (3) The technology still needs to be thoroughly tested by the scientific community |
| PDB ID | Complex Type (Protein:mAptamer) | Polar Contacts (Number) | Interface Area (A2) | Dissociation Constant (nm) | Gibbs Free Energy Change (kJ/mol) | References |
|---|---|---|---|---|---|---|
| 5mxf | Photorhabdus asymbiotica lectin (PHL): alpha-methyl fucoside | 16 | 1165.2 | 1.4 ± 0.21 μm | −48.8 | [32] |
| 1u1y | MS2 coat protein: F5 RNA aptamer | 42 | 3180.6 | 0.6 ± 0.3 | −51.2 | [33] |
| 1exd | Glutamine aminoacyl tRNA synthetase: Glutamine tRNA | 42 | 2599.3 | 0.3 ± 0.1 | −35.9 | [34] |
| 1ooa | NF-kB(p50)2: RNA Aptamer | 27 | 1343.1 | 5.4 ± 2.2 | 0.1 | [35] |
| 2b63 | Complete RNA Polymerase II: RNA inhibitor | 147 | 9311.5 | 33 ± 2 | −91.8 | [36] |
| 4i7y | Human Alpha Thrombin: 27-mer DNA Aptamer | 18 | 1079.5 | 0.7 | −6.1 | [37] |
| 5ew1 | Human thrombin: two DNA aptamers (HD22 and HD1-ΔT3) | 21 | 1116.2 | 4.9 ± 1.6 51.8 ± 5.3 | −10.0 | [38,39] |
| 4dii | Human alpha thrombin: thrombin binding DNA aptamer | 32 | 1086.0 | 33 | −9.4 | [40,41] |
| 3agv | Human IgG: RNA aptamer | 13 | 1092.0 | - | −12.8 | [42,43] |
| 3qlp | Human alpha thrombin: modified thrombin binding DNA aptamer | 39 | 1404.0 | 25 ± 1 | −9.5 | [41,44] |
| 3hxq | Von Willebrand Factor (VWF) A1 Domain: DNA Aptamer ARC1172 | 23 | 1070.8 | 2 | −1.2 | [45,46] |
| 3uzs | G Protein-Coupled Receptor Kinase 2-Heterotrimeric G Protein Beta 1 and Gamma 2 Subunit Complex: C13.28 RNA Aptamer | 13 | 2188.7 | 3.8 ± 1.2 | −41.7 | [47] |
| 3uzt | G Protein-Coupled Receptor Kinase 2: C13.18 RNA Aptamer | 8 | 1096.8 | 35 ± 5 | −4.2 | [47] |
| 4hqx | Human PDGF-BB: Modified DNA aptamer (SOMAmer SL4) | 24 | 1076.8 | 1.20 | −14.4 | [48] |
| 4hqu | Human PDGF-BB: Modified DNA aptamer (SOMAmer SL5) | 24 | 1078.6 | 0.02 | −14.9 | [48] |
| 4lz4 | Human thrombin: TBA (DNA) deletion mutant lacking thymine 3 nucleobase | 43 | 1354.5 | 54.9 | −10.6 | [49] |
| 5hto | Plasmodium Vivax LDH: DNA aptamer pL1 | 14 | 2105.1 | 16.8 ± 0.6 | −37.3 | [50] |
| 4wb2 | Mouse C5a complement anaphylatoxin: mirror-image L-RNA/L-DNA aptamer NOX-D20 | 14 | 420.6 | 0.02 | −3.1 | [51] |
| 3zh2 | Plasmodium falciparum lactate dehydrogenase: DNA aptamer | 13 | 2106.3 | 42 | −37.6 | [52] |
| 4pdb | Bacillus Anthracis Ribosomal Protein S8: RNA Aptamer | 13 | 898.6 | 110 ± 30 | −16.7 | [5] |
| 4ni7 | Human interleukin 6: a modified DNA aptamer (SOMAmer SL1025) | 3 | 336.3 | 0.20 | 3.0 | [53] |
| 4r8i | Chemokine CCL2: Mirror-image RNA Oligonucleotide Aptamer | 10 | 807.3 | 1.4 ± 0.16 | −8.8 | [54] |
| 4zbn | Nerve growth factor: non-helical DNA triplex aptamer | 13 | 1418.9 | 0.21 ± 0.08 | −25.2 | [55] |
| 5cmx | Human alpha thrombin: duplex/quadruplex DNA aptamer | 32 | 1124.0 | 0.56 | −9.0 | [56] |
| 5hrt | Mouse autotaxin: DNA aptamer | 8 | 934.6 | 1.6 | −11.7 | [57] |
| 5do4 | Thrombin: RNA aptamer | 31 | 1173.1 | 0.0081 ± 0.0002 | −8.6 | [58] |
| 5uc6 | IL-1 alpha: naphthyl-modified DNA aptamer | 4 | 598.4 | 7.3 | −1.1 | [59] |
| 6eo6 | Human alpha-thrombin: modified 15-mer DNA aptamer | 36 | 1064.1 | 1.00 | −7.9 | [60] |
| 6sy4 | TetR: TetR-binding RNA-aptamer K1 | 8 | 1615.2 | 5.6 | −31.2 | [61] |
| 6rti | Human glutamate carboxypeptidase II: aptamer A9g | 24 | 2414.7 | - | −25.8 | [62] |
| 6z8w | Human alpha thrombin: thrombin binding aptamer variant (TBA-3G) | 34 | 1066.1 | 9.8 ± 0.6 | −8.1 | [63] |
| 7jtq | Human complement factor B: Slow off-rate modified aptamer | 10 | 894.9 | 0.049 | −3.1 | [64] |
| Principle | Advantages | Limitations | |
|---|---|---|---|
| Isothermal titration calorimetry | Thermodynamics where contact between two molecules results in either exothermic or endothermic | (1) Precise detection of enthalpy change, stoichiometry and binding constant (2) Real-time and dynamic monitoring of the whole process of interaction (3) Application of interaction between proteins and ligands, polysaccharide, small compound | (1) Large quantity and high-quality demand high for samples; (2) The repeatability of identical sample reaction remains poor |
| Surface plasmon resonance | Changes in the local refractive index to detect adsorption onto microarrays | (1) Real-time and dynamic monitoring of the whole process of interaction (2) Maintain the structure and natural activity of the sample (3) The detection process is convenient and swift, with high sensitivity | (1) Sensitive to interference factors such as sample composition and temperature (2) Difficult to distinguish non-specific adsorption (3) Immobilization of one binding partner required |
| Atomic force microscopy | Detection between a ligand-functionalized AFM tip and a receptor-modified substrate | (1) Simple sample preparation; imaging under a lot of conditions (air, liquid, vacuum, physiological status) (2) Operated on one single molecule | (1) The between the tip and the sample could contaminate the tip, adsorb protein molecules or damage the surface of the sample (2) Salt crystallization of samples |
| Flow cytometry | The measurement of light scattered by particles and the fluorescence observed when the particles are passed in a stream through a laser beam. | (1) Tens of thousands of cells can be quickly examined, and the data gathered are processed by a computer (2) It provides quantifiable data from a sample | (1) Flow cytometry instrument is relatively expensive (2) Provides overwhelming information about the samples that may not be always necessary |
| Enzyme-linked oligonucleotide assay (ELONA) | A solid-phase type of enzyme immunoassay to detect the presence of a ligand in a liquid sample | (1) Aptamers can be easily labeled without significant effect on the affinity and specificity of the aptamer (2) Aptamers can provide a constant source of high and uniform quality detection reagent | (1) Sequence labeling required (2) Non-specific binding to the plate, which confuse the enrichment (3) Not suitable for small molecular targets |
| Surface-enhanced Raman spectroscopy | Enhances Raman scattering by molecules adsorbed on rough metal surfaces | (1) Requires relatively lower laser intensity and longer wavelengths (2) Rapid signal acquisition times | (1) Still requires interdisciplinary research effort to develop highly sensitive and reliable system |
| Microscale thermophoresis | Directed movement of molecules along temperature gradients, an effect termed thermophoresis | (1) Low sample consumption (2) Fast experimental procedure (3) Ability to perform measurements in complex samples, such as cell lysates (4) Possibility of labeling-free | (1) Cannot discern 2nd binding site or non-specific binding (2) Labeling with hydrophobic fluorophores required, which can alter binding profile |
| Bio-layer interferometry | A label-free method based on the real-time optical monitoring of biomolecular interactions | Highest throughput | (1) Immoblization of one binding partner required (2) Dissociation phases are imprecise due to analyte rebinding (no flow through system) |
| Modification Strategies | Categories |
|---|---|
| Chemical modifications | (1) Modifications on the inter-nucleotide linkages (2) Modifications on the nucleotide base |
| Strutural modifications | (1) Modifications on secondary structures (2) Modifications on the interactive nucleotides (3) Addition of structural groups |
| Aptamer | Target | Therapy Area | Latest Clinical Trial Phase, Status |
|---|---|---|---|
| ARC1905 | C5 | Age-Related Macular Degeneration Juvenile Macular Degeneration (Stargardt Disease) | Phase 1, completed Phase 2, recruiting |
| Zimura | C5 | Idiopathic Polypoidal Choroidal Vasculopathy | Phase 2, completed |
| Fovista | PDGF BB | Age-Related Macular Degeneration | Phase 1, terminated |
| E10030 plus Lucentis | PDGF | Age-Related Macular Degeneration | Phase 2, completed |
| EYE001 | VEGF | Hippel–Lindau Disease Macular Degeneration Choroidal Neovascularization | Phase 2, completed Phase 2, completed Phase 3, completed |
| Macugen | VEGF165 | Age-Related Macular Degeneration | Phase 3, completed |
| NU172 | Thrombin | Heart disease | Phase 2, recruiting |
| 68Ga-Sgc8 | PTK7 | Colorectal Cancer | Phase 1, unknown |
| ARC1779 | vWF | Von Willebrand Disease Type-2b | Phase 2, completed |
| BT200 | vWF | Von Willebrand Diseases Hemophilia A | Phase 2, completed |
| ApTOLL | TLR4 | Stroke | Phase 1, completed |
| NOX-H94 | Hepcidin peptide hormone | Anemia End-Stage Renal Disease | Phase 2, completed |
| NOX-E36 | CCL2 | Chronic Inflammatory Diseases Type 2 Diabetes Mellitus Systemic Lupus Erythematosus | Phase 1, completed |
| NOX-A12 | CXCL12 | Autologous Stem Cell Transplantation Hematopoietic Stem Cell Transplantation | Phase 1, completed Phase 1, completed |
| ARC19499 | TFPI | Hemophilia | Phase 1, completed |
| AS1411 | Nucleolin | Acute Myeloid Leukemia | Phase 2, terminated |
| AS1411-GNP | Nucleolin | Coronavirus Disease 2019 (COVID-19) | Phase 1, recruiting |
| ACTGRO-777 | Nucleolin | Acute Myelocytic Leukemia Pancreatic Cancer | Phase 2, recruiting Phase 1, recruiting |
| RBM-007 | FGF2 | Exudative Age-Related Macular Degeneration | Phase 2, completed |
| BC-007 | GPCR AAb | Dilated Cardiomyopathy Coronavirus Disease 2019 (COVID-19) | Phase 2, recruiting Phase 1, recruiting |
| REG1 | Coagulation factor IXa | Coronary Artery Disease | Phase 2, completed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; Chen, Z.; Liu, D.; Jiang, H.; Zhang, Z.-K.; Lu, A.; Zhang, B.-T.; Yu, Y.; Zhang, G. Structural Biology for the Molecular Insight between Aptamers and Target Proteins. Int. J. Mol. Sci. 2021, 22, 4093. https://doi.org/10.3390/ijms22084093
Zhang N, Chen Z, Liu D, Jiang H, Zhang Z-K, Lu A, Zhang B-T, Yu Y, Zhang G. Structural Biology for the Molecular Insight between Aptamers and Target Proteins. International Journal of Molecular Sciences. 2021; 22(8):4093. https://doi.org/10.3390/ijms22084093
Chicago/Turabian StyleZhang, Ning, Zihao Chen, Dingdong Liu, Hewen Jiang, Zong-Kang Zhang, Aiping Lu, Bao-Ting Zhang, Yuanyuan Yu, and Ge Zhang. 2021. "Structural Biology for the Molecular Insight between Aptamers and Target Proteins" International Journal of Molecular Sciences 22, no. 8: 4093. https://doi.org/10.3390/ijms22084093
APA StyleZhang, N., Chen, Z., Liu, D., Jiang, H., Zhang, Z. -K., Lu, A., Zhang, B. -T., Yu, Y., & Zhang, G. (2021). Structural Biology for the Molecular Insight between Aptamers and Target Proteins. International Journal of Molecular Sciences, 22(8), 4093. https://doi.org/10.3390/ijms22084093