
VWF, Platelets and the Antiphospholipid Syndrome


Abstract
:1. Introduction
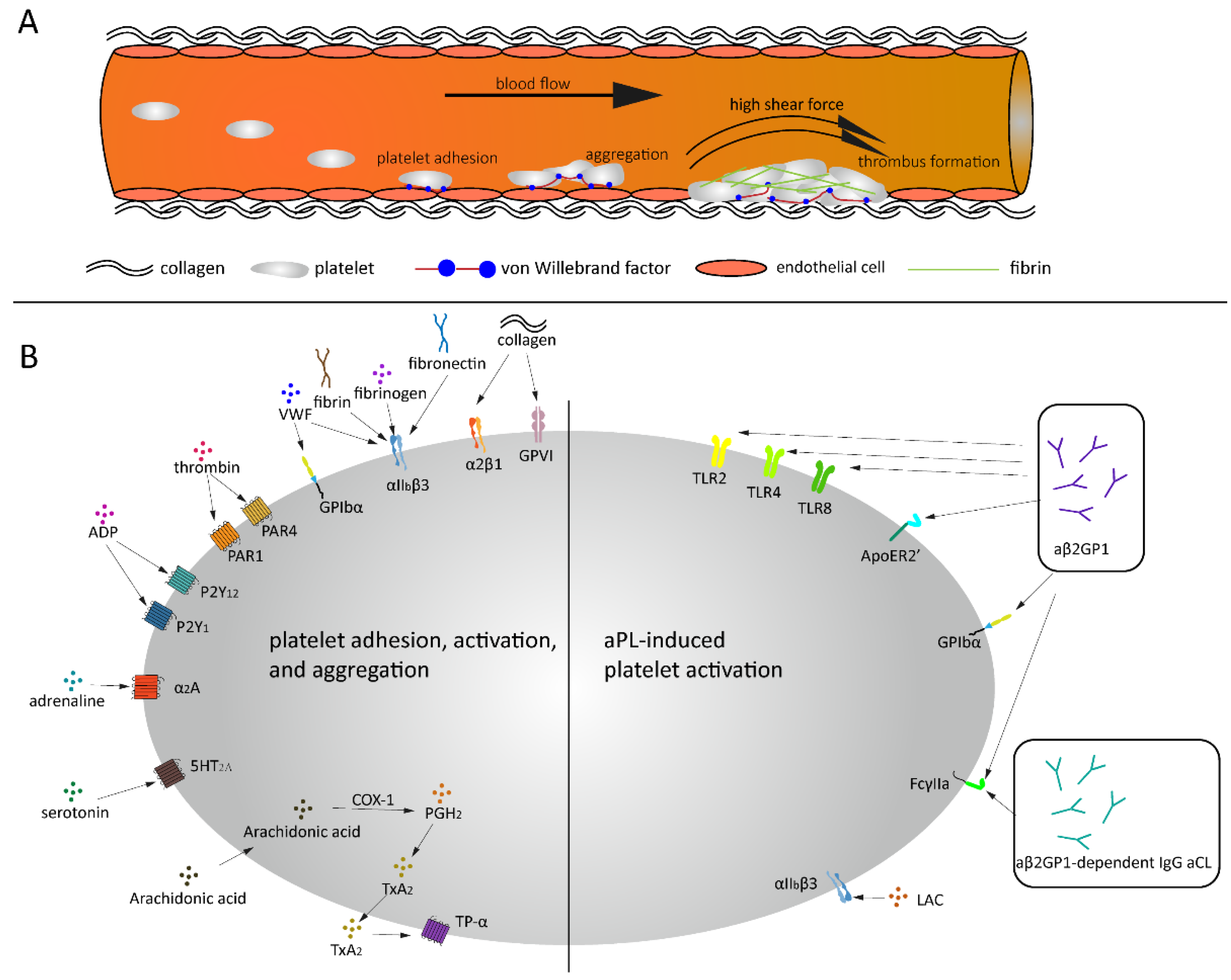
2. Von Willebrand Factor (VWF)
3. Von Willebrand Factor and APS
β2GPI and VWF
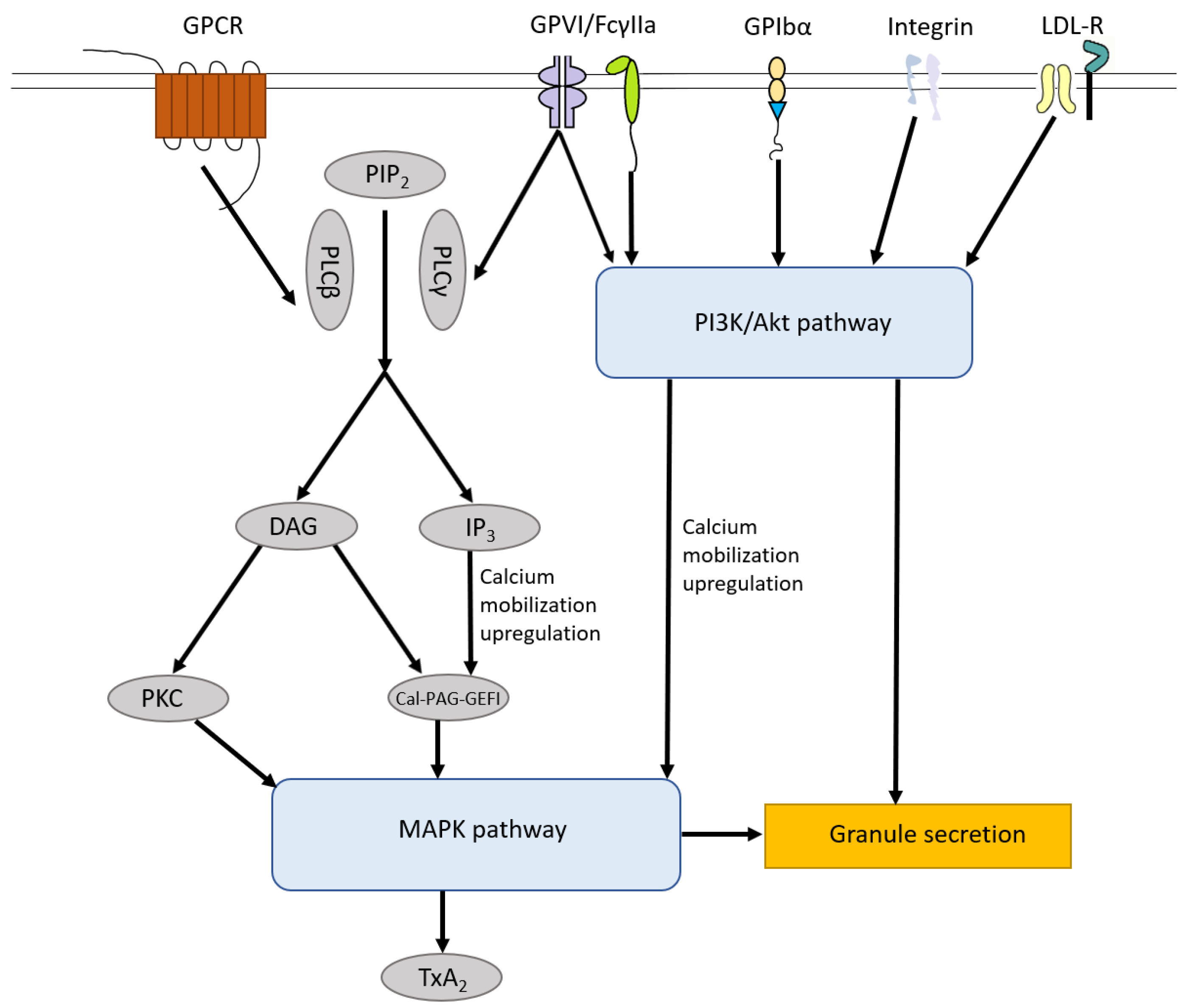
4. Platelet Activation
5. Platelets Activation and APS
5.1. Platelets and LAC
5.2. Platelets and aβ2GPI
5.3. Platelets and aCL
6. Arterial Thrombosis and APS
6.1. LAC and Arterial Thrombosis
6.2. aβ2GPI and Arterial Thrombosis
6.3. aCL and Arterial Thrombosis
6.4. aPS/aPT and Arterial Thrombosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruiz-Irastorza, G.; Crowther, M.; Branch, W.; Khamashta, M.A. Antiphospholipid syndrome. Lancet 2010, 376, 1498–1509. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Cheng, G.Y.; Denas, G.; Pengo, V. Arterial thrombosis in antiphospholipid syndrome (APS): Clinical approach and treatment. A systematic review. Blood Rev. 2020, 100788. [Google Scholar] [CrossRef] [PubMed]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.; PG, D.E.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef]
- Hulstein, J.J.; Lenting, P.J.; de Laat, B.; Derksen, R.H.; Fijnheer, R.; de Groot, P.G. beta2-Glycoprotein I inhibits von Willebrand factor dependent platelet adhesion and aggregation. Blood 2007, 110, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- van der Vorm, L.N.; Visser, R.; Huskens, D.; Veninga, A.; Adams, D.L.; Remijn, J.A.; Hemker, H.C.; Rensma, P.L.; van Horssen, R.; de Laat, B. Circulating active von Willebrand factor levels are increased in chronic kidney disease and end-stage renal disease. Clin. Kidney J. 2020, 13, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Groot, E.; de Groot, P.G.; Fijnheer, R.; Lenting, P.J. The presence of active von Willebrand factor under various pathological conditions. Curr. Opin. Hematol. 2007, 14, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Claus, R.A.; Bockmeyer, C.L.; Budde, U.; Kentouche, K.; Sossdorf, M.; Hilberg, T.; Schneppenheim, R.; Reinhart, K.; Bauer, M.; Brunkhorst, F.M.; et al. Variations in the ratio between von Willebrand factor and its cleaving protease during systemic inflammation and association with severity and prognosis of organ failure. Thromb. Haemost. 2009, 101, 239–247. [Google Scholar]
- Kawecki, C.; Lenting, P.J.; Denis, C.V. von Willebrand factor and inflammation. J. Thromb. Haemost. 2017, 15, 1285–1294. [Google Scholar] [CrossRef]
- O’Sullivan, J.M.; Ward, S.; Lavin, M.; O’Donnell, J.S. von Willebrand factor clearance—biological mechanisms and clinical significance. Br. J. Haematol. 2018, 183, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Swami, A.; Kaur, V. von Willebrand Disease: A Concise Review and Update for the Practicing Physician. Clin. Appl. Thromb. Hemost. 2017, 23, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Nagai, Y.; Kuwabara, C.; Shimizu, R.; Umeki, A.; Yamamoto, T. Acquired von Willebrand Syndrome due to Aortic Valve Stenosis in a Case with Antiphospholipid Antibody. Intern. Med. 2018, 57, 1641–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simurda, T.; Dobrotova, M.; Skornova, I.; Sokol, J.; Kubisz, P.; Stasko, J. Successful Use of a Highly Purified Plasma von Willebrand Factor Concentrate Containing Little FVIII for the Long-Term Prophylaxis of Severe (Type 3) von Willebrand’s Disease. Semin. Thromb. Hemost. 2017, 43, 639–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer Hovinga, J.A.; Coppo, P.; Lämmle, B.; Moake, J.L.; Miyata, T.; Vanhoorelbeke, K. Thrombotic thrombocytopenic purpura. Nat. Rev. Dis. Primers 2017, 3, 17020. [Google Scholar] [CrossRef] [Green Version]
- Brehm, M.A. Von Willebrand factor processing. Hamostaseologie 2017, 37, 59–72. [Google Scholar] [CrossRef]
- Karlaftis, V.; Perera, S.; Monagle, P.; Ignjatovic, V. Importance of post-translational modifications on the function of key haemostatic proteins. Blood Coagul. Fibrinolysis 2016, 27, 1–4. [Google Scholar] [CrossRef]
- Fazavana, J.; Brophy, T.M.; Chion, A.; Cooke, N.; Terraube, V.; Cohen, J.; Parng, C.; Pittman, D.; Cunningham, O.; Lambert, M.; et al. Investigating the clearance of VWF A-domains using site-directed PEGylation and novel N-linked glycosylation. J. Thromb. Haemost. 2020, 18, 1278–1290. [Google Scholar] [CrossRef]
- McKinnon, T.A.; Chion, A.C.; Millington, A.J.; Lane, D.A.; Laffan, M.A. N-linked glycosylation of VWF modulates its interaction with ADAMTS13. Blood 2008, 111, 3042–3049. [Google Scholar] [CrossRef]
- Tischer, A.; Machha, V.R.; Moon-Tasson, L.; Benson, L.M.; Auton, M. Glycosylation sterically inhibits platelet adhesion to von Willebrand factor without altering intrinsic conformational dynamics. J. Thromb. Haemost. 2020, 18, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verweij, C.L.; Diergaarde, P.J.; Hart, M.; Pannekoek, H. Full-length von Willebrand factor (vWF) cDNA encodes a highly repetitive protein considerably larger than the mature vWF subunit. EMBO J. 1986, 5, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Haberichter, S.L. von Willebrand factor propeptide: Biology and clinical utility. Blood 2015, 126, 1753–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadler, J.E. Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 1998, 67, 395–424. [Google Scholar] [CrossRef]
- Randi, A.M.; Smith, K.E.; Castaman, G. von Willebrand factor regulation of blood vessel formation. Blood 2018, 132, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinders, J.H.; de Groot, P.G.; Sixma, J.J.; van Mourik, J.A. Storage and secretion of von Willebrand factor by endothelial cells. Haemostasis 1988, 18, 246–261. [Google Scholar] [CrossRef] [PubMed]
- Slayter, H.; Loscalzo, J.; Bockenstedt, P.; Handin, R.I. Native conformation of human von Willebrand protein. Analysis by electron microscopy and quasi-elastic light scattering. J. Biol. Chem. 1985, 260, 8559–8563. [Google Scholar] [CrossRef]
- Flood, V.H.; Schlauderaff, A.C.; Haberichter, S.L.; Slobodianuk, T.L.; Jacobi, P.M.; Bellissimo, D.B.; Christopherson, P.A.; Friedman, K.D.; Gill, J.C.; Hoffmann, R.G.; et al. Crucial role for the VWF A1 domain in binding to type IV collagen. Blood 2015, 125, 2297–2304. [Google Scholar] [CrossRef] [Green Version]
- Szanto, T.; Vanhoorelbeke, K.; Toth, G.; Vandenbulcke, A.; Toth, J.; Noppe, W.; Deckmyn, H.; Harsfalvi, J. Identification of a VWF peptide antagonist that blocks platelet adhesion under high shear conditions by selectively inhibiting the VWF-collagen interaction. J. Thromb. Haemost. 2009, 7, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Fidalgo, T.; Oliveira, A.; Silva Pinto, C.; Martinho, P.; Ferreira, G.; Salvado, R.; Sevivas, T.; Catarino, C.; Ribeiro, M.L. VWF collagen (types III and VI)-binding defects in a cohort of type 2M VWD patients—a strategy for improvement of a challenging diagnosis. Haemoph. Off. J. World Fed. Hemoph. 2017, 23, e143–e147. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, E.G.; Martijn van der Plas, R.; Kroon, J.; Sixma, J.J.; Gros, P. Crystal structure of the A3 domain of human von Willebrand factor: Implications for collagen binding. Structure 1997, 5, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.A.; Dong, J.F.; Thaggard, J.A.; Cruz, M.A.; López, J.A.; McIntire, L.V. Kinetics of GPIbalpha-vWF-A1 tether bond under flow: Effect of GPIbalpha mutations on the association and dissociation rates. Biophys. J. 2003, 85, 4099–4109. [Google Scholar] [CrossRef] [Green Version]
- Randi, A.M.; Rabinowitz, I.; Mancuso, D.J.; Mannucci, P.M.; Sadler, J.E. Molecular basis of von Willebrand disease type IIB. Candidate mutations cluster in one disulfide loop between proposed platelet glycoprotein Ib binding sequences. J. Clin. Investig. 1991, 87, 1220–1226. [Google Scholar] [CrossRef] [Green Version]
- Sixma, J.J.; Schiphorst, M.E.; Verweij, C.L.; Pannekoek, H. Effect of deletion of the A1 domain of von Willebrand factor on its binding to heparin, collagen and platelets in the presence of ristocetin. Eur. J. Biochem. 1991, 196, 369–375. [Google Scholar] [CrossRef]
- Van der Vorm, L.N.; Li, L.; Huskens, D.; Chayoua, W.; Kelchtermans, H.; de Groot, P.G.; Roest, M.; Remijn, J.A.; de Laat, B. Analytical characterization and reference interval of an enzyme-linked immunosorbent assay for active von Willebrand factor. PLoS ONE 2019, 14, e0211961. [Google Scholar] [CrossRef]
- Furlan, M. Von Willebrand factor: Molecular size and functional activity. Ann. Hematol. 1996, 72, 341–348. [Google Scholar] [CrossRef] [PubMed]
- De Mast, Q.; Groot, E.; Asih, P.B.; Syafruddin, D.; Oosting, M.; Sebastian, S.; Ferwerda, B.; Netea, M.G.; de Groot, P.G.; van der Ven, A.J.A.M.; et al. ADAMTS13 Deficiency with Elevated Levels of Ultra-Large and Active von Willebrand Factor in P. falciparum and P. vivax Malaria. Am. J. Trop. Med. Hyg. 2009, 80, 492–498. [Google Scholar] [CrossRef]
- Sonneveld, M.A.; de Maat, M.P.; Leebeek, F.W. Von Willebrand factor and ADAMTS13 in arterial thrombosis: A systematic review and meta-analysis. Blood Rev. 2014, 28, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Pickens, B.; Mao, Y.; Li, D.; Siegel, D.L.; Poncz, M.; Cines, D.B.; Zheng, X.L. Platelet-delivered ADAMTS13 inhibits arterial thrombosis and prevents thrombotic thrombocytopenic purpura in murine models. Blood 2015, 125, 3326–3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masias, C.; Cataland, S.R. The role of ADAMTS13 testing in the diagnosis and management of thrombotic microangiopathies and thrombosis. Blood 2018, 132, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Verhenne, S.; Denorme, F.; Libbrecht, S.; Vandenbulcke, A.; Pareyn, I.; Deckmyn, H.; Lambrecht, A.; Nieswandt, B.; Kleinschnitz, C.; Vanhoorelbeke, K.; et al. Platelet-derived VWF is not essential for normal thrombosis and hemostasis but fosters ischemic stroke injury in mice. Blood 2015, 126, 1715–1722. [Google Scholar] [CrossRef] [Green Version]
- Brait, V.H.; Miró-Mur, F.; Pérez-de-Puig, I.; Notario, L.; Hurtado, B.; Pedragosa, J.; Gallizioli, M.; Jiménez-Altayó, F.; Arbaizar-Rovirosa, M.; Otxoa-de-Amezaga, A.; et al. CD69 Plays a Beneficial Role in Ischemic Stroke by Dampening Endothelial Activation. Circ. Res. 2019, 124, 279–291. [Google Scholar] [CrossRef]
- Nimjee, S.M.; Dornbos, D., III; Pitoc, G.A.; Wheeler, D.G.; Layzer, J.M.; Venetos, N.; Huttinger, A.; Talentino, S.E.; Musgrave, N.J.; Moody, H.; et al. Preclinical Development of a vWF Aptamer to Limit Thrombosis and Engender Arterial Recanalization of Occluded Vessels. Mol. Ther.J. Am. Soc. Gene Ther. 2019, 27, 1228–1241. [Google Scholar] [CrossRef]
- Sanders, Y.V.; Eikenboom, J.; de Wee, E.M.; van der Bom, J.G.; Cnossen, M.H.; Degenaar-Dujardin, M.E.; Fijnvandraat, K.; Kamphuisen, P.W.; Laros-van Gorkom, B.A.; Meijer, K.; et al. Reduced prevalence of arterial thrombosis in von Willebrand disease. J. Thromb. Haemost. 2013, 11, 845–854. [Google Scholar] [CrossRef]
- Chen, J.; Chung, D.W. Inflammation, von Willebrand factor, and ADAMTS13. Blood 2018, 132, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, N.J.; Dawson, R.A.; Henderson, F.I.; Greaves, M.; Hughes, P. Stimulation of von Willebrand factor antigen release by immunoglobulin from thrombosis prone patients with systemic lupus erythematosus and the anti-phospholipid syndrome. Br. J. Rheumatol. 1993, 32, 123–126. [Google Scholar] [CrossRef]
- Der, H.; Kerekes, G.; Veres, K.; Szodoray, P.; Toth, J.; Lakos, G.; Szegedi, G.; Soltesz, P. Impaired endothelial function and increased carotid intima-media thickness in association with elevated von Willebrand antigen level in primary antiphospholipid syndrome. Lupus 2007, 16, 497–503. [Google Scholar] [CrossRef]
- Wurm, H. beta 2-Glycoprotein-I (apolipoprotein H) interactions with phospholipid vesicles. Int. J. Biochem. 1984, 16, 511–515. [Google Scholar] [CrossRef]
- Ng, C.J.; McCrae, K.R.; Ashworth, K.; Sosa, L.J.; Betapudi, V.; Manco-Johnson, M.J.; Liu, A.; Dong, J.F.; Chung, D.; White-Adams, T.C.; et al. Effects of anti-beta2GPI antibodies on VWF release from human umbilical vein endothelial cells and ADAMTS13 activity. Res. Pract. Thromb. Haemost. 2018, 2, 380–389. [Google Scholar] [CrossRef]
- Hantgan, R.R. Fibrin protofibril and fibrinogen binding to ADP-stimulated platelets: Evidence for a common mechanism. Biochim. Biophys. Acta 1988, 968, 24–35. [Google Scholar] [CrossRef]
- Bennett, J.S.; Vilaire, G. Exposure of platelet fibrinogen receptors by ADP and epinephrine. J. Clin. Investig. 1979, 64, 1393–1401. [Google Scholar] [CrossRef]
- Ni, H.; Denis, C.V.; Subbarao, S.; Degen, J.L.; Sato, T.N.; Hynes, R.O.; Wagner, D.D. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J. Clin. Investig. 2000, 106, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef]
- Schonberger, T.; Ziegler, M.; Borst, O.; Konrad, I.; Nieswandt, B.; Massberg, S.; Ochmann, C.; Jurgens, T.; Seizer, P.; Langer, H.; et al. The dimeric platelet collagen receptor GPVI-Fc reduces platelet adhesion to activated endothelium and preserves myocardial function after transient ischemia in mice. Am. J. Physiol. Cell Physiol. 2012, 303, C757–C766. [Google Scholar] [CrossRef]
- Law, D.A.; DeGuzman, F.R.; Heiser, P.; Ministri-Madrid, K.; Killeen, N.; Phillips, D.R. Integrin cytoplasmic tyrosine motif is required for outside-in alphaIIbbeta3 signalling and platelet function. Nature 1999, 401, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, M.A. Small molecules targeting heterotrimeric G proteins. Eur. J. Pharm. 2018, 826, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S. Activation of Platelet Function Through G Protein–Coupled Receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilatis, D.K.; Hohmann, J.G.; Zeng, H.; Li, F.; Ranchalis, J.E.; Mortrud, M.T.; Brown, A.; Rodriguez, S.S.; Weller, J.R.; Wright, A.C.; et al. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 4903. [Google Scholar] [CrossRef] [Green Version]
- Takeda, S.; Kadowaki, S.; Haga, T.; Takaesu, H.; Mitaku, S. Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett. 2002, 520, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.L.; Norman, J.E.; Morgan, N.V.; Mundell, S.J.; Lordkipanidze, M.; Lowe, G.C.; Daly, M.E.; Simpson, M.A.; Drake, S.; Watson, S.P.; et al. Diversity and impact of rare variants in genes encoding the platelet G protein-coupled receptors. Thromb. Haemost. 2015, 113, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Law, D.A.; Nannizzi-Alaimo, L.; Phillips, D.R. Outside-in integrin signal transduction. Alpha IIb beta 3-(GP IIb IIIa) tyrosine phosphorylation induced by platelet aggregation. J. Biol. Chem. 1996, 271, 10811–10815. [Google Scholar] [CrossRef] [Green Version]
- Sokol, J.; Skerenova, M.; Ivankova, J.; Simurda, T.; Stasko, J. Association of Genetic Variability in Selected Genes in Patients With Deep Vein Thrombosis and Platelet Hyperaggregability. Clin. Appl. Thromb. Hemost. Off. J. Int. Acad. Clin. Appl. Thromb. Hemost. 2018, 24, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Baroni, G.; Banzato, A.; Bison, E.; Denas, G.; Zoppellaro, G.; Pengo, V. The role of platelets in antiphospholipid syndrome. Platelets 2017, 28, 762–766. [Google Scholar] [CrossRef]
- Biasiolo, A.; Pengo, V. Antiphospholipid antibodies are not present in the membrane of gel-filtered platelets of patients with IgG anticardiolipin antibodies, lupus anticoagulant and thrombosis. Blood Coagul. Fibrinolysis 1993, 4, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Khamashta, M.A.; Harris, E.N.; Gharavi, A.E.; Derue, G.; Gil, A.; Vázquez, J.J.; Hughes, G.R. Immune mediated mechanism for thrombosis: Antiphospholipid antibody binding to platelet membranes. Ann. Rheum. Dis. 1988, 47, 849–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forastiero, R.; Martinuzzo, M.; Carreras, L.O.; Maclouf, J. Anti-beta2 glycoprotein I antibodies and platelet activation in patients with antiphospholipid antibodies: Association with increased excretion of platelet-derived thromboxane urinary metabolites. Thromb. Haemost. 1998, 79, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Espinola, R.G.; Pierangeli, S.S.; Gharavi, A.E.; Harris, E.N. Hydroxychloroquine reverses platelet activation induced by human IgG antiphospholipid antibodies. Thromb. Haemost. 2002, 87, 518–522. [Google Scholar] [CrossRef]
- Galli, M.; Cortelazzo, S.; Viero, P.; Finazzi, G.; de Gaetano, G.; Barbui, T. Interaction between platelets and lupus anticoagulant. Eur. J. Haematol. 1988, 41, 88–94. [Google Scholar] [CrossRef]
- Galli, M.; Bevers, E.M.; Comfurius, P.; Barbui, T.; Zwaal, R.F. Effect of antiphospholipid antibodies on procoagulant activity of activated platelets and platelet-derived microvesicles. Br. J. Haematol. 1993, 83, 466–472. [Google Scholar] [CrossRef]
- Joseph, J.E.; Harrison, P.; Mackie, I.J.; Isenberg, D.A.; Machin, S.J. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br. J. Haematol. 2001, 115, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Hell, L.; Lurger, K.; Mauracher, L.M.; Grilz, E.; Reumiller, C.M.; Schmidt, G.J.; Ercan, H.; Koder, S.; Assinger, A.; Basilio, J.; et al. Altered platelet proteome in lupus anticoagulant (LA)-positive patients-protein disulfide isomerase and NETosis as new players in LA-related thrombosis. Exp. Mol. Med. 2020, 52, 66–78. [Google Scholar] [CrossRef]
- Allen, K.L.; Fonseca, F.V.; Betapudi, V.; Willard, B.; Zhang, J.; McCrae, K.R. A novel pathway for human endothelial cell activation by antiphospholipid/anti-β2 glycoprotein I antibodies. Blood 2012, 119, 884–893. [Google Scholar] [CrossRef]
- Lutters, B.C.; Derksen, R.H.; Tekelenburg, W.L.; Lenting, P.J.; Arnout, J.; de Groot, P.G. Dimers of beta 2-glycoprotein I increase platelet deposition to collagen via interaction with phospholipids and the apolipoprotein E receptor 2’. J. Biol. Chem. 2003, 278, 33831–33838. [Google Scholar] [CrossRef] [Green Version]
- Pennings, M.T.; Derksen, R.H.; van Lummel, M.; Adelmeijer, J.; VanHoorelbeke, K.; Urbanus, R.T.; Lisman, T.; de Groot, P.G. Platelet adhesion to dimeric beta-glycoprotein I under conditions of flow is mediated by at least two receptors: Glycoprotein Ibalpha and apolipoprotein E receptor 2’. J. Thromb. Haemost. 2007, 5, 369–377. [Google Scholar] [CrossRef]
- Satta, N.; Dunoyer-Geindre, S.; Reber, G.; Fish, R.J.; Boehlen, F.; Kruithof, E.K.; de Moerloose, P. The role of TLR2 in the inflammatory activation of mouse fibroblasts by human antiphospholipid antibodies. Blood 2007, 109, 1507–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, Y.; Hurst, J.; Lorenz, M.; Prinz, N.; Clemens, N.; Drechsler, M.D.; Bauer, S.; Chapman, J.; Shoenfeld, Y.; Blank, M.; et al. Human antiphospholipid antibodies induce TNFalpha in monocytes via Toll-like receptor 8. Immunobiology 2010, 215, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Hollerbach, A.; Muller-Calleja, N.; Ritter, S.; Hauser, F.; Canisius, A.; Orning, C.; Jurk, K.; Lackner, K.J. Platelet Activation by Antiphospholipid Antibodies Depends on Epitope Specificity and is Prevented by mTOR Inhibitors. Thromb. Haemost. 2019. [Google Scholar] [CrossRef]
- Nimpf, J.; Wurm, H.; Kostner, G.M. Beta 2-glycoprotein-I (apo-H) inhibits the release reaction of human platelets during ADP-induced aggregation. Atherosclerosis 1987, 63, 109–114. [Google Scholar] [CrossRef]
- Fu, H.; Zhao, J.; Xu, L.; Liu, K.; Wang, Y.; Chen, H.; Han, W.; Wang, J.; Wang, F.; Huang, X.; et al. Reduced β2-GPI is associated with increased platelet aggregation and activation in patients with prolonged isolated thrombocytopenia after allo-HSCT. Sci. China Life Sci. 2019, 62, 921–929. [Google Scholar] [CrossRef]
- Zhang, W.; Gao, F.; Lu, D.; Sun, N.; Yin, X.; Jin, M.; Liu, Y. Anti-β2 glycoprotein I antibodies in complex with β2 glycoprotein I induce platelet activation via two receptors: Apolipoprotein E receptor 2’ and glycoprotein I bα. Front. Med. 2016, 10, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Vega-Ostertag, M.; Casper, K.; Swerlick, R.; Ferrara, D.; Harris, E.N.; Pierangeli, S.S. Involvement of p38 MAPK in the up-regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheum. 2005, 52, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhang, G.; Ouyang, H.; Zhang, P.; Chen, Y.; Wang, R.; Zhou, H. Effects of β2/aβ2 on oxLDL-induced CD36 activation in THP-1 macrophages. Life Sci. 2019, 239, 117000. [Google Scholar] [CrossRef]
- Shao, F.; Miao, Y.; Zhang, Y.; Han, L.; Ma, X.; Deng, J.; Jiang, C.; Kong, W.; Xu, Q.; Feng, J.; et al. B cell-derived anti-beta 2 glycoprotein I antibody contributes to hyperhomocysteinemia-aggravated abdominal aortic aneurysm. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shoenfeld, Y.; Blank, M.; Cervera, R.; Font, J.; Raschi, E.; Meroni, P.L. Infectious origin of the antiphospholipid syndrome. Ann. Rheum. Dis. 2006, 65, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Finazzi, G.; Bevers, E.M.; Barbui, T. Kaolin clotting time and dilute Russell’s viper venom time distinguish between prothrombin-dependent and beta 2-glycoprotein I-dependent antiphospholipid antibodies. Blood 1995, 86, 617–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, R.K.; Adler, L.; Hickman, A.R.; Hageman, J.R. Anticardiolipin antibody-positive serum enhances endothelial cell platelet-activating factor production. Am. J. Obstet. Gynecol. 1991, 165, 1748–1752. [Google Scholar] [CrossRef]
- Sammaritano, L.R.; Ng, S.; Sobel, R.; Lo, S.K.; Simantov, R.; Furie, R.; Kaell, A.; Silverstein, R.; Salmon, J.E. Anticardiolipin IgG subclasses: Association of IgG2 with arterial and/or venous thrombosis. Arthritis Rheum. 1997, 40, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Nisar, S.P.; Jones, M.L.; Cunningham, M.R.; Mumford, A.D.; Mundell, S.J. Rare platelet GPCR variants: What can we learn? Br. J. Pharmacol. 2015, 172, 3242–3253. [Google Scholar] [CrossRef] [Green Version]
- Kamato, D.; Thach, L.; Bernard, R.; Chan, V.; Zheng, W.; Kaur, H.; Brimble, M.; Osman, N.; Little, P.J. Structure, Function, Pharmacology, and Therapeutic Potential of the G Protein, Gα/q,11. Front. Cardiovasc. Med. 2015, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Bergmeier, W.; Stefanini, L. Platelet ITAM signaling. Curr. Opin. Hematol. 2013, 20, 445–450. [Google Scholar] [CrossRef]
- Brandt, K.J.; Kruithof, E.K.; de Moerloose, P. Receptors involved in cell activation by antiphospholipid antibodies. Thromb. Res. 2013, 132, 408–413. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, N.C.; Anderson, R.C.; McDermott, K.W. Reelin: Diverse roles in central nervous system development, health and disease. Int. J. Biochem. Cell Biol. 2019, 112, 72–75. [Google Scholar] [CrossRef]
- Urbanus, R.T.; Siegerink, B.; Roest, M.; Rosendaal, F.R.; de Groot, P.G.; Algra, A. Antiphospholipid antibodies and risk of myocardial infarction and ischaemic stroke in young women in the RATIO study: A case-control study. Lancet. Neurol. 2009, 8, 998–1005. [Google Scholar] [CrossRef]
- Arnaud, L.; Mathian, A.; Devilliers, H.; Ruffatti, A.; Tektonidou, M.; Forastiero, R.; Pengo, V.; Lambert, M.; Lefevre, G.; Martinez-Zamora, M.A.; et al. Patient-level analysis of five international cohorts further confirms the efficacy of aspirin for the primary prevention of thrombosis in patients with antiphospholipid antibodies. Autoimmun. Rev. 2015, 14, 192–200. [Google Scholar] [CrossRef]
- Saidi, S.; Mahjoub, T.; Almawi, W.Y. Lupus anticoagulants and anti-phospholipid antibodies as risk factors for a first episode of ischemic stroke. J. Thromb. Haemost. 2009, 7, 1075–1080. [Google Scholar] [CrossRef]
- Pasoto, S.G.; Chakkour, H.P.; Natalino, R.R.; Viana, V.S.; Bueno, C.; Lianza, A.C.; de Andrade, J.L.; Neto, M.L.; Fuller, R.; Bonfa, E. Lupus anticoagulant: A marker for stroke and venous thrombosis in primary Sjögren’s syndrome. Clin. Rheumatol. 2012, 31, 1331–1338. [Google Scholar] [CrossRef]
- De Mast, Q.; Molhoek, J.E.; van der Ven, A.J.; Gray, W.K.; de Groot, P.G.; Jusabani, A.; Mugusi, F.; Urbanus, R.T.; Walker, R.W. Antiphospholipid Antibodies and the Risk of Stroke in Urban and Rural Tanzania: A Community-Based Case-Control Study. Stroke 2016, 47, 2589–2595. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Akashi, Y.J.; Soma, Y. Silent myocardial infarction subsequent to cutaneous polyarteritis nodosa in a patient with positive lupus anticoagulant. J. Am. Acad. Dermatol. 2011, 65, 442–443. [Google Scholar] [CrossRef]
- Landi, G.; Calloni, M.V.; Grazia Sabbadini, M.; Mannuccio Mannucci, P.; Candelise, L. Recurrent ischemic attacks in two young adults with lupus anticoagulant. Stroke 1983, 14, 377–379. [Google Scholar] [CrossRef] [Green Version]
- Mills, T.J.; Safford, R.E.; Kazmier, F.J. Myocardial infarction, persistent coronary artery thrombosis and lupus anticoagulant. Int. J. Cardiol. 1988, 21, 190–194. [Google Scholar] [CrossRef]
- Murai, K.; Sakata, K.; Gamou, T.; Nagata, Y.; Tada, H.; Shimojima, M.; Okada, H.; Hayashi, K.; Kawashiri, M.A. Acute myocardial infarction in a patient positive for lupus anticoagulant: A case report. BMC Cardiovasc. Disord. 2019, 19, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nojima, J.; Suehisa, E.; Akita, N.; Toku, M.; Fushimi, R.; Tada, H.; Kuratsune, H.; Machii, T.; Kitani, T.; Amino, N. Risk of arterial thrombosis in patients with anticardiolipin antibodies and lupus anticoagulant. Br. J. Haematol. 1997, 96, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, Q.; Lega, J.C.; Mismetti, P.; Chapelle, C.; Wahl, D.; Cathébras, P.; Laporte, S. Risk of venous and arterial thrombosis according to type of antiphospholipid antibodies in adults without systemic lupus erythematosus: A systematic review and meta-analysis. Autoimmun. Rev. 2014, 13, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Borges, R.B.; Bodanese, L.C.; Mühlen, C.A.; Repetto, G.; Viehe, M.; Norman, G.L.; Staub, H.L. Anti-beta2-glycoprotein I autoantibodies and metabolic syndrome. Arq. Bras. Cardiol. 2011, 96, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Kahles, T.; Humpich, M.; Steinmetz, H.; Sitzer, M.; Lindhoff-Last, E. Phosphatidylserine IgG and beta-2-glycoprotein I IgA antibodies may be a risk factor for ischaemic stroke. Rheumatology 2005, 44, 1161–1165. [Google Scholar] [CrossRef] [Green Version]
- Ranzolin, A.; Bohn, J.M.; Norman, G.L.; Manenti, E.; Bodanese, L.C.; von Mühlen, C.A.; Staub, H.L. Anti-beta2-glycoprotein I antibodies as risk factors for acute myocardial infarction. Arq. Bras. Cardiol. 2004, 83, 137–140. [Google Scholar] [CrossRef] [Green Version]
- Staub, H.L.; von Muhlen, C.A.; Norman, G.L. Beta2-glycoprotein I IgA antibodies and ischaemic stroke. Rheumatology 2006, 45, 645–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachostergios, P.J.; Dufresne, F. Acute renal infarction associated with homozygous methylenetetrahydrofolate reductase mutation C677T and IgA beta-2-glycoprotein antibodies. Blood Coagul. Fibrinolysis 2015, 26, 583–585. [Google Scholar] [CrossRef]
- Arad, A.; Proulle, V.; Furie, R.A.; Furie, B.C.; Furie, B. β₂-Glycoprotein-1 autoantibodies from patients with antiphospholipid syndrome are sufficient to potentiate arterial thrombus formation in a mouse model. Blood 2011, 117, 3453–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wu, Z.; Zhang, W.; Zhang, F.; Li, Y.; Liu, Y. Clinical performance of non-criteria antibodies to phospholipids in Chinese patients with antiphospholipid syndrome. Clin. Chim. Acta 2019, 495, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.; Willis, R.; Romay-Penabad, Z.; Ruiz-Limón, P.; Martínez-Martínez, L.A.; Jatwani, S.; Jajoria, P.; Seif, A.; Alarcón, G.S.; Papalardo, E.; et al. Value of isolated IgA anti-β2 -glycoprotein I positivity in the diagnosis of the antiphospholipid syndrome. Arthritis Rheum. 2013, 65, 3186–3193. [Google Scholar] [CrossRef] [Green Version]
- Tortosa, C.; Cabrera-Marante, O.; Serrano, M.; Martínez-Flores, J.A.; Pérez, D.; Lora, D.; Morillas, L.; Paz-Artal, E.; Morales, J.M.; Pleguezuelo, D.; et al. Incidence of thromboembolic events in asymptomatic carriers of IgA anti ß2 glycoprotein-I antibodies. PLoS ONE 2017, 12, e0178889. [Google Scholar] [CrossRef] [PubMed]
- Urbanski, G.; Yelnik, C.M.; Maillard, H.; Launay, D.; Dubucquoi, S.; Hachulla, E.; Hatron, P.Y.; Lambert, M. Antiphospholipid Syndrome With Isolated Isotype M Anticardiolipin and/or Anti-B2GPI Antibody Is Associated With Stroke. Stroke 2018, 49, 2770–2772. [Google Scholar] [CrossRef]
- Brey, R.L.; Abbott, R.D.; Curb, J.D.; Sharp, D.S.; Ross, G.W.; Stallworth, C.L.; Kittner, S.J. beta(2)-Glycoprotein 1-dependent anticardiolipin antibodies and risk of ischemic stroke and myocardial infarction: The honolulu heart program. Stroke 2001, 32, 1701–1706. [Google Scholar] [CrossRef] [Green Version]
- Matyja-Bednarczyk, A.; Swadźba, J.; Iwaniec, T.; Sanak, M.; Dziedzina, S.; Ćmiel, A.; Musiał, J. Risk factors for arterial thrombosis in antiphospholipid syndrome. Thromb. Res. 2014, 133, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Bucci, T.; Triggiani, M.; Ames, P.R.J.; Parrotto, S.; Violi, F.; Pignatelli, P.; Farcomeni, A. Immunoglobulin G (IgG) anticardiolipin antibodies and recurrent cardiovascular events. A systematic review and Bayesian meta-regression analysis. Autoimmun. Rev. 2019, 18, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.R.; Dier, K.J.; Lopez, D.; Merrill, J.T.; Fink, C.A. Anti-beta 2-glycoprotein I and antiphosphatidylserine antibodies are predictors of arterial thrombosis in patients with antiphospholipid syndrome. Am. J. Clin. Pathol. 2004, 121, 142–149. [Google Scholar] [CrossRef]
- Nojima, J.; Kuratsune, H.; Suehisa, E.; Kitani, T.; Iwatani, Y.; Kanakura, Y. Strong correlation between the prevalence of cerebral infarction and the presence of anti-cardiolipin/beta2-glycoprotein I and anti-phosphatidylserine/prothrombin antibodies--Co-existence of these antibodies enhances ADP-induced platelet activation in vitro. Thromb. Haemost. 2004, 91, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Okuma, H.; Kitagawa, Y.; Ishikawa, T.; Takagi, S. Study of phosphatidylserine-dependent anti-prothrombin antibody in cerebral infarction. Intern. Med. 2009, 48, 1351–1355. [Google Scholar] [CrossRef]
- Sciascia, S.; Sanna, G.; Murru, V.; Roccatello, D.; Khamashta, M.A.; Bertolaccini, M.L. Anti-prothrombin (aPT) and anti-phosphatidylserine/prothrombin (aPS/PT) antibodies and the risk of thrombosis in the antiphospholipid syndrome. A systematic review. Thromb. Haemost. 2014, 111, 354–364. [Google Scholar] [CrossRef]
- Kirchhof, K.; Welzel, T.; Mecke, C.; Zoubaa, S.; Sartor, K. Differentiation of white, mixed, and red thrombi: Value of CT in estimation of the prognosis of thrombolysis phantom study. Radiology 2003, 228, 126–130. [Google Scholar] [CrossRef]
- Lippi, G.; Favaloro, E.J. Venous and Arterial Thromboses: Two Sides of the Same Coin? Semin. Thromb. Hemost. 2018, 44, 239–248. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Criteria |
|
| Laboratory Criteria |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Ninivaggi, M.; Chayoua, W.; de Laat, B. VWF, Platelets and the Antiphospholipid Syndrome. Int. J. Mol. Sci. 2021, 22, 4200. https://doi.org/10.3390/ijms22084200
Huang S, Ninivaggi M, Chayoua W, de Laat B. VWF, Platelets and the Antiphospholipid Syndrome. International Journal of Molecular Sciences. 2021; 22(8):4200. https://doi.org/10.3390/ijms22084200
Chicago/Turabian StyleHuang, Shengshi, Marisa Ninivaggi, Walid Chayoua, and Bas de Laat. 2021. "VWF, Platelets and the Antiphospholipid Syndrome" International Journal of Molecular Sciences 22, no. 8: 4200. https://doi.org/10.3390/ijms22084200
APA StyleHuang, S., Ninivaggi, M., Chayoua, W., & de Laat, B. (2021). VWF, Platelets and the Antiphospholipid Syndrome. International Journal of Molecular Sciences, 22(8), 4200. https://doi.org/10.3390/ijms22084200