Pathomechanisms in the Kidneys in Selected Protozoan Parasitic Infections

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
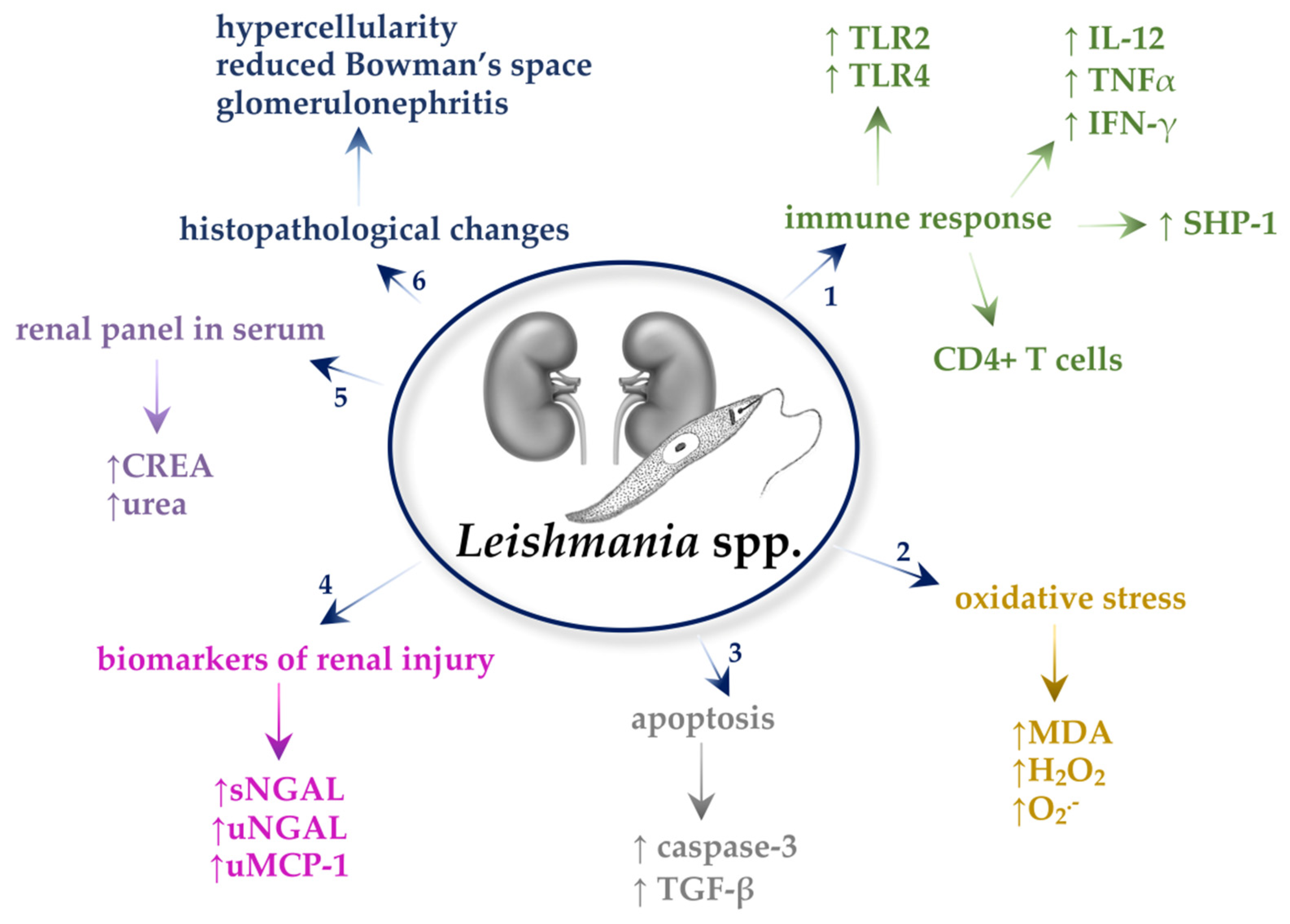
2. Leishmania spp.
2.1. Immune Response
2.2. Oxidative Stress
2.3. Apoptosis
2.4. Transforming Growth Factor β (TGF-β)
2.5. Neutrophil Gelatinase-Associated Lipocalin (NGAL)
2.6. Kidney Injury Molecule 1 (KIM-1)
2.7. Monocyte Chemoattractant Protein-1 (MCP-1)
2.8. Biochemical Parameters in the Serum and Urine
2.9. Histopathological Changes
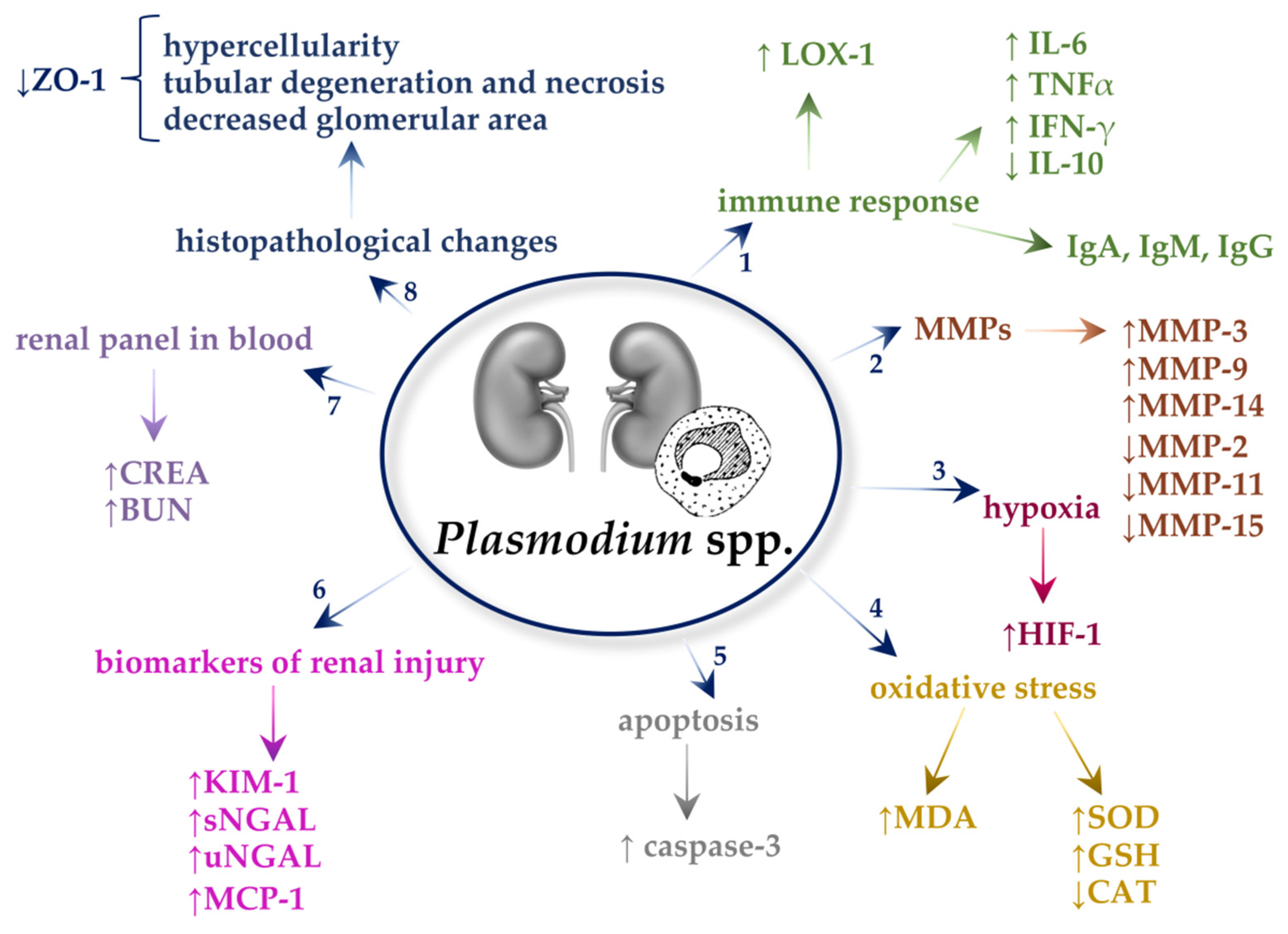
3. Plasmodium spp.
3.1. Immune Responses
3.2. Matrix Metalloproteinases (MMPs)
3.3. Oxidative Stress
3.4. Apoptosis
3.5. Hypoxia
3.6. Neutrophil Gelatinase-Associated Lipocalin (NGAL)
3.7. Kidney Injury Molecule-1 (KIM-1)
3.8. Monocyte Chemoattractant Protein-1 (MCP-1)
3.9. Renal Panel in the Blood
3.10. Histopathological Changes
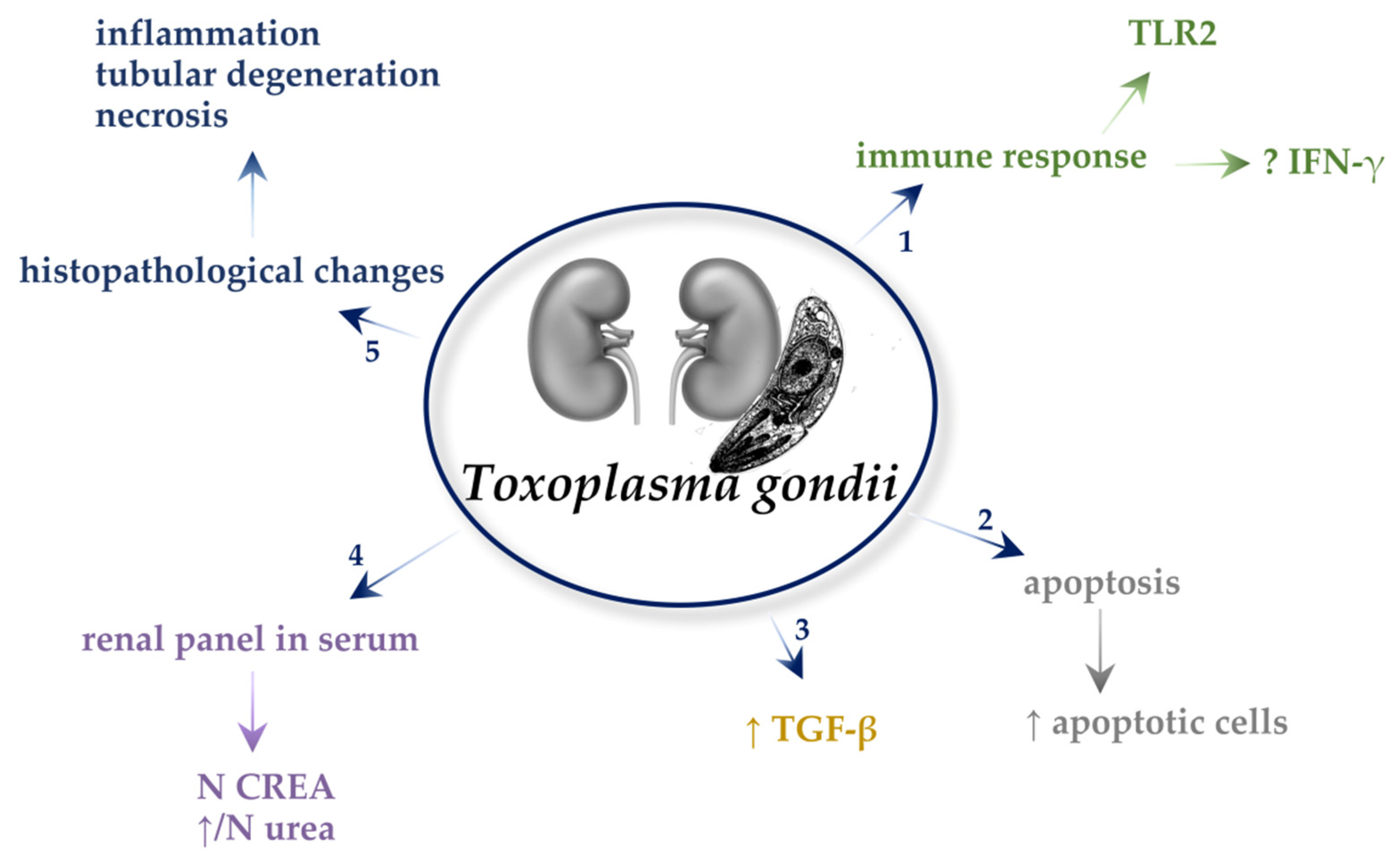
4. Toxoplasma gondii
4.1. Immune Responses
4.2. Oxidative Stress
4.3. Apoptosis
4.4. Transforming Growth Factor β (TGF-β)
4.5. Biochemical Parameters in the Serum
4.6. Histopathological Changes
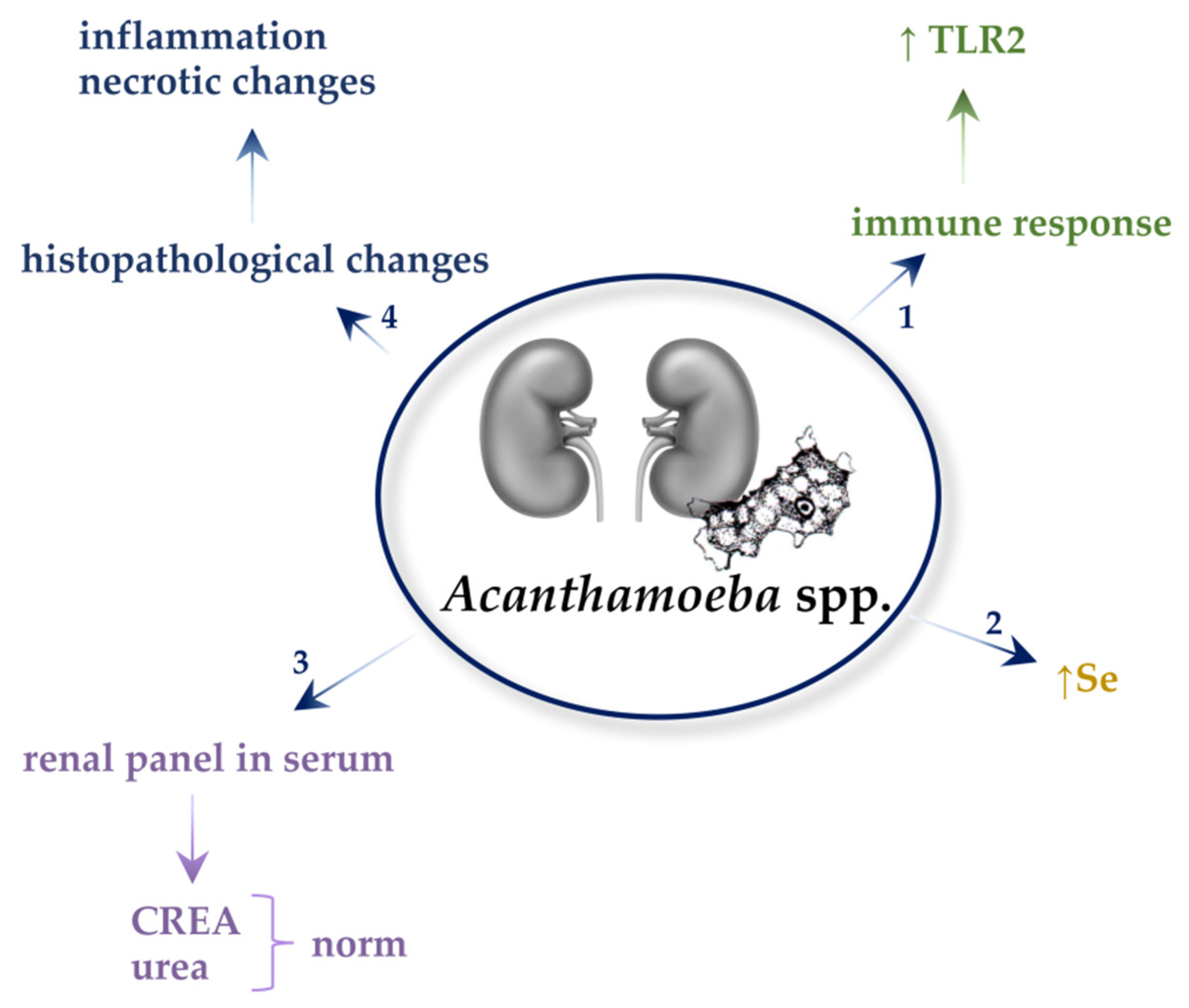
5. Acanthamoeba spp.
5.1. Immune Responses
5.2. Biochemical Parameters in the Serum and Urine
5.3. Selenium (Se) Concentration
5.4. Histopathological Changes
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication—worldwide more than 850 million individuals have kidney diseases. Kidney Int. 2019, 96, 1048–1050. [Google Scholar] [CrossRef]
- Barsoum, R.S. Parasitic Kidney Disease: Milestones in the Evolution of Our Knowledge. Am. J. Kidney Dis. 2013, 61, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Iturbe, B.; Burdmann, E.A.; Barsoum, R.S. Glomerular Diseases Associated with Infection. Compr. Clin. Nephrol. 2010, 662–674. [Google Scholar] [CrossRef]
- Manesh, R.M.; Safa, A.H.; Sharafi, S.M.; Jafari, R.; Bahadoran, M.; Yousefi, M.; Nasri, H.; Darani, H.Y. Parasites and chronic renal failure. J. Ren. Inj. Prev. 2014, 3, 87–90. [Google Scholar]
- Van Velthuysen, M.-L.F.; Florquin, S. Glomerulopathy Associated with Parasitic Infections. Clin. Microbiol. Rev. 2000, 13, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Pace, D. Leishmaniasis. J. Infect. 2014, 69, S10–S18. [Google Scholar] [CrossRef]
- Sharma, U.; Singh, S. Insect vectors of Leishmania: Distribution, physiology and their control. J. Vector Borne Dis. 2008, 45, 255–272. [Google Scholar]
- Thakur, S.; Joshi, J.; Kaur, S. Leishmaniasis diagnosis: An update on the use of parasitological, immunological and molecular methods. J. Parasit. Dis. 2020, 44, 253–272. [Google Scholar] [CrossRef]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; WHO Leishmaniasis Control Team. Leishmaniasis Worldwide and Global Estimates of Its Incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef]
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: A review. F1000Research 2017, 6, 750. [Google Scholar] [CrossRef]
- Clementi, A.; Battaglia, G.; Floris, M.; Castellino, P.; Ronco, C.; Cruz, D.N. Renal involvement in leishmaniasis: A review of the literature. Clin. Kidney J. 2011, 4, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Akira, S. Toll-Like Receptors. Curr. Protoc. Immunol. 2015, 109, 14.12.1–14.12.10. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Couser, W.G.; Johnson, R.J. The etiology of glomerulonephritis: Roles of infection and autoimmunity. Kidney Int. 2014, 86, 905–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertowski, S.; Lipa, P.; Morawska, I.; Niedźwiedzka-Rystwej, P.; Bębnowska, D.; Hrynkiewicz, R.; Grywalska, E.; Roliński, J.; Załuska, W. Toll-Like Receptor as a Potential Biomarker in Renal Diseases. Int. J. Mol. Sci. 2020, 21, 6712. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Kitaura, J.; Izawa, K.; Kaitani, A.; Komeno, Y.; Nakamura, M.; Yamazaki, S.; Enomoto, Y.; Oki, T.; Akiba, H.; et al. TIM1 is an endogenous ligand for LMIR5/CD300b: LMIR5 deficiency ameliorates mouse kidney ischemia/reperfusion injury. J. Exp. Med. 2010, 207, 1501–1511. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.-J. Toll-Like Receptors and Danger Signaling in Kidney Injury. J. Am. Soc. Nephrol. 2010, 21, 1270–1274. [Google Scholar] [CrossRef] [Green Version]
- Rosin, D.L.; Okusa, M.D. Dangers Within: DAMP Responses to Damage and Cell Death in Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 416–425. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Tiwari, N.; Gedda, M.R.; Haque, R.; Singh, R.K. Leishmania donovani infection activates Toll-like receptor 2, 4 expressions and Transforming growth factor-beta mediated apoptosis in renal tissues. Braz. J. Infect. Dis. 2017, 21, 545–549. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [Green Version]
- Costa, F.A.L.; Prianti, M.G.; Silva, T.C.; Silva, S.M.M.S.; Guerra, J.L.; Goto, H. T cells, adhesion molecules and modulation of apoptosis in visceral leishmaniasis glomerulonephritis. BMC Infect. Dis. 2010, 10, 112. [Google Scholar] [CrossRef] [Green Version]
- Junior, G.B.D.S.; Barros, E.J.G.; Daher, E.D.F. Kidney involvement in leishmaniasis—A review. Braz. J. Infect. Dis. 2014, 18, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Wang, H.; Koles, N.L.; Zhang, A.; Aronson, N.E. Leishmania infantum-chagasi activates SHP-1 and reduces NFAT5/TonEBP activity in the mouse kidney inner medulla. Am. J. Physiol. Physiol. 2014, 307, F516–F524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, W.M.; Ruley, J.K. NK cells and their receptors. Reprod. Biomed. 2008, 16, 173–191. [Google Scholar] [CrossRef]
- Vara, D.; Pula, G. Reactive Oxygen Species: Physiological Roles in the Regulation of Vascular Cells. Curr. Mol. Med. 2014, 14, 1103–1125. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Gyurászová, M.; Gurecká, R.; Bábíčková, J.; Tóthová, Ľ. Oxidative Stress in the Pathophysiology of Kidney Disease: Implications for Noninvasive Monitoring and Identification of Biomarkers. Oxidative Med. Cell. Longev. 2020, 2020, 5478708. [Google Scholar] [CrossRef] [Green Version]
- Priante, G.; Gianesello, L.; Ceol, M.; Del Prete, D.; Anglani, F. Cell Death in the Kidney. Int. J. Mol. Sci. 2019, 20, 3598. [Google Scholar] [CrossRef] [Green Version]
- Kapczuk, P.; Kosik-Bogacka, D.; Kupnicka, P.; Metryka, E.; Simińska, D.; Rogulska, K.; Skórka-Majewicz, M.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. The Influence of Selected Gastrointestinal Parasites on Apoptosis in Intestinal Epithelial Cells. Biomolecules 2020, 10, 674. [Google Scholar] [CrossRef]
- Renehan, A.G.; Booth, C.; Potten, C.S. What is apoptosis, and why is it important? BMJ 2001, 322, 1536–1538. [Google Scholar] [CrossRef] [Green Version]
- Susin, S.A.; Daugas, E.; Ravagnan, L.; Samejima, K.; Zamzami, N.; Loeffler, M.; Costantini, P.; Ferri, K.F.; Irinopoulou, T.; Prévost, M.-C.; et al. Two Distinct Pathways Leading to Nuclear Apoptosis. J. Exp. Med. 2000, 192, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a Human Protein Homologous to C. elegans CED-4, Participates in Cytochrome c–Dependent Activation of Caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef] [Green Version]
- de Caestecker, M. The transforming growth factor-β superfamily of receptors. Cytokine Growth Factor Rev. 2004, 15, 1–11. [Google Scholar] [CrossRef]
- Gu, Y.-Y.; Liu, X.-S.; Huang, X.-R.; Yu, X.-Q.; Lan, H.-Y. Diverse Role of TGF-β in Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 123. [Google Scholar] [CrossRef]
- Alves, A.F.; Pereira, R.A.; de Andrade, H.M.; Mosser, D.M.; Tafuri, W.L. Immunohistochemical study of renal fibropoiesis associated with dogs naturally and experimentally infected with two different strains of Leishmania (L.) infantum. Int. J. Exp. Pathol. 2019, 100, 222–233. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Griffin, K.A.; Lan, R.; Geng, H.; Saikumar, P.; Bidani, A.K. Acute kidney injury: A springboard for progression in chronic kidney disease. Am. J. Physiol. Physiol. 2010, 298, F1078–F1094. [Google Scholar] [CrossRef] [Green Version]
- Greijer, A.E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef]
- Singer, E.; Markó, L.; Paragas, N.; Barasch, J.; Dragun, D.; Müller, D.N.; Budde, K.; Schmidt-Ott, K.M. Neutrophil gelatinase-associated lipocalin: Pathophysiology and clinical applications. Acta Physiol. 2013, 207, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Ricci, Z.; Ronco, C. Today’s Approach to the Critically Ill Patient with Acute Kidney Injury. Blood Purif. 2009, 27, 127–134. [Google Scholar] [CrossRef]
- Di Carlo, A. Evaluation of neutrophil gelatinase-associated lipocalin (NGAL), matrix metalloproteinase-9 (MMP-9) and their complex MMP-9/NGAL in sera and urine of patients with kidney tumors. Oncol. Lett. 2013, 5, 1677–1681. [Google Scholar] [CrossRef] [Green Version]
- Bolignano, D.; Donato, V.; Coppolino, G.; Campo, S.; Buemi, A.; Lacquaniti, A.; Buemi, M. Neutrophil Gelatinase–Associated Lipocalin (NGAL) as a Marker of Kidney Damage. Am. J. Kidney Dis. 2008, 52, 595–605. [Google Scholar] [CrossRef]
- Peris, M.P.; Morales, M.; Ares-Gómez, S.; Esteban-Gil, A.; Gómez-Ochoa, P.; Gascón, M.; Moreno, B.; Castillo, J.A. Neutrophil Gelatinase-Associated Lipocalin (NGAL) Is Related with the Proteinuria Degree and the Microscopic Kidney Findings in Leishmania-Infected Dogs. Microorganisms 2020, 8, 1966. [Google Scholar] [CrossRef]
- Meneses, G.C.; Daher, E.D.F.; Junior, G.B.D.S.; Bezerra, G.F.; Da Rocha, T.P.; De Azevedo, I.E.P.; Libório, A.B.; Martins, A.M.C. Visceral leishmaniasis-associated nephropathy in hospitalised Brazilian patients: New insights based on kidney injury biomarkers. Trop. Med. Int. Health 2018, 23, 1046–1057. [Google Scholar] [CrossRef]
- Han, W.K.; Bailly, V.; Abichandani, R.; Thadhani, R.; Bonventre, J.V. Kidney Injury Molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002, 62, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Kubrak, T.; Podgórski, R.; Aebisher, D.; Gala-Błądzińska, A. The significance of NGAL and KIM-1 proteins for diagnosis of acute kidney injury (AKI) in clinical practice. Eur. J. Clin. Exp. Med. 2018, 16, 28–33. [Google Scholar] [CrossRef]
- Pennemans, V.; Rigo, J.-M.; Faes, C.; Reynders, C.; Penders, J.; Swennen, Q. Establishment of reference values for novel urinary biomarkers for renal damage in the healthy population: Are age and gender an issue? Clin. Chem. Lab. Med. 2013, 51, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Brooks, C.R.; Xiao, S.; Sabbisetti, V.; Yeung, M.Y.; Hsiao, L.-L.; Ichimura, T.; Kuchroo, V.; Bonventre, J.V. KIM-1–mediated phagocytosis reduces acute injury to the kidney. J. Clin. Investig. 2015, 125, 1620–1636. [Google Scholar] [CrossRef] [PubMed]
- Van Timmeren, M.M.; Bakker, S.J.L.; Vaidya, V.S.; Bailly, V.; Schuurs, T.A.; Damman, J.; Stegeman, C.A.; Bonventre, J.V.; Van Goor, H. Tubular kidney injury molecule-1 in protein-overload nephropathy. Am. J. Physiol. Physiol. 2006, 291, F456–F464. [Google Scholar] [CrossRef] [PubMed]
- Van Timmeren, M.M.; Heuvel, M.C.V.D.; Bailly, V.; Bakker, S.J.L.; Van Goor, H.; Stegeman, C.A. Tubular kidney injury molecule-1 (KIM-1) in human renal disease. J. Pathol. 2007, 212, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.-L.; Liu, S.-X.; Chen, Y.-H.; Yan, L.-J.; Li, H.; Xuan, H.-J.; Liang, Y.-Z.; Shi, W. Combination of urinary kidney injury molecule-1 and interleukin-18 as early biomarker for the diagnosis and progressive assessment of acute kidney injury following cardiopulmonary bypass surgery: A prospective nested case–control study. Biomarkers 2010, 15, 332–339. [Google Scholar] [CrossRef]
- Liang, X.-L.; Shi, W. Beyond Early Diagnosis: Prognostic Biomarkers for Monitoring Acute Kidney Injury. Hong Kong J. Nephrol. 2010, 12, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Nogare, A.L.; Veronese, F.V.; Carpio, V.N.; Montenegro, R.M.; Pedroso, J.A.; Pegas, K.L.; Gonçalves, L.F.; Manfro, R.C. Kidney injury molecule-1 expression in human kidney transplants with interstitial fibrosis and tubular atrophy. BMC Nephrol. 2015, 16, 19. [Google Scholar] [CrossRef] [Green Version]
- Lobato, G.; Lobato, M.; Thomé, F.; Veronese, F. Performance of urinary kidney injury molecule-1, neutrophil gelati-nase-associated lipocalin, and N-acetyl-β-D-glucosaminidase to predict chronic kidney disease progression and adverse out-comes. Braz. J. Med. Biol. Res. 2017, 50, e6106. [Google Scholar] [CrossRef] [Green Version]
- Han, W.K.; Alinani, A.; Wu, C.-L.; Michaelson, D.; Loda, M.; McGovern, F.J.; Thadhani, R.; Bonventre, J.V. Human Kidney Injury Molecule-1 Is a Tissue and Urinary Tumor Marker of Renal Cell Carcinoma. J. Am. Soc. Nephrol. 2005, 16, 1126–1134. [Google Scholar] [CrossRef]
- Tam, F.W.K.; Ong, A.C.M. Renal monocyte chemoattractant protein-1: An emerging universal biomarker and therapeutic target for kidney diseases? Nephrol. Dial. Transpl. 2019, 198–203. [Google Scholar] [CrossRef]
- Tesch, G.H. MCP-1/CCL2: A new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am. J. Physiol. Physiol. 2008, 294, F697–F701. [Google Scholar] [CrossRef] [PubMed]
- Haller, H.; Bertram, A.; Nadrowitz, F.; Menne, J. Monocyte chemoattractant protein-1 and the kidney. Curr. Opin. Nephrol. Hypertens. 2016, 25, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.I.; Bennett, M.R.; Mina, R.; Suzuki, M.; Petri, M.; Kiani, A.N.; Pendl, J.; Witte, D.P.; Ying, J.; Rovin, B.H.; et al. Association of noninvasively measured renal protein biomarkers with histologic features of lupus nephritis. Arthritis Rheum. 2012, 64, 2687–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, M.J.C.; Junior, G.B.S.; Sampaio, A.M.; Montenegro, B.L.; Alves, M.P.; Henn, G.A.L.; Rocha, H.A.L.; Meneses, G.C.; Martins, A.M.C.; Daher, E.F. Preliminary Study on Tubuloglomerular Dysfunction and Evidence of Renal Inflammation in Patients with Visceral Leishmaniasis. Am. J. Trop. Med. Hyg. 2014, 91, 908–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowda, S.; Desai, P.B.; Kulkarni, S.S.; Hull, V.V.; Math, A.A.K.; Vernekar, S.N. Markers of renal function tests. N. Am. J. Med. Sci. 2010, 2, 170–173. [Google Scholar]
- Elnojomi, N.A.A.; Musa, A.M.; Younis, B.M.; Elfaki, M.E.E.; El-Hassan, A.M.; Khalil, E.A.G. Surrogate markers of subtle renal injury in patients with visceral leishmaniasis. Saudi J. Kidney Dis. Transpl. 2010, 21, 872–875. [Google Scholar] [PubMed]
- Filho, N.S.; Ferreira, T.M.A.; Costa, J.M. Envolvimento da função renal em pacientes com leishmaniose visceral (calazar). Rev. Soc. Bras. Med. Trop. 2003, 36, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalremruata, A.; Jeyaraj, S.; Engleitner, T.; Joanny, F.; Lang, A.; Bélard, S.; Mombo-Ngoma, G.; Ramharter, M.; Kremsner, P.G.; Mordmüller, B.; et al. Species and genotype diversity of Plasmodium in malaria patients from Gabon analysed by next generation sequencing. Malar. J. 2017, 16, 1–11. [Google Scholar] [CrossRef]
- Junior, G.B.D.S.; Pinto, J.R.; Barros, E.J.G.; Farias, G.M.N.; Daher, E.D.F.; Da Silva, G.B. Kidney involvement in malaria: An update. Rev. Inst. Med. Trop. São Paulo 2017, 59, e53. [Google Scholar] [CrossRef] [Green Version]
- Marks, M.; Gupta-Wright, A.; Doherty, J.F.; Singer, M.; Walker, D. Managing malaria in the intensive care unit. Br. J. Anaesth. 2014, 113, 910–921. [Google Scholar] [CrossRef] [Green Version]
- Sacomboio, E.N.M.; Sebastião, C.D.S.; Tchivango, A.T.; Pecoits-Filho, R.; Calice-Silva, V. Does parasitemia level increase the risk of acute kidney injury in patients with malaria? Results from an observational study in Angola. Sci. Afr. 2020, 7, e00232. [Google Scholar] [CrossRef]
- Kher, V.; Srisawat, N.; Noiri, E.; Gharbi, M.B.; Shetty, M.S.; Yang, L.; Bagga, A.; Chakravarthi, R.; Mehta, R. Prevention and Therapy of Acute Kidney Injury in the Developing World. Kidney Int. Rep. 2017, 2, 544–558. [Google Scholar] [CrossRef] [Green Version]
- Pongponratn, E.; Aikawa, M.; Punpoowong, B.; Riganti, M. Microvascular Sequestration of Parasitized Erythrocytes in Human Falciparum Malaria: A Pathological Study. Am. J. Trop. Med. Hyg. 1991, 44, 168–175. [Google Scholar] [CrossRef]
- Brown, D.D.; Solomon, S.; Lerner, D.; del Rio, M. Malaria and acute kidney injury. Pediatr. Nephrol. 2019, 35, 603–608. [Google Scholar] [CrossRef]
- Mikkelsen, R.B.; Tanabe, K.; Wallach, D.F. Membrane potential of Plasmodium-infected erythrocytes. J. Cell Biol. 1982, 93, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhang, J.; Wu, H.; Li, L.; Yang, C.; Song, S.; Peng, P.; Shao, M.; Zhang, M.; Zhao, J.; et al. Lectin-like oxidized low-density lipoprotein receptor-1 facilitates metastasis of gastric cancer through driving epithelial-mesenchymal transition and PI3K/Akt/GSK3β activation. Sci. Rep. 2017, 7, 45275. [Google Scholar] [CrossRef]
- Ando, K.; Fujita, T. Role of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in the development of hypertensive organ damage. Clin. Exp. Nephrol. 2004, 8, 178–182. [Google Scholar] [CrossRef]
- Lu, J.; Mitra, S.; Wang, X.; Khaidakov, M.; Mehta, J.L. Oxidative Stress and Lectin-Like Ox-LDL-Receptor LOX-1 in Atherogenesis and Tumorigenesis. Antioxid. Redox Signal 2011, 15, 2301–2333. [Google Scholar] [CrossRef]
- Song, G.; Tian, H.; Liu, J.; Zhang, H.; Sun, X.; Qin, S. H2 inhibits TNF-α-induced lectin-like oxidized LDL receptor-1 expression by inhibiting nuclear factor κB activation in endothelial cells. Biotechnol. Lett. 2011, 33, 1715–1722. [Google Scholar] [CrossRef]
- Murase, T.; Kume, N.; Korenaga, R.; Ando, J.; Sawamura, T.; Masaki, T.; Kita, T. Fluid shear stress transcriptionally induces lectin-like oxidized LDL receptor-1 in vascular endothelial cells. Circ. Res. 1998, 83, 328–333. [Google Scholar] [CrossRef] [Green Version]
- Elias, R.M.; Corrêa-Costa, M.; Barreto, C.R.; Silva, R.C.; Hayashida, C.Y.; Castoldi, A.; Gonçalves, G.M.; Braga, T.T.; Barboza, R.; Rios, F.J.; et al. Oxidative Stress and Modification of Renal Vascular Permeability Are Associated with Acute Kidney Injury during P. berghei ANKA Infection. PLoS ONE 2012, 7, e44004. [Google Scholar] [CrossRef]
- Bosch, S.S.; Kronenberger, T.; Meissner, K.A.; Zimbres, F.M.; Stegehake, D.; Izui, N.M.; Schettert, I.; Liebau, E.; Wrenger, C. Oxidative Stress Control by Apicomplexan Parasites. BioMed Res. Int. 2015, 2015, 351289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinniah, R.; Rui-Mei, L.; Kara, A. Up-regulation of cytokines in glomerulonephritis associated with murine malaria infection. Int. J. Exp. Pathol. 2001, 80, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M. Molecular Determinants of Metalloproteinase Substrate Specificity: Matrix Metalloproteinase Substrate Binding Domains, Modules, and Exosites. Mol. Biotechnol. 2002, 22, 51–86. [Google Scholar] [CrossRef]
- Trojanek, J. Matrix metalloproteinases and their tissue inhibitors. Postepy Biochem. 2012, 58, 353–362. [Google Scholar] [PubMed]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipka, D.; Boratyński, J. Metalloproteinases. Structure and function. Post. Hig. Med. Dosw. 2008, 62, 328–336. [Google Scholar]
- Wright, J.W.; Harding, J.W. Contributions of Matrix Metalloproteinases to Neural Plasticity, Habituation, Associative Learning and Drug Addiction. Neural Plast. 2009, 2009, 579382. [Google Scholar] [CrossRef] [Green Version]
- Zakiyanov, O.; Kalousová, M.; Zima, T.; Tesař, V. Matrix Metalloproteinases in Renal Diseases: A Critical Appraisal. Kidney Blood Press. Res. 2019, 44, 298–330. [Google Scholar] [CrossRef]
- Tan, R.J.; Liu, Y. Matrix metalloproteinases in kidney homeostasis and diseases. Am. J. Physiol. Physiol. 2012, 302, F1351–F1361. [Google Scholar] [CrossRef] [Green Version]
- Lelongt, B.; Trugnan, G.; Murphy, G.; Ronco, P.M. Matrix Metalloproteinases MMP2 and MMP9 Are Produced in Early Stages of Kidney Morphogenesis but Only MMP9 Is Required for Renal Organogenesis In Vitro. J. Cell Biol. 1997, 136, 1363–1373. [Google Scholar] [CrossRef]
- Arnould, C.; Lelièvre-Pégorier, M.; Ronco, P.; Lelongt, B. MMP9 Limits Apoptosis and Stimulates Branching Morphogenesis During Kidney Development. J. Am. Soc. Nephrol. 2009, 20, 2171–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengatta, S.; Arnould, C.; Letavernier, E.; Monge, M.; de Préneuf, H.M.; Werb, Z.; Ronco, P.; Lelongt, B. MMP9 and SCF Protect from Apoptosis in Acute Kidney Injury. J. Am. Soc. Nephrol. 2009, 20, 787–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Limbu, M.H.; Wang, Z.; Liu, J.; Liu, L.; Zhang, X.; Chen, P.; Liu, B. MMP-2 and 9 in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18, 776. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-R.; Kuo, W.-H.; Hsieh, Y.-S.; Yang, S.-F.; Lin, C.-C.; Lee, M.-L.; Lian, J.-D.; Chu, S.-C. Circulating matrix metalloproteinase-2 is associated with cystatin C level, posttransplant duration, and diabetes mellitus in kidney transplant recipients. Transl. Res. 2008, 151, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ke, B.; Fan, C.; Yang, L.; Fang, X. Matrix Metalloproteinases-7 and Kidney Fibrosis. Front. Physiol. 2017, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Steen, P.E.V.D.; Van Aelst, I.; Starckx, S.; Maskos, K.; Opdenakker, G.; Pagenstecher, A. Matrix metalloproteinases, tissue inhibitors of MMPs and TACE in experimental cerebral malaria. Lab. Investig. 2006, 86, 873–888. [Google Scholar] [CrossRef]
- Punsawad, C.; Viriyavejakul, P. Increased expression of kidney injury molecule-1 and matrix metalloproteinase-3 in severe Plasmodium falciparum malaria with acute kidney injury. Int. J. Clin. Exp. Pathol. 2017, 10, 7856–7864. [Google Scholar]
- Lim, A.I.; Chan, L.Y.; Lai, K.N.; Tang, S.C.; Chow, C.W.; Lam, M.F.; Leung, J.C. Distinct role of matrix metalloproteinase-3 in kidney injury molecule-1 shedding by kidney proximal tubular epithelial cells. Int. J. Biochem. Cell Biol. 2012, 44, 1040–1050. [Google Scholar] [CrossRef]
- Sanchavanakit, N.; Saengtong, W.; Manokawinchoke, J.; Pavasant, P. TNF-α stimulates MMP-3 production via PGE2 signalling through the NF-kB and p38 MAPK pathway in a murine cementoblast cell line. Arch. Oral Biol. 2015, 60, 1066–1074. [Google Scholar] [CrossRef]
- Sharma, L.; Kaur, J.; Rishi, P.; Shukla, G. Plasmodium berghei: Influence of infection on the oxidant and antioxidants levels in pregnant BALB/c mice. Exp. Parasitol. 2012, 131, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Nanda, R.; Mishra, P.K.; Das, U.K.; Rout, S.B.; Mohapatra, P.C.; Panda, A. Evaluating role of oxidative stress in determining the pathogenesis of falciparum malaria induced acute renal failure. Indian J. Clin. Biochem. 2004, 19, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Wichapoon, B.; Punsawad, C.; Viriyavejakul, P. Expression of cleaved caspase-3 in renal tubular cells in Plasmodium falciparum malaria patients. Nephrology 2016, 22, 79–84. [Google Scholar] [CrossRef]
- Eckardt, K.-U.; Bernhardt, W.W.; Weidemann, A.; Warnecke, C.; Rosenberger, C.; Wiesener, M.M.; Willam, C. Role of hypoxia in the pathogenesis of renal disease. Kidney Int. 2005, 68, S46–S51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landázuri, M.O.; Vara-Vega, A.; Vitón, M.; Cuevas, Y.; del Peso, L. Analysis of HIF-prolyl hydroxylases binding to substrates. Biochem. Biophys. Res. Commun. 2006, 351, 313–320. [Google Scholar] [CrossRef]
- Woodford, J.; Yeo, T.W.; Piera, K.A.; Butler, K.; Weinberg, J.B.; McCarthy, J.S.; Anstey, N.M.; Barber, B.E. Early Endothelial Activation Precedes Glycocalyx Degradation and Microvascular Dysfunction in Experimentally Induced Plasmodium falciparum and Plasmodium vivax Infection. Infect. Immun. 2020, 88, 00895-19. [Google Scholar] [CrossRef] [PubMed]
- Van Wolfswinkel, M.E.; Koopmans, L.C.; Hesselink, D.A.; Hoorn, E.J.; Koelewijn, R.; van Hellemond, J.J.; van Genderen, P.J.J. Neutrophil gelatinase-associated lipocalin (NGAL) predicts the occurrence of malaria-induced acute kidney injury. Malar. J. 2016, 15, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plewes, K.; Royakkers, A.A.; Hanson, J.; Hasan, M.U.; Alam, S.; Ghose, A.; Maude, R.J.; Stassen, P.M.; Charunwatthana, P.; Lee, S.J.; et al. Correlation of biomarkers for parasite burden and immune activation with acute kidney injury in severe falciparum malaria. Malar. J. 2014, 13, 91. [Google Scholar] [CrossRef] [Green Version]
- Wichapoon, B.; Punsawad, C.; Chaisri, U.; Viriyavejakul, P. Glomerular changes and alterations of zonula occludens-1 in the kidneys of Plasmodium falciparum malaria patients. Malar. J. 2014, 13, 176. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, B.R.; Siliciano, J.D.; Mooseker, M.S.; Goodenough, D.A. Identification of ZO-1: A high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J. Cell Biol. 1986, 103, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Montoya, J.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Mizani, A.; Alipour, A.; Sharif, M.; Sarvi, S.; Amouei, A.; Shokri, A.; Rahimi, M.-T.; Hosseini, S.A.; Daryani, A. Toxoplasmosis seroprevalence in Iranian women and risk factors of the disease: A systematic review and meta-analysis. Trop. Med. Health 2017, 45, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, J. History of the discovery of the life cycle of Toxoplasma gondii. Int. J. Parasitol. 2009, 39, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Devleesschauwer, B.; Liu, M.; Li, J.; Wu, Y.; van der Giessen, J.W.B.; Opsteegh, M. Seroprevalence of Toxoplasma gondii in pregnant women and livestock in the mainland of China: A systematic review and hierarchical meta-analysis. Sci. Rep. 2018, 8, 6218. [Google Scholar] [CrossRef]
- Milne, G.; Webster, J.P.; Walker, M. Toxoplasma gondii: An Underestimated Threat? Trends Parasitol. 2020, 36, 959–969. [Google Scholar] [CrossRef]
- Dadimoghaddam, Y.; Daryani, A.; Sharif, M.; Ahmadpour, E.; Hossienikhah, Z. Tissue tropism and parasite burden of Toxoplasma gondii RH strain in experimentally infected mice. Asian Pac. J. Trop. Med. 2014, 7, 521–524. [Google Scholar] [CrossRef]
- Martina, M.-N.; Cervera, C.; Esforzado, N.; Linares, L.; Torregrosa, V.; Sanclemente, G.; Hoyo, I.; Cofan, F.; Oppenheimer, F.; Miro, J.M.; et al. Toxoplasma gondii primary infection in renal transplant recipients. Two case reports and literature review. Transpl. Int. 2010, 24, e6–e12. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S. The close relationship between toxoplasmosis and kidney function. Rev. Inst. Med. Trop. São Paulo 2012, 54, 318. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Aosai, F.; Mun, H.-S.; Norose, K.; Akira, S.; Iwakura, Y.; Yano, A. The Role of IFN-γ and Toll-Like Receptors in Nephropathy Induced by Toxoplasma gondii Infection. Microbiol. Immunol. 2004, 48, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2003, 75, 163–189. [Google Scholar] [CrossRef]
- Norose, K.; Mun, H.S.; Aosai, F.; Chen, M.; Hata, H.; Tagawa, Y.; Iwakura, Y.; Yano, A. Organ infectivity of Toxoplasma gondii in interferon-gamma knockout mice. J. Parasitol. 2001, 87, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Türkoğlu, Ş.A.; Yaman, K.; Orallar, H.; Camsari, C.; Karabörk, Ş.; Ayaz, E. Acute toxoplasmosis and antioxidant levels in the liver, kidney and brain of rats. Ann. Parasitol. 2018, 64, 241–247. [Google Scholar] [PubMed]
- Gharadaghi, Y.; Shojaee, S.; Khaki, A.; Hatef, A.; Ashtiani, H.R.A.; Rastegar, H.; Fathiazad, F. Modulating effect of Allium cepa on kidney apoptosis caused by Toxoplasma gondii. Adv. Pharm. Bull. 2012, 2, 1–6. [Google Scholar] [PubMed]
- Isaka, Y. Targeting TGF-β Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, A.V.; Gois, M.B.; Lera, K.R.J.L.; Miranda-Sapla, M.M.; Falkowski-Temporini, G.J.; Bezerril, J.E.; Zanusso-Junior, G.; Ferraz, F.N.; Da Silva, S.S.; Aleixo, D.L.; et al. Treatment with Lycopodium clavatum 200dH Intensifies Kidney and Liver Injury in Mice Infected with Toxoplasma gondii. Arch. Immunol. Ther. Exp. 2020, 68, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Al-Kaysi, A.M.; Eid, R.A.A.; Fahmy, B.G.A. Biochemical studies on the effect of Toxoplasma infection on liver and kidney functions in mice. Egypt J. Comp. Pathol. Clin. Pathol. 2010, 23, 174–185. [Google Scholar]
- Mattos, D.P.B.G.; Menezes, R.C.; Freire, R.B.; Martorelli, R.A.; Coelho, J.M.C.O.; Armendoeira, M.R.R. Histopatology and immunohistochemistry of C57BL/6 mice infected with Toxoplasma gondii ME-49 strain and fed with mycotoxins. Rev. Bras. Ci Vet. 2009, 16, 27–32. [Google Scholar] [CrossRef]
- Visvesvara, G.S.; Moura, H.; Schuster, F.L. Pathogenic and opportunistic free-living amoebae: Acanthamoeba spp., Balamuthia mandrillaris, Naegleria fowleri, and Sappinia diploidea. FEMS Immunol. Med. Microbiol. 2007, 50, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Kot, K.; Łanocha-Arendarczyk, N.A.; Kosik-Bogacka, D.I. Amoebas from the genus Acanthamoeba and their pathogenic properties. Ann. Parasitol. 2019, 64, 299–308. [Google Scholar]
- Khan, N.A. Acanthamoeba: Biology and increasing importance in human health. FEMS Microbiol. Rev. 2006, 30, 564–595. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmus, K.R.; Jones, D.B.; Matoba, A.Y.; Hamill, M.B.; Pflugfelder, S.C.; Weikert, M.P. Bilateral Acanthamoeba Keratitis. Am. J. Ophthalmol. 2008, 145, 193–197.e1. [Google Scholar] [CrossRef]
- Lorenzo-Morales, J.; Khan, N.A.; Walochnik, J. An update on Acanthamoeba keratitis: Diagnosis, pathogenesis and treatment. Parasite 2015, 22, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łanocha-Arendarczyk, N.; Kosik-Bogacka, D.; Zaorski, W.; Kot, K.; Galant, K.; Łanocha, A. Pathogenic Free-Living Amoeba. Adv. Microbiol. 2019, 56, 106–112. [Google Scholar] [CrossRef]
- Kot, K.; Łanocha-Arendarczyk, N.; Kosik-Bogacka, D. Immunopathogenicity of Acanthamoeba spp. in the Brain and Lungs. Int. J. Mol. Sci. 2021, 22, 1261. [Google Scholar] [CrossRef]
- Trabelsi, H.; Dendana, F.; Sellami, A.; Cheikhrouhou, F.; Neji, S.; Makni, F.; Ayadi, A. Pathogenic free-living amoebae: Epidemiology and clinical review. Pathol. Biol. 2012, 60, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Król-Turmińska, K.; Olender, A. Human infections caused by free-living amoebae. Ann. Agric. Environ. Med. 2017, 24, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Ringsted, J.; Jager, B.V.; Suk, D.; Visvesvara, G.S.; Rlngsted, Ø.; Vlsvesvara, G.S. Probable Acanthamoeba Meningoencephalitis in a Korean Child. Am. J. Clin. Pathol. 1976, 66, 723–730. [Google Scholar] [CrossRef]
- Górnik, K.; Kuźna-Grygiel, W. Histological studies of selected organs of mice experimentally infected with Acanthamoeba spp. Folia Morphol. 2005, 64, 161–167. [Google Scholar]
- Kot, K.; Kosik-Bogacka, D.; Wojtkowiak-Giera, A.; Kolasa-Wołosiuk, A.; Łanocha-Arendarczyk, N. The expression of TLR2 and TLR4 in the kidneys and heart of mice infected with Acanthamoeba spp. Parasites Vectors 2020, 13, 1–11. [Google Scholar] [CrossRef]
- Steinberg, J.P.; Galindo, R.L.; Kraus, E.S.; Ghanem, K.G. Disseminated Acanthamebias is in a Renal Transplant Recipient with Osteomyelitis and Cutaneous Lesions: Case Report and Literature Review. Clin. Infect. Dis. 2002, 35, e43–e49. [Google Scholar] [CrossRef] [PubMed]
- Brondfield, M.N.; Reid, M.J.; Rutishauser, R.L.; Cope, J.R.; Tang, J.; Ritter, J.M.; Matanock, A.; Ali, I.; Doernberg, S.B.; Hilts-Horeczko, A.; et al. Disseminated Acanthamoeba infection in a heart transplant recipient treated successfully with a miltefosine-containing regimen: Case report and review of the literature. Transpl. Infect. Dis. 2017, 19, e12661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łanocha-Arendarczyk, N.; Baranowska-Bosiacka, I.; Kot, K.; Pilarczyk, B.; Tomza-Marciniak, A.; Kabat-Koperska, J.; Kosik-Bogacka, D. Biochemical Profile, Liver and Kidney Selenium (Se) Status during Acanthamoebiasis in a Mouse Model. Folia Biol. 2018, 66, 33–40. [Google Scholar] [CrossRef]
- Jelicks, L.A.; De Souza, A.P.; Araújo-Jorge, T.C.; Tanowitz, H.B. Would selenium supplementation aid in therapy for Chagas Disease? Trends Parasitol. 2011, 27, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Lanocha-Arendarczyk, N.; Kosik-Bogacka, D.I.; Prokopowicz, A.; Kalisinska, E.; Sokolowski, S.; Karaczun, M.; Zietek, P.; Podlasińska, J.; Pilarczyk, B.; Tomza-Marciniak, A.; et al. The Effect of Risk Factors on the Levels of Chemical Elements in the Tibial Plateau of Patients with Osteoarthritis following Knee Surgery. BioMed Res. Int. 2015, 2015, 650282. [Google Scholar] [CrossRef] [Green Version]
- Lener, M.R.; Scott, R.J.; Wiechowska-Kozłowska, A.; Serrano-Fernández, P.; Baszuk, P.; Jaworska-Bieniek, K.; Sukiennicki, G.; Marciniak, W.; Muszyńska, M.; Kładny, J.; et al. Serum Concentrations of Selenium and Copper in Patients Diagnosed with Pancreatic Cancer. Cancer Res. Treat. 2016, 48, 1056–1064. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Yang, S. Inhibitory Effect of Selenium on Cryptosporidium parvum Infection In Vitro and In Vivo. Biol. Trace Elem. Res. 2002, 90, 261–272. [Google Scholar] [CrossRef]
- Silva-Jardim, I.; Thiemann, O. Biological Implications of Selenium and its Role in Trypanosomiasis Treatment. Curr. Med. Chem. 2014, 21, 1772–1780. [Google Scholar] [CrossRef]
- Frade, M.T.; de Melo, L.F.; Pessoa, C.R.; de Araújo, J.L.; Fighera, R.A.; Souza, A.P.; Uzal, F.; Dantas, A.F. Systemic acanthamoebiasis associated with canine distemper in dogs in the semiarid region of Paraíba, Brazil. Pesqui. Vet. Bras. 2015, 35, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Dubey, J.; Benson, J.; Blakeley, K.; Booton, G.; Visvesvara, G. Disseminated Acanthamoeba sp. infection in a dog. Vet. Parasitol. 2005, 128, 183–187. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kot, K.; Łanocha-Arendarczyk, N.; Ptak, M.; Łanocha, A.; Kalisińska, E.; Kosik-Bogacka, D. Pathomechanisms in the Kidneys in Selected Protozoan Parasitic Infections. Int. J. Mol. Sci. 2021, 22, 4209. https://doi.org/10.3390/ijms22084209
Kot K, Łanocha-Arendarczyk N, Ptak M, Łanocha A, Kalisińska E, Kosik-Bogacka D. Pathomechanisms in the Kidneys in Selected Protozoan Parasitic Infections. International Journal of Molecular Sciences. 2021; 22(8):4209. https://doi.org/10.3390/ijms22084209
Chicago/Turabian StyleKot, Karolina, Natalia Łanocha-Arendarczyk, Michał Ptak, Aleksandra Łanocha, Elżbieta Kalisińska, and Danuta Kosik-Bogacka. 2021. "Pathomechanisms in the Kidneys in Selected Protozoan Parasitic Infections" International Journal of Molecular Sciences 22, no. 8: 4209. https://doi.org/10.3390/ijms22084209
APA StyleKot, K., Łanocha-Arendarczyk, N., Ptak, M., Łanocha, A., Kalisińska, E., & Kosik-Bogacka, D. (2021). Pathomechanisms in the Kidneys in Selected Protozoan Parasitic Infections. International Journal of Molecular Sciences, 22(8), 4209. https://doi.org/10.3390/ijms22084209