
Pharmacologic Control of CAR T Cells




Abstract
:1. Introduction
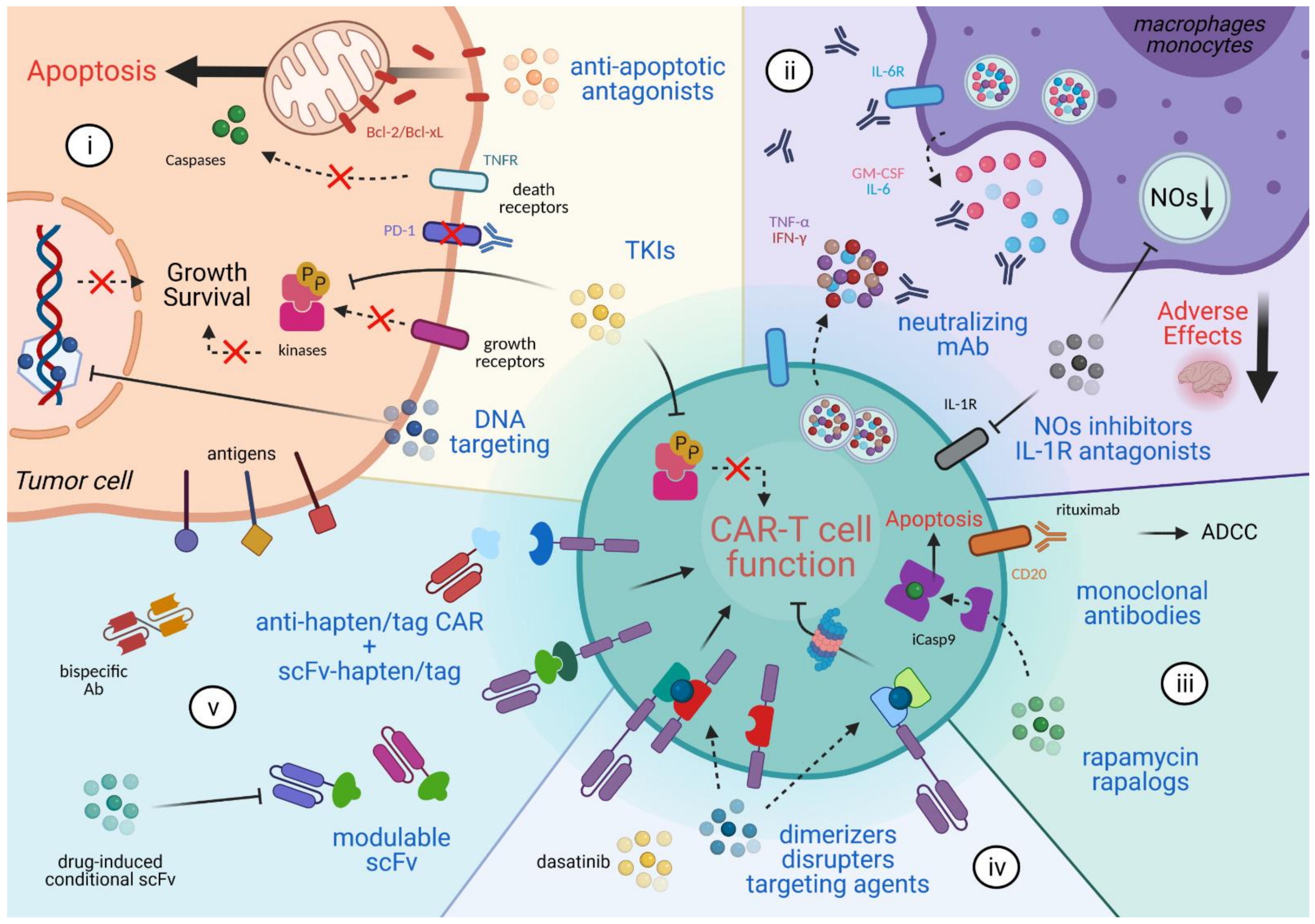
2. Combinatorial Anti-Cancer Approaches
3. Mitigating the Adverse Effects
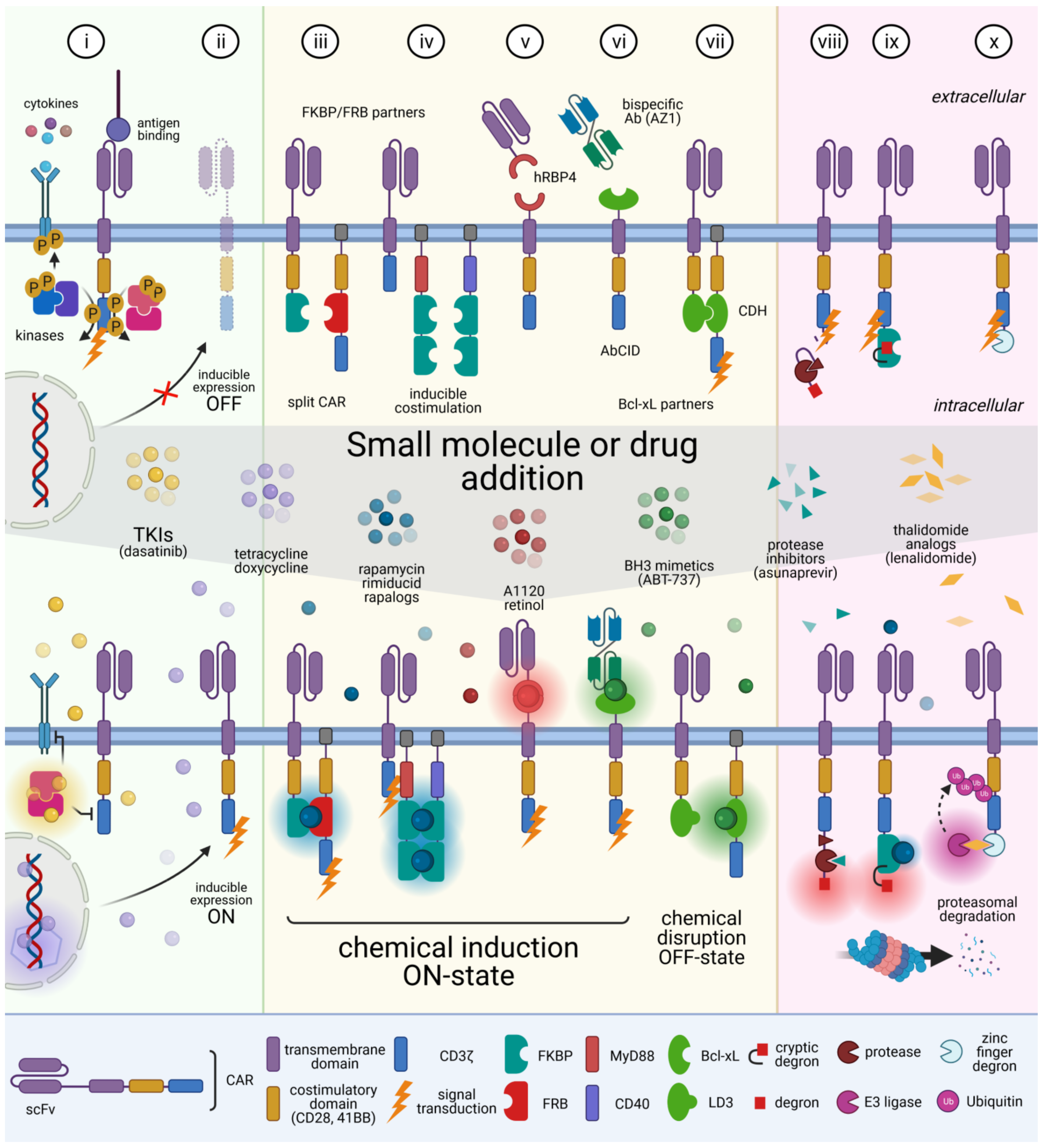
4. CAR T Cell Elimination
5. Reversible Spatio-Temporal Control of CAR
6. Modulating CAR Specificity
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-cell therapy in multiple myeloma: Can we do better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Schubert, M.L.; Wang, L.; He, B.; Neuber, B.; Dreger, P.; Muller-Tidow, C.; Schmitt, M. Chimeric Antigen Receptor (CAR) T Cell Therapy in Acute Myeloid Leukemia (AML). J. Clin. Med. 2019, 8, 200. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, N.; Shi, H. Driving better and safer HER2-specific CARs for cancer therapy. Oncotarget 2017, 8, 62730–62741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Karlsson, H.; Lindqvist, A.C.; Fransson, M.; Paul-Wetterberg, G.; Nilsson, B.; Essand, M.; Nilsson, K.; Frisk, P.; Jernberg-Wiklund, H.; Loskog, A. Combining CAR T cells and the Bcl-2 family apoptosis inhibitor ABT-737 for treating B-cell malignancy. Cancer Gene Ther. 2013, 20, 386–393. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Torres-Collado, A.X.; Jazirehi, A.R. Overcoming Resistance of Human Non-Hodgkin’s Lymphoma to CD19-CAR CTL Therapy by Celecoxib and Histone Deacetylase Inhibitors. Cancers 2018, 10, 200. [Google Scholar] [CrossRef] [Green Version]
- Dufva, O.; Koski, J.; Maliniemi, P.; Ianevski, A.; Klievink, J.; Leitner, J.; Polonen, P.; Hohtari, H.; Saeed, K.; Hannunen, T.; et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 2020, 135, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef] [Green Version]
- Moon, E.K.; Wang, L.C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Pure, E.; Milone, M.C.; et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kaur, G.; Sankin, A.I.; Chen, F.; Guan, F.; Zang, X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Sandau, M.M.; Kohlmeier, J.E.; Woodland, D.L.; Jameson, S.C. IL-15 regulates both quantitative and qualitative features of the memory CD8 T cell pool. J. Immunol. 2010, 184, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Koya, R.C.; Mok, S.; Otte, N.; Blacketor, K.J.; Comin-Anduix, B.; Tumeh, P.C.; Minasyan, A.; Graham, N.A.; Graeber, T.G.; Chodon, T.; et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012, 72, 3928–3937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deniger, D.C.; Kwong, M.L.; Pasetto, A.; Dudley, M.E.; Wunderlich, J.R.; Langhan, M.M.; Lee, C.R.; Rosenberg, S.A. A Pilot Trial of the Combination of Vemurafenib with Adoptive Cell Therapy in Patients with Metastatic Melanoma. Clin. Cancer Res. 2017, 23, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Gargett, T.; Fraser, C.K.; Dotti, G.; Yvon, E.S.; Brown, M.P. BRAF and MEK inhibition variably affect GD2-specific chimeric antigen receptor (CAR) T-cell function in vitro. J. Immunother. 2015, 38, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urak, R.; Walter, M.; Lim, L.; Wong, C.W.; Budde, L.E.; Thomas, S.; Forman, S.J.; Wang, X. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J. Immunother. Cancer 2017, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, J.; Hirayama, A.V.; Purushe, J.; Hay, K.A.; Lymp, J.; Li, D.H.; Yeung, C.C.S.; Sheih, A.; Pender, B.S.; Hawkins, R.M.; et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood 2020, 135, 1650–1660. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, M.B.; Riviere, I.; Senechal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Palomba, M.L.; Halton, E.; et al. Safety and tolerability of conditioning chemotherapy followed by CD19-targeted CAR T cells for relapsed/refractory CLL. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Li, Z.; Zhang, R.; Mu, J.; Jiang, Y.; Deng, Q.; Sun, L. Synergistic effect of ibrutinib and CD19 CAR-T cells on Raji cells in vivo and in vitro. Cancer Sci. 2020, 111, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ding, J.; Lu, M.; Liu, H.; Miao, Y.; Li, L.; Wang, G.; Zheng, J.; Pei, D.; Zhang, Q. CAIX-specific CAR-T Cells and Sunitinib Show Synergistic Effects Against Metastatic Renal Cancer Models. J. Immunother. 2020, 43, 16–28. [Google Scholar] [CrossRef]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3(-)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef]
- Fasslrinner, F.; Arndt, C.; Koristka, S.; Feldmann, A.; Altmann, H.; von Bonin, M.; Schmitz, M.; Bornhauser, M.; Bachmann, M. Midostaurin abrogates CD33-directed UniCAR and CD33-CD3 bispecific antibody therapy in acute myeloid leukaemia. Br. J. Haematol. 2019, 186, 735–740. [Google Scholar] [CrossRef]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Hofmann, T.J.; Gershenson, Z.; Levine, B.L.; Grupp, S.A.; Teachey, D.T.; Barrett, D.M. Monocyte lineage-derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy 2017, 19, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [Green Version]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Chen, F.; Teachey, D.T.; Pequignot, E.; Frey, N.; Porter, D.; Maude, S.L.; Grupp, S.A.; June, C.H.; Melenhorst, J.J.; Lacey, S.F. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR T cell therapy. J. Immunol. Methods 2016, 434, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Hay, K.A.; Hanafi, L.A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; Lopez, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, C.D.; Richards, S.M.; Kinsey, S.E.; Lilleyman, J.; Vora, A.; Eden, T.O.; Medical Research Council Childhood Leukaemia Working Party. Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: Results of the UK Medical Research Council ALL97 randomized trial. Br. J. Haematol. 2005, 129, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachdeva, M.; Duchateau, P.; Depil, S.; Poirot, L.; Valton, J. Granulocyte-macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J. Biol. Chem. 2019, 294, 5430–5437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennell, C.A.; Barnum, J.L.; McDonald-Hyman, C.S.; Panoskaltsis-Mortari, A.; Riddle, M.J.; Xiong, Z.; Loschi, M.; Thangavelu, G.; Campbell, H.M.; Storlie, M.D.; et al. Human CD19-Targeted Mouse T Cells Induce B Cell Aplasia and Toxicity in Human CD19 Transgenic Mice. Mol. Ther. 2018, 26, 1423–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Staedtke, V.; Bai, R.Y.; Kim, K.; Darvas, M.; Davila, M.L.; Riggins, G.J.; Rothman, P.B.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature 2018, 564, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Bordignon, C.; Bonini, C.; Verzeletti, S.; Nobili, N.; Maggioni, D.; Traversari, C.; Giavazzi, R.; Servida, P.; Zappone, E.; Benazzi, E.; et al. Transfer of the HSV-tk gene into donor peripheral blood lymphocytes for in vivo modulation of donor anti-tumor immunity after allogeneic bone marrow transplantation. Hum. Gene Ther. 1995, 6, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Bonini, C.; Ferrari, G.; Verzeletti, S.; Servida, P.; Zappone, E.; Ruggieri, L.; Ponzoni, M.; Rossini, S.; Mavilio, F.; Traversari, C.; et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 1997, 276, 1719–1724. [Google Scholar] [CrossRef]
- Sato, T.; Neschadim, A.; Konrad, M.; Fowler, D.H.; Lavie, A.; Medin, J.A. Engineered human tmpk/AZT as a novel enzyme/prodrug axis for suicide gene therapy. Mol. Ther. 2007, 15, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Tiberghien, P.; Cahn, J.Y.; Brion, A.; Deconinck, E.; Racadot, E.; Herve, P.; Milpied, N.; Lioure, B.; Gluckman, E.; Bordigoni, P.; et al. Use of donor T-lymphocytes expressing herpes-simplex thymidine kinase in allogeneic bone marrow transplantation: A phase I-II study. Hum. Gene Ther. 1997, 8, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Marin, V.; Cribioli, E.; Philip, B.; Tettamanti, S.; Pizzitola, I.; Biondi, A.; Biagi, E.; Pule, M. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum. Gene Ther. Methods 2012, 23, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Flowers, M.E.; Warren, E.H.; Riddell, S.R. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood 2006, 107, 2294–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomis, D.C.; Marktel, S.; Bonini, C.; Traversari, C.; Gilman, M.; Bordignon, C.; Clackson, T. A Fas-based suicide switch in human T cells for the treatment of graft-versus-host disease. Blood 2001, 97, 1249–1257. [Google Scholar] [CrossRef]
- Tey, S.K.; Dotti, G.; Rooney, C.M.; Heslop, H.E.; Brenner, M.K. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol. Blood Marrow Transplant. 2007, 13, 913–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straathof, K.C.; Pule, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Di Stasi, A.; Tey, S.K.; Krance, R.A.; Martinez, C.; Leung, K.S.; Durett, A.G.; Wu, M.F.; Liu, H.; Leen, A.M.; et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood 2014, 123, 3895–3905. [Google Scholar] [CrossRef] [Green Version]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2017, 25, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budde, L.E.; Berger, C.; Lin, Y.; Wang, J.; Lin, X.; Frayo, S.E.; Brouns, S.A.; Spencer, D.M.; Till, B.G.; Jensen, M.C.; et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS ONE 2013, 8, e82742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavrou, M.; Philip, B.; Traynor-White, C.; Davis, C.G.; Onuoha, S.; Cordoba, S.; Thomas, S.; Pule, M. A Rapamycin-Activated Caspase 9-Based Suicide Gene. Mol. Ther. 2018, 26, 1266–1276. [Google Scholar] [CrossRef] [Green Version]
- Serafini, M.; Manganini, M.; Borleri, G.; Bonamino, M.; Imberti, L.; Biondi, A.; Golay, J.; Rambaldi, A.; Introna, M. Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum. Gene Ther. 2004, 15, 63–76. [Google Scholar] [CrossRef]
- Introna, M.; Barbui, A.M.; Bambacioni, F.; Casati, C.; Gaipa, G.; Borleri, G.; Bernasconi, S.; Barbui, T.; Golay, J.; Biondi, A.; et al. Genetic modification of human T cells with CD20: A strategy to purify and lyse transduced cells with anti-CD20 antibodies. Hum. Gene Ther. 2000, 11, 611–620. [Google Scholar] [CrossRef]
- Fehse, B.; Richters, A.; Putimtseva-Scharf, K.; Klump, H.; Li, Z.; Ostertag, W.; Zander, A.R.; Baum, C. CD34 splice variant: An attractive marker for selection of gene-modified cells. Mol. Ther. 2000, 1, 448–456. [Google Scholar] [CrossRef]
- Vogler, I.; Newrzela, S.; Hartmann, S.; Schneider, N.; von Laer, D.; Koehl, U.; Grez, M. An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol. Ther. 2010, 18, 1330–1338. [Google Scholar] [CrossRef]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chang, W.C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Kao, R.L.; Truscott, L.C.; Chiou, T.T.; Tsai, W.; Wu, A.M.; De Oliveira, S.N. A Cetuximab-Mediated Suicide System in Chimeric Antigen Receptor-Modified Hematopoietic Stem Cells for Cancer Therapy. Hum. Gene Ther. 2019, 30, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Riviere, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramello, M.C.; Benzaid, I.; Kuenzi, B.M.; Lienlaf-Moreno, M.; Kandell, W.M.; Santiago, D.N.; Pabon-Saldana, M.; Darville, L.; Fang, B.; Rix, U.; et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.; Svensson, E.; Gigg, C.; Jarvius, M.; Olsson-Stromberg, U.; Savoldo, B.; Dotti, G.; Loskog, A. Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors. PLoS ONE 2015, 10, e0144787. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, P.L.; Keam, S.J. Dasatinib: A review of its use in the treatment of chronic myeloid leukaemia and Philadelphia chromosome-positive acute lymphoblastic leukaemia. Drugs 2011, 71, 1771–1795. [Google Scholar] [CrossRef]
- Tokarski, J.S.; Newitt, J.A.; Chang, C.Y.; Cheng, J.D.; Wittekind, M.; Kiefer, S.E.; Kish, K.; Lee, F.Y.; Borzillerri, R.; Lombardo, L.J.; et al. The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006, 66, 5790–5797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blake, S.; Hughes, T.P.; Mayrhofer, G.; Lyons, A.B. The Src/ABL kinase inhibitor dasatinib (BMS-354825) inhibits function of normal human T-lymphocytes in vitro. Clin. Immunol. 2008, 127, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Ouwehand, I.; Giannini, A.L.; Thomas, N.S.; Dibb, N.J.; Bijlmakers, M.J. Lck is a key target of imatinib and dasatinib in T-cell activation. Leukemia 2010, 24, 896–900. [Google Scholar] [CrossRef] [Green Version]
- Schade, A.E.; Schieven, G.L.; Townsend, R.; Jankowska, A.M.; Susulic, V.; Zhang, R.; Szpurka, H.; Maciejewski, J.P. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood 2008, 111, 1366–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.W.; Lynn, R.C.; Sotillo, E.; Lattin, J.; Xu, P.; Mackall, C.L. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 2019, 3, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Sakemura, R.; Terakura, S.; Watanabe, K.; Julamanee, J.; Takagi, E.; Miyao, K.; Koyama, D.; Goto, T.; Hanajiri, R.; Nishida, T.; et al. A Tet-On Inducible System for Controlling CD19-Chimeric Antigen Receptor Expression upon Drug Administration. Cancer Immunol. Res. 2016, 4, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.Y.; Wei, D.; Liu, Z.K.; Yong, Y.L.; Wei, W.; Zhang, Z.Y.; Lv, J.J.; Zhang, Z.; Chen, Z.N.; Bian, H. Doxycycline Inducible Chimeric Antigen Receptor T Cells Targeting CD147 for Hepatocellular Carcinoma Therapy. Front. Cell Dev. Biol. 2019, 7, 233. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; He, D.; Li, C.; Wang, H.; Yang, G. Development of Inducible CD19-CAR T Cells with a Tet-On System for Controlled Activity and Enhanced Clinical Safety. Int. J. Mol. Sci. 2018, 19, 3455. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, W.H.; Gay, J.; Martin, U.; Garrett, T.E.; Horton, H.M.; Certo, M.T.; Blazar, B.R.; Morgan, R.A.; Gregory, P.D.; Jarjour, J.; et al. Sensitive and adaptable pharmacological control of CAR T cells through extracellular receptor dimerization. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, Z.B.; Martinko, A.J.; Nguyen, D.P.; Wells, J.A. Human antibody-based chemically induced dimerizers for cell therapeutic applications. Nat. Chem. Biol. 2018, 14, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Zajc, C.U.; Dobersberger, M.; Schaffner, I.; Mlynek, G.; Puhringer, D.; Salzer, B.; Djinovic-Carugo, K.; Steinberger, P.; De Sousa Linhares, A.; Yang, N.J.; et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc. Natl. Acad. Sci. USA 2020, 117, 14926–14935. [Google Scholar] [CrossRef]
- Foster, A.E.; Mahendravada, A.; Shinners, N.P.; Chang, W.C.; Crisostomo, J.; Lu, A.; Khalil, M.; Morschl, E.; Shaw, J.L.; Saha, S.; et al. Regulated Expansion and Survival of Chimeric Antigen Receptor-Modified T Cells Using Small Molecule-Dependent Inducible MyD88/CD40. Mol. Ther. 2017, 25, 2176–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, T.; DeRose, R.; Suarez, A.; Ueno, T.; Chen, M.; Sun, T.P.; Wolfgang, M.J.; Mukherjee, C.; Meyers, D.J.; Inoue, T. Rapid and orthogonal logic gating with a gibberellin-induced dimerization system. Nat. Chem. Biol. 2012, 8, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Fegan, A.; White, B.; Carlson, J.C.; Wagner, C.R. Chemically controlled protein assembly: Techniques and applications. Chem. Rev. 2010, 110, 3315–3336. [Google Scholar] [CrossRef]
- Kelly, P.N.; Grabow, S.; Delbridge, A.R.; Adams, J.M.; Strasser, A. Prophylactic treatment with the BH3 mimetic ABT-737 impedes Myc-driven lymphomagenesis in mice. Cell Death Differ. 2013, 20, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Giordano-Attianese, G.; Gainza, P.; Gray-Gaillard, E.; Cribioli, E.; Shui, S.; Kim, S.; Kwak, M.J.; Vollers, S.; Corria Osorio, A.J.; Reichenbach, P.; et al. A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nat. Biotechnol. 2020, 38, 426–432. [Google Scholar] [CrossRef]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra240. [Google Scholar] [CrossRef]
- Raina, K.; Crews, C.M. Chemical inducers of targeted protein degradation. J. Biol. Chem. 2010, 285, 11057–11060. [Google Scholar] [CrossRef] [Green Version]
- Banaszynski, L.A.; Chen, L.C.; Maynard-Smith, L.A.; Ooi, A.G.; Wandless, T.J. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 2006, 126, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Fukagawa, T.; Takisawa, H.; Kakimoto, T.; Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 2009, 6, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Bonger, K.M.; Chen, L.C.; Liu, C.W.; Wandless, T.J. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat. Chem. Biol. 2011, 7, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.K.; Jacobs, C.L.; Huo, Y.; Yang, J.; Krumm, S.A.; Plemper, R.K.; Tsien, R.Y.; Lin, M.Z. Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 2015, 11, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Tkach, D.; Busser, B.W.; Temburni, S.; Valton, J.; Duclert, A.; Poirot, L.; Depil, S.; Duchateau, P. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. 2019, 19, 44. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Kang, C.H.; Choi, S.U.; Kim, Y.; Hwang, J.Y.; Jeong, H.G.; Park, C.H. A Chemical Switch System to Modulate Chimeric Antigen Receptor T Cell Activity through Proteolysis-Targeting Chimaera Technology. ACS Synth. Biol. 2020, 9, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.A.; Wang, L.C.; Moon, E.K.; Khire, U.R.; Albelda, S.M.; Milone, M.C. Ligand-Induced Degradation of a CAR Permits Reversible Remote Control of CAR T Cell Activity In Vitro and In Vivo. Mol. Ther 2020, 28, 1600–1613. [Google Scholar] [CrossRef]
- Jan, M.; Scarfo, I.; Larson, R.C.; Walker, A.; Schmidts, A.; Guirguis, A.A.; Gasser, J.A.; Slabicki, M.; Bouffard, A.A.; Castano, A.P.; et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Maynard-Smith, L.A.; Chen, L.C.; Banaszynski, L.A.; Ooi, A.G.; Wandless, T.J. A directed approach for engineering conditional protein stability using biologically silent small molecules. J. Biol. Chem. 2007, 282, 24866–24872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Qiu, S.; Chen, J.; Jiang, S.; Chen, W.; Jiang, J.; Wang, F.; Si, W.; Shu, Y.; Wei, P.; et al. Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 2020, 53, 456–470. [Google Scholar] [CrossRef]
- Sievers, Q.L.; Petzold, G.; Bunker, R.D.; Renneville, A.; Slabicki, M.; Liddicoat, B.J.; Abdulrahman, W.; Mikkelsen, T.; Ebert, B.L.; Thoma, N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Wu, Y.; Allen, M.E.; Pan, Y.; Kyriakakis, P.; Lu, S.; Chang, Y.J.; Wang, X.; Chien, S.; Wang, Y. Engineering light-controllable CAR T cells for cancer immunotherapy. Sci. Adv. 2020, 6, eaay9209. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Lopez, R.A.; Yu, W.; Cabral, K.A.; Creasey, O.A.; Lopez Pazmino, M.D.P.; Tonai, Y.; De Guzman, A.; Makela, A.; Saksela, K.; Gartner, Z.J.; et al. T cell circuits that sense antigen density with an ultrasensitive threshold. Science 2021, 371, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J., Jr. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Ma, J.S.; Yun, H.; Cao, Y.; Kim, J.Y.; Chi, V.; Wang, D.; Woods, A.; Sherwood, L.; Caballero, D.; et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J. Am. Chem. Soc. 2015, 137, 2832–2835. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.S.; Kim, J.Y.; Kazane, S.A.; Choi, S.H.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E450–E458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.J.; Chu, H.; Wheeler, L.W.; Nelson, M.; Westrick, E.; Matthaei, J.F.; Cardle, I.I.; Johnson, A.; Gustafson, J.; Parker, N.; et al. Preclinical Evaluation of Bispecific Adaptor Molecule Controlled Folate Receptor CAR-T Cell Therapy With Special Focus on Pediatric Malignancies. Front. Oncol. 2019, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef] [Green Version]
- Koristka, S.; Cartellieri, M.; Arndt, C.; Bippes, C.C.; Feldmann, A.; Michalk, I.; Wiefel, K.; Stamova, S.; Schmitz, M.; Ehninger, G.; et al. Retargeting of regulatory T cells to surface-inducible autoantigen La/SS-B. J. Autoimmun. 2013, 42, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.; von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M.P. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, A.; Hoffmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Arndt, C.; Rodrigues Loureiro, L.; Kittel-Boselli, E.; Mitwasi, N.; Kegler, A.; et al. Versatile chimeric antigen receptor platform for controllable and combinatorial T cell therapy. Oncoimmunology 2020, 9, 1785608. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, A.; Arndt, C.; Bergmann, R.; Loff, S.; Cartellieri, M.; Bachmann, D.; Aliperta, R.; Hetzenecker, M.; Ludwig, F.; Albert, S.; et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget 2017, 8, 31368–31385. [Google Scholar] [CrossRef] [Green Version]
- Fasslrinner, F.; Arndt, C.; Feldmann, A.; Koristka, S.; Loureiro, L.R.; Schmitz, M.; Jung, G.; Bornhaeuser, M.; Bachmann, M. Targeting the FMS-like Tyrosin Kinase 3 with the Unicar System: Preclinical Comparison of Murine and Humanized Single-Chain Variable Fragment-Based Targeting Modules. Blood 2019, 134, 5614. [Google Scholar] [CrossRef]
- Albert, S.; Arndt, C.; Feldmann, A.; Bergmann, R.; Bachmann, D.; Koristka, S.; Ludwig, F.; Ziller-Walter, P.; Kegler, A.; Gartner, S.; et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology 2017, 6, e1287246. [Google Scholar] [CrossRef]
- Yang, F.; Wen, W.; Qin, W. Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies. Int. J. Mol. Sci. 2016, 18, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Viaud, S.; Ma, J.S.Y.; Hardy, I.R.; Hampton, E.N.; Benish, B.; Sherwood, L.; Nunez, V.; Ackerman, C.J.; Khialeeva, E.; Weglarz, M.; et al. Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc. Natl. Acad. Sci. USA 2018, 115, E10898–E10906. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438. [Google Scholar] [CrossRef] [Green Version]
- Lohmueller, J.J.; Ham, J.D.; Kvorjak, M.; Finn, O.J. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology 2017, 7, e1368604. [Google Scholar] [CrossRef] [PubMed]
- Grote, S.; Mittelstaet, J.; Baden, C.; Chan, K.C.; Seitz, C.; Schlegel, P.; Kaiser, A.; Handgretinger, R.; Schleicher, S. Adapter chimeric antigen receptor (AdCAR)-engineered NK-92 cells: An off-the-shelf cellular therapeutic for universal tumor targeting. Oncoimmunology 2020, 9, 1825177. [Google Scholar] [CrossRef]
- Landgraf, K.E.; Williams, S.R.; Steiger, D.; Gebhart, D.; Lok, S.; Martin, D.W.; Roybal, K.T.; Kim, K.C. convertibleCARs: A chimeric antigen receptor system for flexible control of activity and antigen targeting. Commun. Biol. 2020, 3, 296. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, A.R.; Owens, M.N.; Geyer, C.R. Modular Chimeric Antigen Receptor Systems for Universal CAR T Cell Retargeting. Int. J. Mol. Sci. 2020, 21, 7222. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.W.; Horn, J.R. An anti-hapten camelid antibody reveals a cryptic binding site with significant energetic contributions from a nonhypervariable loop. Protein Sci. 2011, 20, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Pascua, E.; Lindquist, K.C.; Kimberlin, C.; Deng, X.; Mak, Y.S.L.; Melton, Z.; Johnson, T.O.; Lin, R.; Boldajipour, B.; et al. Direct control of CAR T cells through small molecule-regulated antibodies. Nat. Commun. 2021, 12, 710. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Deng, B.; Yin, Z.; Lin, Y.; An, L.; Liu, D.; Pan, J.; Yu, X.; Chen, B.; Wu, T.; et al. Combination of CD19 and CD22 CAR-T cell therapy in relapsed B-cell acute lymphoblastic leukemia after allogeneic transplantation. Am. J. Hematol. 2021. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Year | CAR Target | CAR Design * | Cancer or Model | Small Molecule or Drug | Target | Observations | Ref. |
|---|---|---|---|---|---|---|---|
| Combinatorial anti-cancer approaches | |||||||
| 2013 | CD19 | scFv-CD28-CD3ζ | B ALL | ABT-737; ABT-263 (navitoclax) | Bcl-2 family members | Restore intrinsic apoptosis in tumor cells; Enhance CAR T cells efficacy | [17] |
| 2018 | CD19 | N/A | NHL | suberoylanilide hydroxamic acid and LBH589; Celecoxib | Histone deacetylase; cyclo- oxygenase-2 | Enhance CAR T cells cytotoxicity | [19] |
| 2020 | CD19 | scFv-CD28-CD3ζ | B ALL | >500 small molecules | Multiple | primary: birinapant, AT-406, LCL-161 (SMAC mimetics/inhibitor of apoptotic antagonists); secondary: bryostatin-1 (PKC activator), idasanutlin and nutlin-3 (MDM2 inhibitors), etoposide and teniposide (topoisomerase 2 inhibitors); Enhance CAR T cells cytotoxicity | [20] |
| 2013 | HER-2 | scFv-CD28-CD3ζ | HER-2+ PD-1+ tumor cells | anti-PD-1 mAb | PD-1 | Enhance CAR T cell function; decrease MDSCs frequency | [24] |
| 2017 | CD19 | scFv-CD28-CD3ζ | B ALL | Akt inhibitor VIII | Akt | Akt signaling inhibition during CAR T cell expansion improve antitumor efficacy | [39] |
| 2013 | CD19 | scFv-CD28-CD3ζ | CLL | ibrutinib | Bruton’s tyrosine kinase | Improve CAR T cells engraftment, tumor clearance and mice survival | [41] |
| 2020 | CAIX | scFv-41BB-CD3ζ | RCC (lung metastasis) | sunitinib | Multiple kinases | Up-regulate CAIX in tumor cells; decrease MDSCs frequency | [44] |
| 2018 | FLT3 | scFv-CD28/41BB -CD3ζ | AML | crenolanib | FLT3 kinase | Synergize anti-leukemia effect | [45] |
| Mitigating the adverse effects | |||||||
| 2016 | CD19 | scFv-41BB-CD3ζ | B ALL | etanercept, infliximab | TNF-α | Reduce toxicity | [50] |
| 2019 | CD19 | scFv-41BB-CD3ζ | B ALL | lenzilumab | GM-CSF | Suppress CRS and neurotoxicity | [62] |
| 2018 | CD19 | scFv-CD28-CD3ζ | B lymphoma | anti-IL-6 and anti-IFN-γ mAb | IL-6 and IFN-γ | Reduce toxicity | [64] |
| 2018 | CD19, CD44v6 | scFv-CD28-CD3ζ | B ALL | anakinra, tocilizumab | IL-1 receptor antagonist,IL-6 | Suppress CRS and neurotoxicity; Extend leukemia-free survival | [65] |
| 2018 | CD19 | scFv-CD28-CD3ζ | B ALL | anakinra; L-NIL and 1400W | IL-1 receptor antagonist, iNOs inhibitors | Inhibit macrophage-derived products (NOs, IL-1 and IL-6); Suppress CRS-related mortality | [66] |
| 2018 | CD19 | scFv-CD28-41BB -CD3ζ | B lymphoma | metyrosine | catecholamines | Protect mice from lethal complications of CRS | [67] |
| CAR T cell elimination | |||||||
| 2017 | CD19 | iCasp9-2A-tNGFR -2A-scFv-41BB -CD3ζ | B lymphoma | AP1903 (rimiducid) | FKBP/FRB Inducible caspase 9 (iCasp9) | Eliminate CAR T cells in a dose-dependent manner | [79] |
| 2018 | CD19 | rapaCasp9-2A -RQR8-2A-scFv -41BB-CD3ζ | B ALL and lymphoma | AP20187, rapamycin | FKBP/FRB Inducible caspase 9 (rapaCasp9) | Eliminate CAR T cells in vivo | [82] |
| 2014 | GD2 | N/A | N/A | rituximab | CD20 epitope fused to CD8 stalk (RQR8, also contain tCD34) | Enable CAR T cells selection, cell tracking (tCD34) and deletion (CD20) | [87] |
| Reversible spatio-temporal control of CAR | |||||||
| 2019 | CD19 | scFv-CD28/41BB -CD3ζ-2A-EGFRt | B lymphoma | dasatinib | SRC kinases | Reversibly suppress CAR T cell cytotoxicity, cytokine secretion, and proliferation; protect from CRS | [99] |
| 2019 | CD19 | scFv-CD28/41BB -CD3ζ | B ALL | dasatinib | SRC kinases | Reversibly suppress CAR T cell cytotoxicity, cytokine secretion, and proliferation | [100] |
| 2015 | CD19, Meso | scFv-41BB-FKBP + DAP10-41BB -FRB-CD3ζ | CD19+ or Meso+ K562 | rapalog, (gibberellic acid) | FKBP/FRB- (or GID1/GAI) -based CAR dimerization | ON-switch CAR: control the timing, location, and dosage of CAR T cell activity; mitigate toxicity | [104] |
| 2018 | CD19 | soluble bispecific scFv-Fab(AZ1) + BclxL-41BB -CD3ζ | CD19+ K562 | ABT-737 | Fab(AZ1) specific for Bcl-xL only in the presence of ABT-737 | Drug-dependent CAR T cell activation | [106] |
| 2020 | CD19 | scFv-IgG1Fc -hRBP4 + RS3-IgG1Fc -CD28-CD3ζ | B ALL | A1120 | hRBP4 and hRBP4 binders (RS3) | Drug-dependent regulation of CAR T cells activity | [107] |
| 2017 | PSCA, GD2, CD123 | iMC: MyD88-CD40 -(FKBP)x2-2A -ΔCD19 iMC-2A-scFv -CD3ζ scFv -CD28/41BB/OX40-CD3ζ | prostate, melanoma, AML | AP1903 (rimiducid) | FKBP/FRB -based dimerization of MyD88/CD40 (iMC) | Enhance CAR T cell proliferation and antitumor activity | [108] |
| 2020 | PSMA | scFv-CD28-LD3+ DAP10-CD28 -BclxL-CD3ζ | prostate | A-1155463, A-1331852 (BH3 mimetic) | LD3/Bcl-xL -based CAR dimerization | STOP-CAR: dynamically and reversibly inactivate CAR T cells | [112] |
| 2019 | CD22 | scFv-41BB-CD3ζ -NS3cleaving_site -NS3protease -degron | B lymphoma | Asunaprevir | HCV NS3 protease | Switch-OFF CAR (SWIFF-CAR): constitutive CAR degron proteolysis; Asunaprevir-dependent CAR degradation | [120] |
| 2020 | CD19 | scFv-CD28-BD2 -CD3ζ | B ALL | ARV-771 or ARV-825 (retinol) | bromodomain (BD of brd4) | Induce drug-dependent CAR degradation; Reversibly suppress CAR T cells | [121] |
| 2020 | GD2 | scFv -41BB/KIR2DS2 -CD3ζ-[FKBP -degron]LIDdomain | FAP+ Mesothelioma | Shield-1 | LID domain -based CAR degradation | Induce drug-dependent CAR degradation; temporarily reduce CAR T cells activity | [122] |
| 2021 | CD19 | OFF-switch: scFc-41BB-CD3ζ -C2H2degron ON-switch: CD8-CD28-CRBN-CD3ζ + scFv-CD28 -C2H2degron | B ALL and lymphoma | thalidomide analogs | C2H2, CRBN | OFF-switch: thalidomide analog-induced CAR proteasomal degradation, limit inflammatory cytokine production while retaining antitumor efficacy ON-switch: thalidomide analog-induced split CAR dimerization, drug-dependent antitumor activity | [123] |
| Modulating CAR specificity | |||||||
| 2016 | FITC | scFv-41BB-CD3ζ | B ALL and lymphoma | FITC-modified anti-CD19 and anti-CD22 antibodies | CD19, CD22 | Enable CAR-switch combinations; potent and dose-dependent antitumor activity | [135] |
| 2016 | 5B9 epitope of La/SS-B | scFv-CD28-CD3ζ | AML (others) | 5B9-tagged anti-CD33 and anti-CD123 antibodies | CD33, CD123 | UniCAR T (Universal): Redirect CAR in a time- and target-dependent manner; potent anti-AML activity | [139] |
| 2018 | HER-2, Axl, Meso | zipFv: scFv -EEleucine_zipper zipCAR: RRleucine_zipper -CD28-41BB-CD3ζ | HER-2+, Axl+, Meso+ K562 | Soluble zipFv | Membrane -bound zipCAR | SUPRA-CAR: control signaling, fine-tune T cell activation, mitigate toxicity and allow multiple antigens sensing | [147] |
| 2021 | CD33, EGFR | scFv-41BB-CD3ζ | AML, GBM | Methotrexate | Conditional scFvs | Drug-induced decrease of CAR T cells affinity and cytotoxicity; reversible | [153] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caulier, B.; Enserink, J.M.; Wälchli, S. Pharmacologic Control of CAR T Cells. Int. J. Mol. Sci. 2021, 22, 4320. https://doi.org/10.3390/ijms22094320
Caulier B, Enserink JM, Wälchli S. Pharmacologic Control of CAR T Cells. International Journal of Molecular Sciences. 2021; 22(9):4320. https://doi.org/10.3390/ijms22094320
Chicago/Turabian StyleCaulier, Benjamin, Jorrit M. Enserink, and Sébastien Wälchli. 2021. "Pharmacologic Control of CAR T Cells" International Journal of Molecular Sciences 22, no. 9: 4320. https://doi.org/10.3390/ijms22094320
APA StyleCaulier, B., Enserink, J. M., & Wälchli, S. (2021). Pharmacologic Control of CAR T Cells. International Journal of Molecular Sciences, 22(9), 4320. https://doi.org/10.3390/ijms22094320