
Delineating the Tnt1 Insertion Landscape of the Model Legume Medicago truncatula cv. R108 at the Hi-C Resolution Using a Chromosome-Length Genome Assembly

 ,
,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
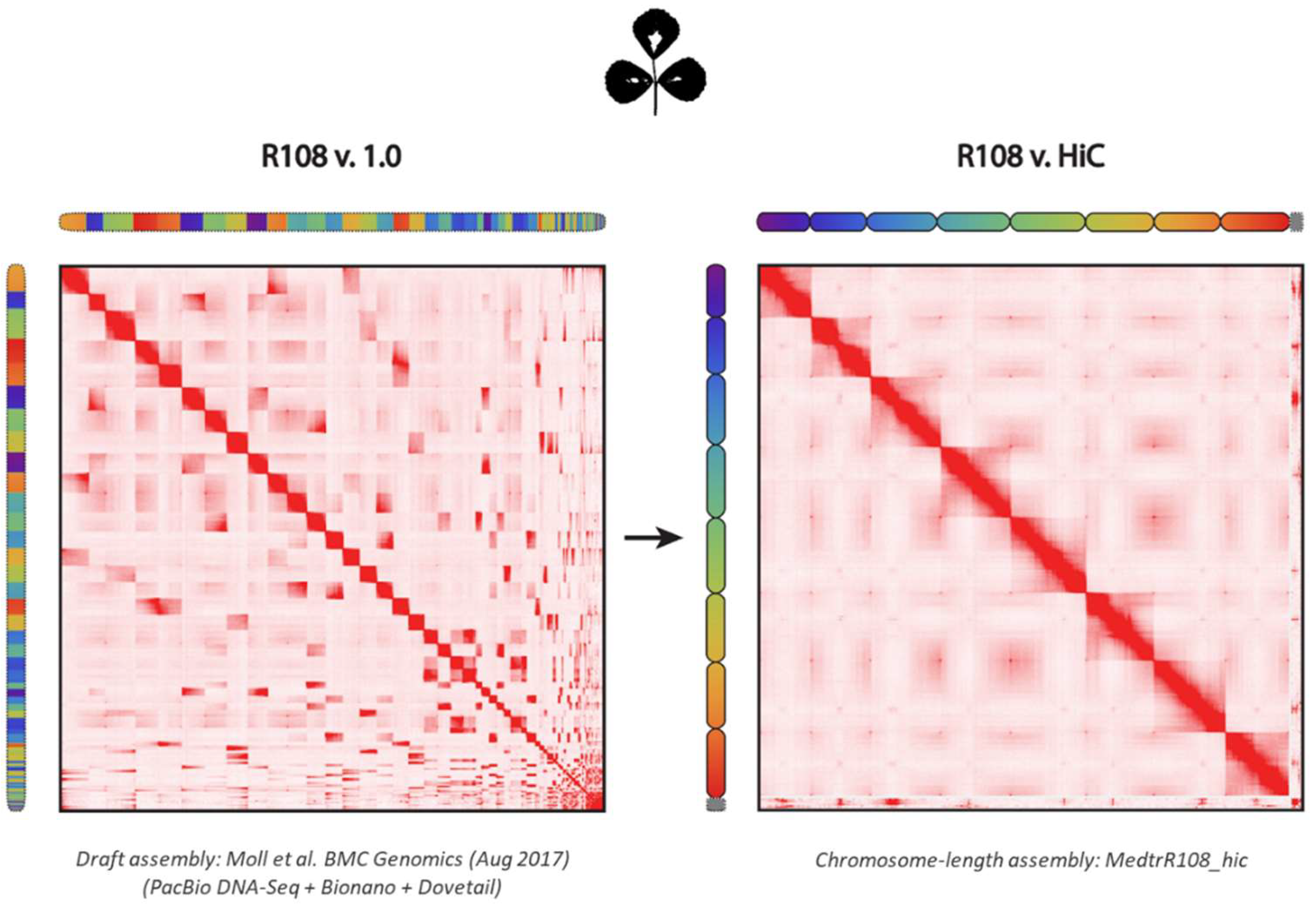
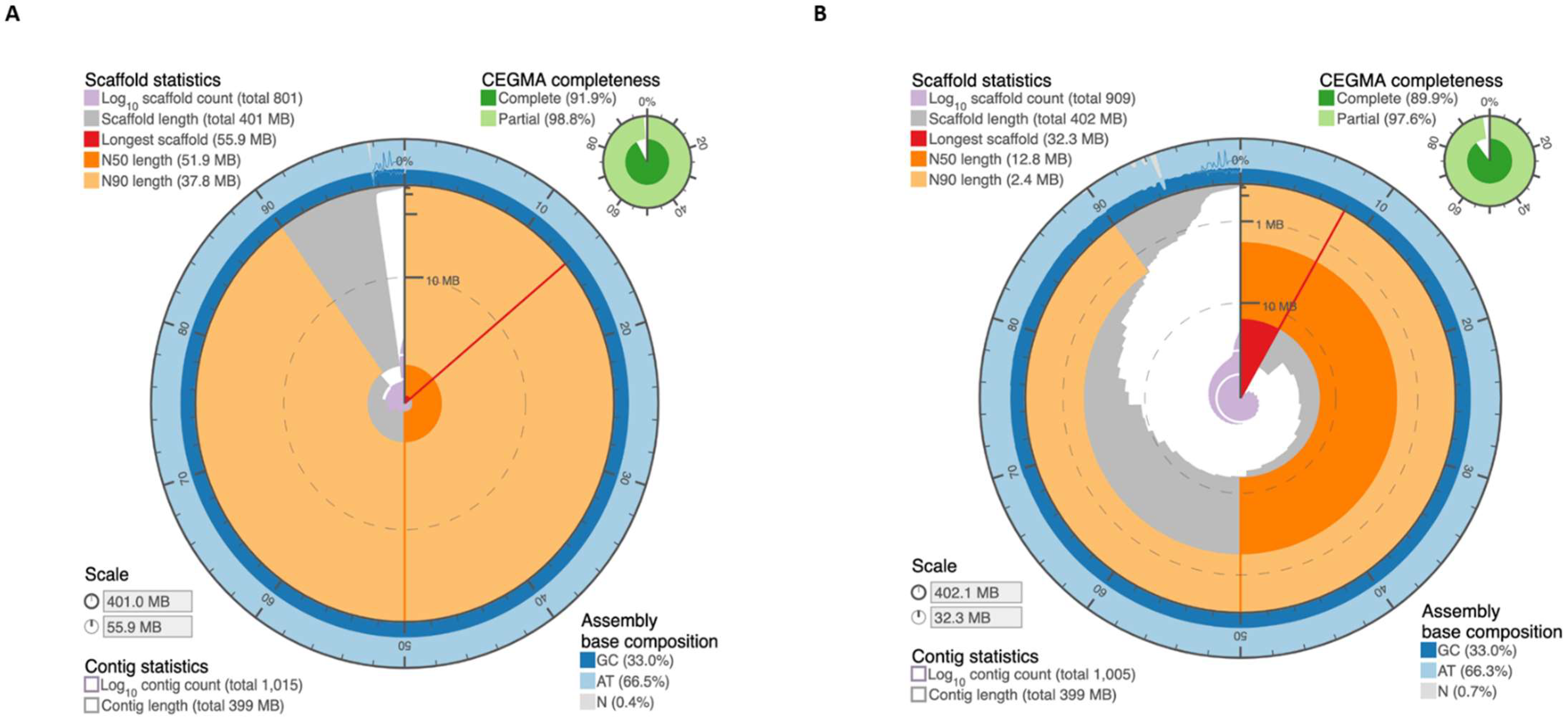
2.1. Assembly of M. truncatula Accession R108 with Chromosome-Length Scaffolds
2.2. Genome Annotation and Functional Characterization
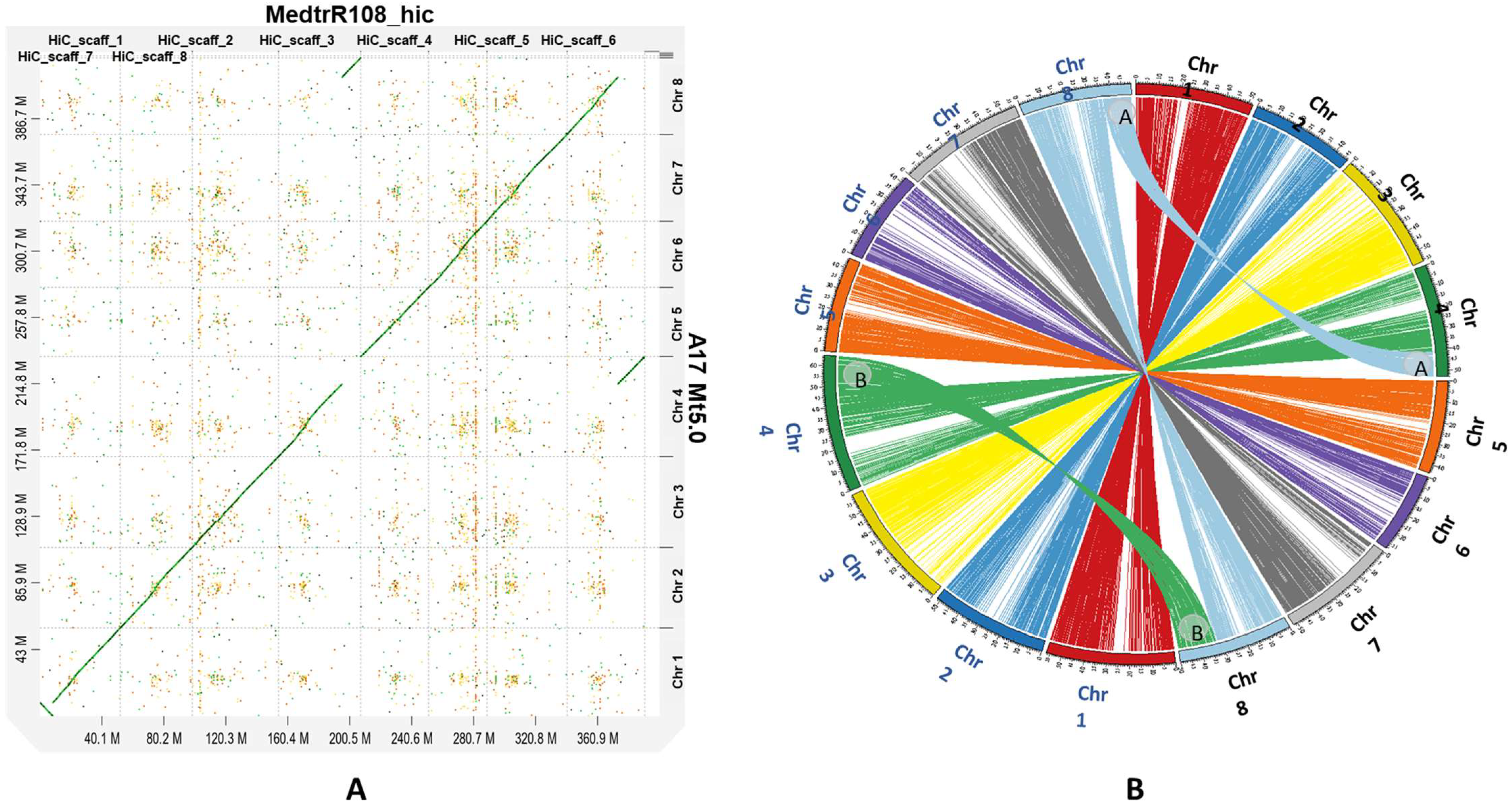
2.3. Synteny Analysis and Chromosomal Translocation
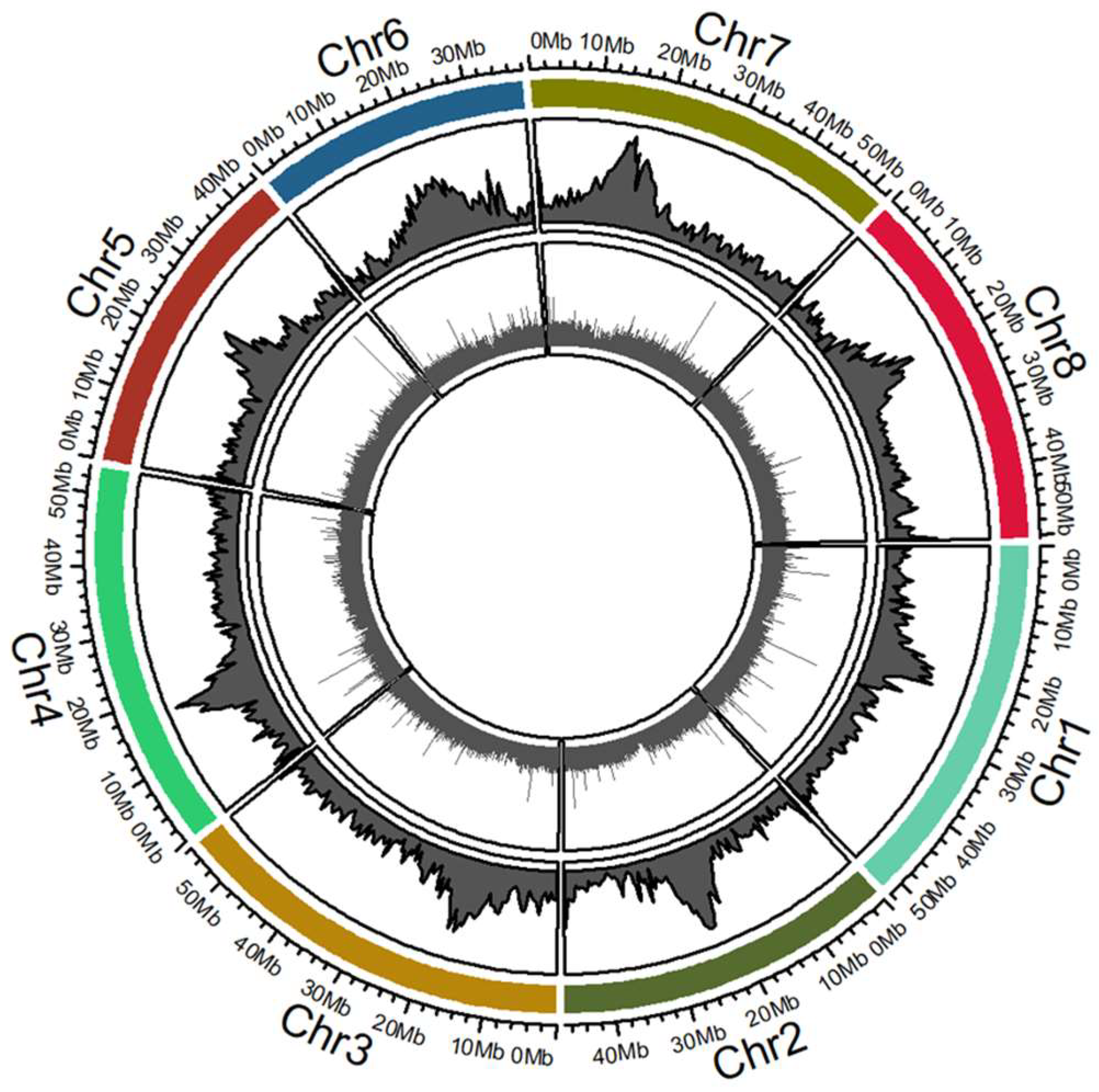
2.4. Mapping Tnt1 Insertion Sites in the M. truncatula R108 Hi-C Genome Assembly
2.5. Comparison of Tnt1 Insertions Using M. truncatula R108 Hi-C or A17 v5.0 Genic Regions and Functional Annotation of Genes with Insertions
2.6. Tnt1 Insertions in Genes in the Syntenic Regions
3. Discussion
4. Materials and Methods
4.1. Hi-C Library Preparation and Sequencing
4.2. Genome Assembly
4.3. Genome Annotations
4.4. Assessment of Genome Assembly and Annotation Quality
4.5. Functional Annotation of the Predicted Genes
4.6. Genome Alignment and Detection of Chromosomal Translocation
4.7. Mapping of Tnt1 Insertion Lines and Functional Gene Group Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tilman, D.; Balzer, C.; Hill, J.; Befort, B.L. Global food demand and the sustainable intensification of agriculture. Proc. Natl. Acad. Sci USA 2011, 108, 20260–20264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikowo, R.; Mapfumo, P.; Nyamugafata, P.; Giller, K.E. Maize productivity and mineral N dynamics following different soil fertility management practices on a depleted sandy soil in Zimbabwe. Agric. Ecosyst. Environ. 2004, 102, 119–131. [Google Scholar] [CrossRef]
- Saha, N.; Mandal, B. Soil health—A precondition for crop production. In Microbial STRATEgies for Crop Improvement; Springer: Cham, Switzerland, 2009; pp. 161–184. [Google Scholar]
- Tang, H.B.; Krishnakumar, V.; Bidwell, S.; Rosen, B.; Chan, A.N.; Zhou, S.G.; Gentzbittel, L.; Childs, K.L.; Yandell, M.; Gundlach, H.; et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Michno, J.M.; Wang, X.B.; Liu, J.Q.; Curtin, S.J.; Kono, T.J.Y.; Stupar, R.M. CRISPR/Cas mutagenesis of soybean and Medicago truncatula using a new web-tool and a modified Cas9 enzyme. Genet. Modif. Crop Food 2015, 6, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Cermak, T.; Curtin, S.J.; Gil-Humanes, J.; Cegan, R.; Kono, T.J.Y.; Konecna, E.; Belanto, J.J.; Starker, C.G.; Mathre, J.W.; Greenstein, R.L.; et al. a multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 2017, 29, 1196–1217. [Google Scholar] [CrossRef] [Green Version]
- Curtin, S.J.; Xiong, Y.; Michno, J.M.; Campbell, B.W.; Stec, A.O.; Cermak, T.; Starker, C.; Voytas, D.F.; Eamens, A.L.; Stupar, R.M. CRISPR/Cas9 and TALENs generate heritable mutations for genes involved in small RNA processing of Glycine max and Medicago truncatula. Plant Biotechnol. J. 2018, 16, 1125–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.Y.; Hou, Y.L.; Wang, H.; Ji, R.H.; Liu, B.; Wen, J.Q.; Niu, L.F.; Lin, H. Targeted mutagenesis by CRISPR/Cas9 system in the model legume Medicago truncatula. Plant Cell Rep. 2017, 36, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Wang, X.; Li, X.M.; Xu, M.Y.; Li, H.Y.; Abla, M.; Sun, H.G.; Wei, S.J.; Feng, J.C.; Zhou, Y.J. Long-read sequencing and de novo genome assembly of Ammopiptanthus nanus, a desert shrub. Gigascience 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Tadege, M.; Wen, J.Q.; He, J.; Tu, H.D.; Kwak, Y.; Eschstruth, A.; Cayrel, A.; Endre, G.; Zhao, P.X.; Chabaud, M.; et al. Large-scale insertional mutagenesis using the Tnt1 retrotransposon in the model legume Medicago truncatula. Plant J. 2008, 54, 335–347. [Google Scholar] [CrossRef]
- Young, N.D.; Debelle, F.; Oldroyd, G.E.D.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.X.; Gouzy, J.; Schoof, H.; et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Gill, U.S.; Nandety, R.S.; Kwon, S.; Mehta, P.; Dickstein, R.; Udvardi, M.K.; Mysore, K.S.; Wen, J.Q. Genome-wide analysis of flanking sequences reveals that Tnt1 insertion is positively correlated with gene methylation in Medicago truncatula. Plant J. 2019, 98, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecrix, Y.; Staton, S.E.; Sallet, E.; Lelandais-Brere, C.; Moreau, S.; Carrere, S.; Blein, T.; Jardinaud, M.F.; Latrasse, D.; Zouine, M.; et al. Whole-genome landscape of Medicago truncatula symbiotic genes. Nat. Plants 2018, 4, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.; Smith, L.; La Thangue, N.B. Chk2 activates E2F-1 in response to DNA damage. Nat. Cell Biol. 2003, 5, 401–409. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.L.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [Green Version]
- Moll, K.M.; Zhou, P.; Ramaraj, T.; Fajardo, D.; Devitt, N.P.; Sadowsky, M.J.; Stupar, R.M.; Tiffin, P.; Miller, J.R.; Young, N.D.; et al. Strategies for optimizing BioNano and Dovetail explored through a second reference quality assembly for the legume model, Medicago truncatula. BMC Genom. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Dudchenko, O.; Shamim, M.S.; Batra, S.S.; Durand, N.C.; Musial, N.T.; Mostofa, R.; Pham, M.; St Hilaire, B.G.; Yao, W.; Stamenova, E. The juicebox assembly tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. Biorxiv 2018, 254797. [Google Scholar]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinform. 2007, 23, 1061–1067. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodstein, D.M.; Shu, S.Q.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [Green Version]
- Kamphuis, L.G.; Williams, A.H.; D’Souza, N.K.; Pfaff, T.; Ellwood, S.R.; Groves, E.J.; Singh, K.B.; Oliver, R.P.; Lichtenzveig, J. The Medicago truncatula reference accession A17 has an aberrant chromosomal configuration. New Phytol. 2007, 174, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Liu, Y.; Yan, H.Y.; You, Q.; Yi, X.; Du, Z.; Xu, W.Y.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Campbell, M.S.; Law, M.Y.; Holt, C.; Stein, J.C.; Moghe, G.D.; Hufnagel, D.E.; Lei, J.K.; Achawanantakun, R.; Jiao, D.; Lawrence, C.J.; et al. MAKER-P: A tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 2014, 164, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.J.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Whole-Genome Annotation with BRAKER. Methods Mol. Biol. 2019, 1962, 65–95. [Google Scholar]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008, 9. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Voytas, D.F.; Gao, C.X. Precision genome engineering and agriculture: Opportunities and regulatory challenges. PLoS Biol. 2014, 12. [Google Scholar] [CrossRef] [PubMed]
- Wolabu, T.W.; Park, J.-J.; Chen, M.; Cong, L.; Ge, Y.; Jiang, Q.; Debnath, S.; Li, G.; Wen, J.; Wang, Z. Improving the genome editing efficiency of CRISPR/Cas9 in Arabidopsis and Medicago truncatula. Planta 2020, 252, 1–14. [Google Scholar] [CrossRef]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. juicebox provides a visualization system for hi-c contact maps with unlimited zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [Green Version]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution hi-c experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert Hubley, A.S. RepeatModeler Open-1.0, Version 1.0. 2008.
- Robert Hubley, A.S. RepeatMasker Open-4.0, Version 4.0, 2013–2015.
- Lukashin, A.V.; Borodovsky, M. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, Ii215–Ii225. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc, G.P.D. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slater, G.S.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, D543–D546. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.Z.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Cabanettes, F.; Klopp, C. D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 2018, 6, e4958. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.B.; Bowers, J.E.; Wang, X.Y.; Ming, R.; Alam, M.; Paterson, A.H. Perspective—Synteny and collinearity in plant genomes. Science 2008, 320, 486–488. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics | MedtrR108_hic |
|---|---|
| Draft scaffolds | |
| Base pairs | 399,348,955 |
| Number of contigs | 1005 |
| Contig N50 | 5,925,378 |
| Number of scaffolds | 909 |
| Scaffold N50 | 12,848,239 |
| Chromosome-length scaffolds | |
| Base pairs | 390,045,474 |
| Number of contigs | 209 |
| Contig N50 | 6,045,855 |
| Number of scaffolds | 8 |
| Scaffold N50 | 51,860,634 |
| Small scaffolds | |
| Base pairs | 5,840,890 |
| Number of contigs | 248 |
| Contig N50 | 24,000 |
| Number of scaffolds | 236 |
| Scaffold N50 | 24,736 |
| Tiny scaffolds | |
| Base pairs | 3,462,591 |
| Number of contigs | 558 |
| Contig N50 | 9246 |
| Number of scaffolds | 557 |
| Scaffold N50 | 9246 |
| Mapping Description | No of FSTs | % of Total FSTs |
|---|---|---|
| FSTs mapped to Chromosome 1 | 27,902 | 12.61 |
| FSTs mapped to Chromosome 2 | 24,559 | 11.1 |
| FSTs mapped to Chromosome 3 | 27,679 | 12.51 |
| FSTs mapped to Chromosome 4 | 26,975 | 12.19 |
| FSTs mapped to Chromosome 5 | 25,313 | 11.44 |
| FSTs mapped to Chromosome 6 | 16,433 | 7.43 |
| FSTs mapped to Chromosome 7 | 25,451 | 11.5 |
| FSTs mapped to Chromosome 8 | 27,115 | 12.25 |
| Total mapped to 8 chromosomes | 201,427 | 91.03 |
| Total mapped to non Chr scaffolds | 1361 | 0.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, P.; Lui, C.; Dudchenko, O.; Nandety, R.S.; Hurgobin, B.; Pham, M.; Lieberman Aiden, E.; Wen, J.; Mysore, K.S. Delineating the Tnt1 Insertion Landscape of the Model Legume Medicago truncatula cv. R108 at the Hi-C Resolution Using a Chromosome-Length Genome Assembly. Int. J. Mol. Sci. 2021, 22, 4326. https://doi.org/10.3390/ijms22094326
Kaur P, Lui C, Dudchenko O, Nandety RS, Hurgobin B, Pham M, Lieberman Aiden E, Wen J, Mysore KS. Delineating the Tnt1 Insertion Landscape of the Model Legume Medicago truncatula cv. R108 at the Hi-C Resolution Using a Chromosome-Length Genome Assembly. International Journal of Molecular Sciences. 2021; 22(9):4326. https://doi.org/10.3390/ijms22094326
Chicago/Turabian StyleKaur, Parwinder, Christopher Lui, Olga Dudchenko, Raja Sekhar Nandety, Bhavna Hurgobin, Melanie Pham, Erez Lieberman Aiden, Jiangqi Wen, and Kirankumar S Mysore. 2021. "Delineating the Tnt1 Insertion Landscape of the Model Legume Medicago truncatula cv. R108 at the Hi-C Resolution Using a Chromosome-Length Genome Assembly" International Journal of Molecular Sciences 22, no. 9: 4326. https://doi.org/10.3390/ijms22094326
APA StyleKaur, P., Lui, C., Dudchenko, O., Nandety, R. S., Hurgobin, B., Pham, M., Lieberman Aiden, E., Wen, J., & Mysore, K. S. (2021). Delineating the Tnt1 Insertion Landscape of the Model Legume Medicago truncatula cv. R108 at the Hi-C Resolution Using a Chromosome-Length Genome Assembly. International Journal of Molecular Sciences, 22(9), 4326. https://doi.org/10.3390/ijms22094326