
Deubiquitinases: Modulators of Different Types of Regulated Cell Death

Abstract
:1. Introduction
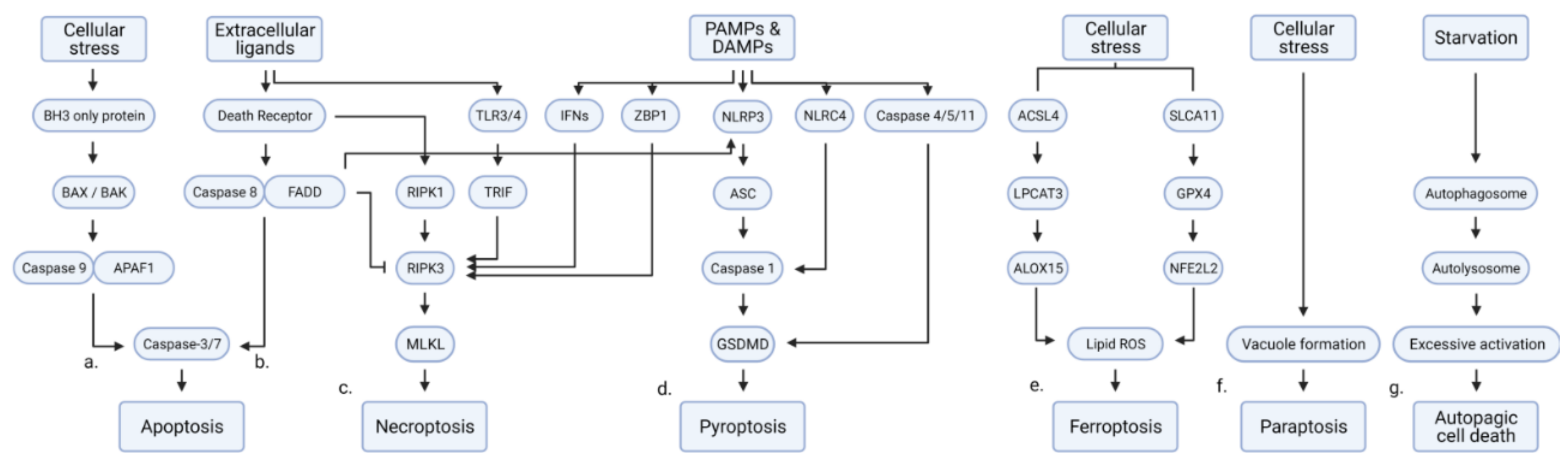
1.1. Regulated Cell Death (RCD)
1.2. Apoptosis
1.3. Necroptosis
1.4. Pyroptosis
1.5. Ferroptosis
1.6. Paraptosis
1.7. Autophagy-Dependent Cell Death
2. DUBs
3. DUBs Regulating Diverse RCD
3.1. USP5
3.2. USP7 (HAUSP)
3.3. USP8
3.4. USP10
3.5. USP11
3.6. USP15
3.7. USP18
3.8. USP20
3.9. USP22
3.10. CYLD
3.11. A20
3.12. OTULIN
3.13. OTUB1
3.14. BAP1
3.15. UCHL1
3.16. UCHL5/USP14
3.17. BRCC36
3.18. STAMBPL1
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Yang, X. Proteases for cell suicide: Functions and regulation of caspases. Microbiol. Mol. Biol. Rev. 2000, 64, 821–846. [Google Scholar] [CrossRef] [Green Version]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Goldschneider, D.; Mehlen, P. Dependence receptors: A new paradigm in cell signaling and cancer therapy. Oncogene 2010, 29, 1865–1882. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, K. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiol. Immunol. 2020, 64, 252–269. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, R.; Tan, H. Role of pyroptosis in cardiovascular diseases and its therapeutic implications. Int. J. Biol. Sci. 2019, 15, 1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Jiang, J.; Gao, Y.; Shi, T.; Zhu, X.; Zhang, K.; Lu, K.; Xue, B. Research progress of the relationship between pyroptosis and disease. Am. J. Transl. Res. 2018, 10, 2213. [Google Scholar]
- Py, B.F.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, X.; Yang, Q.; Chen, J.; Huang, Q.; Yao, L.; Yan, D.; Wu, J.; Zhang, P.; Tang, D. Broad spectrum deubiquitinase inhibition induces both apoptosis and ferroptosis in cancer cells. Front. Oncol. 2020, 10, 949. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. The emerging role of paraptosis in tumor cell biology: Perspectives for cancer prevention and therapy with natural compounds. Biochim. Biophys. Acta BBA Rev. Cancer 2020, 1873, 188338. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, X.; Chen, J.; Yang, Q.; Yang, L.; Xu, D.; Zhang, P.; Wang, X.; Liu, J. Hinokitiol copper complex inhibits proteasomal deubiquitination and induces paraptosis-like cell death in human cancer cells. Eur. J. Pharmacol. 2017, 815, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Coulson, J.M.; Urbé, S. Cellular functions of the DUBs. J. Cell Sci. 2012, 125, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanpude, P.; Bhattacharya, S.; Dey, A.K.; Maiti, T.K. Deubiquitinating enzymes in cellular signaling and disease regulation. IUBMB Life 2015, 67, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.; Komander, D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, T.; Shin, S.C.; Song, E.J.; Kim, E.E. Regulation of Deubiquitinating Enzymes by Post-Translational Modifications. Int. J. Mol. Sci. 2020, 21, 4028. [Google Scholar] [CrossRef]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9, 190147. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; Wang, X. From discovery to bedside: Targeting the ubiquitin system. Cell Chem. Biol. 2019, 26, 156–177. [Google Scholar] [CrossRef]
- Kwasna, D.; Rehman, S.A.A.; Natarajan, J.; Matthews, S.; Madden, R.; De Cesare, V.; Weidlich, S.; Virdee, S.; Ahel, I.; Gibbs-Seymour, I. Discovery and characterization of ZUFSP/ZUP1, a distinct deubiquitinase class important for genome stability. Mol. Cell 2018, 70, 150–164.e6. [Google Scholar] [CrossRef] [Green Version]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Díaz, C.; Ikeda, F. Roles of ubiquitin in autophagy and cell death. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2019; pp. 125–135. [Google Scholar]
- Bhattacharya, S.; Ghosh, M.K. Cell death and deubiquitinases: Perspectives in cancer. BioMed Res. Int. 2014, 2014, 435197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, L.; Wu, J.; Sokirniy, I.; Nguyen, P.; Bregnard, T.; Weinstock, J.; Mattern, M.; Bezsonova, I.; Hancock, W.W. Active site-targeted covalent irreversible inhibitors of USP7 impair the functions of Foxp3+ T-regulatory cells by promoting ubiquitination of Tip60. PLoS ONE 2017, 12, e0189744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Wang, S.; Tong, J.; Jiang, S.; Yang, Y.; Zhang, Z.; Xu, Y.; Zeng, Y.; Cao, B.; Moran, M.F. The deubiquitinase USP7 stabilizes Maf proteins to promote myeloma cell survival. J. Biol. Chem. 2020, 295, 2084–2096. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Peng, Y.; Zhang, J.; Zhang, Y.; Roy, M.; Han, X.; Xiao, X.; Sun, S.; Liu, H.; Nie, L. Deubiquitylase USP7 regulates human terminal erythroid differentiation by stabilizing GATA1. Haematologica 2019, 104, 2178–2187. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q.R.; He, Q.; Zhao, S.; Zhang, G.A.; Wang, Y. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, e47563. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, W.; Lv, D.; Zhang, X.; Zhang, X.; Ortiz, Y.T.; Budamagunta, V.; Campisi, J.; Zheng, G.; Zhou, D. Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell 2020, 19, e13117. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Li, M.; Hu, M.; Shi, Y.; Gu, W. The p53–Mdm2–HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene 2007, 26, 7262–7266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Oh, T.-I.; Um, K.B.; Yoon, H.; Son, J.; Kim, B.M.; Kim, H.-I.; Kim, H.; Kim, Y.J.; Lee, C.-S. Small-molecule inhibitors of USP7 induce apoptosis through oxidative and endoplasmic reticulum stress in cancer cells. Biochem. Biophys. Res. Commun. 2016, 470, 181–186. [Google Scholar] [CrossRef]
- Tavana, O.; Sun, H.; Gu, W. Targeting HAUSP in both p53 wildtype and p53-mutant tumors. Cell Cycle 2018, 17, 823–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaman, M.M.-U.; Nomura, T.; Takagi, T.; Okamura, T.; Jin, W.; Shinagawa, T.; Tanaka, Y.; Ishii, S. Ubiquitination-deubiquitination by the TRIM27-USP7 complex regulates tumor necrosis factor alpha-induced apoptosis. Mol. Cell. Biol. 2013, 33, 4971–4984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vugmeyster, Y.; Borodovsky, A.; Maurice, M.M.; Maehr, R.; Furman, M.H.; Ploegh, H.L. The ubiquitin–proteasome pathway in thymocyte apoptosis: Caspase-dependent processing of the deubiquitinating enzyme USP7 (HAUSP). Mol. Immunol. 2002, 39, 431–441. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, J.; Xu, C.; Zhang, S.; Bian, S.; Jiang, F.; Ni, W.; Qu, L.; Lu, C.; Ni, R. Ubiquitin-specific protease 7 is a drug-able target that promotes hepatocellular carcinoma and chemoresistance. Cancer Cell Int. 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mungamuri, S.K.; Qiao, R.F.; Yao, S.; Manfredi, J.J.; Gu, W.; Aaronson, S.A. USP7 enforces heterochromatinization of p53 target promoters by protecting SUV39H1 from MDM2-mediated degradation. Cell Rep. 2016, 14, 2528–2537. [Google Scholar] [CrossRef] [Green Version]
- Agathanggelou, A.; Smith, E.; Davies, N.J.; Kwok, M.; Zlatanou, A.; Oldreive, C.E.; Mao, J.; Da Costa, D.; Yadollahi, S.; Perry, T. USP7 inhibition alters homologous recombination repair and targets CLL cells independently of ATM/p53 functional status. Blood J. Am. Soc. Hematol. 2017, 130, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhang, Y.; Wang, T.; Zhang, J.; Zhou, Z.; Sun, Y.; Wang, S.; Shi, Y.; Luan, X.; Zhang, Y. The USP7 inhibitor P5091 induces cell death in ovarian cancers with different P53 status. Cell. Physiol. Biochem. 2017, 43, 1755–1766. [Google Scholar] [CrossRef]
- Fan, Y.; Cheng, J.; Vasudevan, S.; Dou, J.; Zhang, H.; Patel, R.; Ma, I.; Rojas, Y.; Zhao, Y.; Yu, Y. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell Death Dis. 2013, 4, e867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Liu, Y.; Gao, Y.; Yuan, B.; Qi, X.; Fu, Y.; Zhu, Q.; Cao, T.; Zhang, S.; Yin, L. USP7 is a novel Deubiquitinase sustaining PLK1 protein stability and regulating chromosome alignment in mitosis. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Kolluri, K.K.; Pennycuick, A.; Nigro, E.; Alrifai, D.; Borg, E.; Falzon, M.; Shah, K.; Kumar, N.; Janes, S.M. BAP1 and YY1 regulate expression of death receptors in malignant pleural mesothelioma. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kumar, R.; Taylor, M.; Miao, B.; Ji, Z.; Njauw, J.C.; Jönsson, G.; Frederick, D.T.; Tsao, H. BAP1 has a survival role in cutaneous melanoma. J. Investig. Dermatol. 2015, 135, 1089–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sime, W.; Niu, Q.; Abassi, Y.; Masoumi, K.C.; Zarrizi, R.; Køhler, J.B.; Kjellström, S.; Lasorsa, V.A.; Capasso, M.; Fu, H. BAP1 induces cell death via interaction with 14-3-3 in neuroblastoma. Cell Death Dis. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Dai, F.; Lee, H.; Zhang, Y.; Zhuang, L.; Yao, H.; Xi, Y.; Xiao, Z.-D.; You, M.J.; Li, W.; Su, X. BAP1 inhibits the ER stress gene regulatory network and modulates metabolic stress response. Proc. Natl. Acad. Sci. USA 2017, 114, 3192–3197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S. BAP1 regulates IP3R3-mediated Ca 2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guazzelli, A.; Meysami, P.; Bakker, E.; Demonacos, C.; Giordano, A.; Krstic-Demonacos, M.; Mutti, L. BAP1 status determines the sensitivity of malignant mesothelioma cells to gemcitabine treatment. Int. J. Mol. Sci. 2019, 20, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Zhao, C.; Li, S.; Wang, J.; Zhou, Q.; Sun, J.; Ding, Q.; Liu, M.; Ding, G. EZH2 Expression is increased in BAP1-mutant renal clear cell carcinoma and is related to poor prognosis. J. Cancer 2018, 9, 3787. [Google Scholar] [CrossRef]
- He, M.; Chaurushiya, M.S.; Webster, J.D.; Kummerfeld, S.; Reja, R.; Chaudhuri, S.; Chen, Y.-J.; Modrusan, Z.; Haley, B.; Dugger, D.L. Intrinsic apoptosis shapes the tumor spectrum linked to inactivation of the deubiquitinase BAP1. Science 2019, 364, 283–285. [Google Scholar] [PubMed]
- Zhang, Y.; Koppula, P.; Gan, B. Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell Cycle 2019, 18, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmotto, M.; Monteleone, D.; Vasciaveo, V.; Repetto, I.E.; Manassero, G.; Tabaton, M.; Tamagno, E. The decrease of Uch-L1 activity is a common mechanism responsible for Aβ 42 accumulation in Alzheimer’s and vascular disease. Front. Aging Neurosci. 2017, 9, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, H.-L.; Zhang, X.-L.; Zhang, Y.-L.; Xie, X.; Xia, Y.-L.; Du, J.; Li, H.-H. The deubiquitinase UCHL1 regulates cardiac hypertrophy by stabilizing epidermal growth factor receptor. Sci. Adv. 2020, 6, eaax4826. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Ding, X.; Huang, J.; Xue, M.; Zhang, J.; Wang, Q.; Yu, H.; Wang, Y.; Zhao, F.; Wang, H. The deubiquitinating enzyme UCHL1 negatively regulates the immunosuppressive capacity and survival of multipotent mesenchymal stromal cells. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, K.; Lee, Y.Y.; Choo, O.-S.; Jang, J.H.; Choung, Y.-H. Downregulated UCHL1 accelerates gentamicin-induced auditory cell death via autophagy. Mol. Neurobiol. 2019, 56, 7433–7447. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, H.; Hu, Y.; Fang, Y.; Qi, C.; Huang, J.; Cai, X.; Wu, H.; Ding, X.; Zhang, Z. High glucose-induced apoptosis and necroptosis in podocytes is regulated by UCHL1 via RIPK1/RIPK3 pathway. Exp. Cell Res. 2019, 382, 111463. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Sikorska, M.; Leblanc, J.; Walker, P.; Liu, Q. Oxidative stress regulated expression of ubiquitin Carboxyl-terminal Hydrolase-L1: Role in cell survival. Apoptosis 2006, 11, 1049–1059. [Google Scholar] [CrossRef]
- Zhang, Y.-P.; Zhu, Y.-B.; Duan, D.D.; Fan, X.-M.; He, Y.; Su, J.-W.; Liu, Y.-L. Serum UCH-L1 as a novel biomarker to predict neuronal apoptosis following deep hypothermic circulatory arrest. Int. J. Med. Sci. 2015, 12, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tao, Q.; Jin, H.; Van Hasselt, A.; Poon, F.F.; Wang, X.; Zeng, M.-S.; Jia, W.-H.; Zeng, Y.-X.; Chan, A.T. The tumor suppressor UCHL1 forms a complex with p53/MDM2/ARF to promote p53 signaling and is frequently silenced in nasopharyngeal carcinoma. Clin. Cancer Res. 2010, 16, 2949–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, J.; Wang, Y.-L.; Setsuie, R.; Sekiguchi, S.; Sato, Y.; Sakurai, M.; Noda, M.; Aoki, S.; Yoshikawa, Y.; Wada, K. Two closely related ubiquitin C-terminal hydrolase isozymes function as reciprocal modulators of germ cell apoptosis in cryptorchid testis. Am. J. Pathol. 2004, 165, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Mochida, K.; Wang, Y.-L.; Sekiguchi, S.; Sankai, T.; Aoki, S.; Ogura, A.; Yoshikawa, Y.; Wada, K. Ubiquitin C-terminal hydrolase L-1 is essential for the early apoptotic wave of germinal cells and for sperm quality control during spermatogenesis. Biol. Reprod. 2005, 73, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, K.; Zigrino, P.; Witt, A.; Schell, M.; Ackermann, L.; Broxtermann, P.; Schüll, S.; Andree, M.; Coutelle, O.; Yazdanpanah, B. Ubiquitin C-terminal hydrolase-L1 potentiates cancer chemosensitivity by stabilizing NOXA. Cell Rep. 2013, 3, 881–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisan, T.; Coppotelli, G.; Dryselius, R.; Masucci, M.G. Ubiquitin C-terminal hydrolase-L1 interacts with adhesion complexes and promotes cell migration, survival, and anchorage independent growth. FASEB J. 2012, 26, 5060–5070. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Li, L.; Yin, X.; Yuan, C.; Tan, C.; Su, X.; Xiong, L.; Putti, T.C.; Oberst, M.; Kelly, K. The ubiquitin peptidase UCHL1 induces G0/G1 cell cycle arrest and apoptosis through stabilizing p53 and is frequently silenced in breast cancer. PLoS ONE 2012, 7, e29783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costes, S.; Gurlo, T.; Rivera, J.F.; Butler, P.C. UCHL1 deficiency exacerbates human islet amyloid polypeptide toxicity in β-cells: Evidence of interplay between the ubiquitin/proteasome system and autophagy. Autophagy 2014, 10, 1004–1014. [Google Scholar] [CrossRef]
- Jin, C.; Yu, W.; Lou, X.; Zhou, F.; Han, X.; Zhao, N.; Lin, B. UCHL1 is a putative tumor suppressor in ovarian cancer cells and contributes to cisplatin resistance. J. Cancer 2013, 4, 662. [Google Scholar] [CrossRef]
- Wu, H.; Ying, W.; Wang, W.; Li, W.; Feng, X. HIF1α and HIF2α mediated UCHL1 upregulation in hypoxia-induced neuronal injury following neuronal hypoxic ischemic encephalopathy. Int. J. Clin. Exp. Pathol. 2016, 9, 2677–2685. [Google Scholar]
- Wang, S.; Juan, J.; Zhang, Z.; Du, Y.; Xu, Y.; Tong, J.; Cao, B.; Moran, M.F.; Zeng, Y.; Mao, X. Inhibition of the deubiquitinase USP5 leads to c-Maf protein degradation and myeloma cell apoptosis. Cell Death Dis. 2017, 8, e3058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaistha, B.P.; Krattenmacher, A.; Fredebohm, J.; Schmidt, H.; Behrens, D.; Widder, M.; Hackert, T.; Strobel, O.; Hoheisel, J.D.; Gress, T.M. The deubiquitinating enzyme USP5 promotes pancreatic cancer via modulating cell cycle regulators. Oncotarget 2017, 8, 66215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potu, H.; Kandarpa, M.; Peterson, L.F.; Donato, N.J.; Talpaz, M. Tumor necrosis factor related apoptosis inducing ligand (TRAIL) regulates deubiquitinase USP5 in tumor cells. Oncotarget 2019, 10, 5745. [Google Scholar] [CrossRef]
- Fan, X.; Huang, Q.; Ye, X.; Lin, Y.; Chen, Y.; Lin, X.; Qu, J. Drosophila USP5 controls the activation of apoptosis and the Jun N-terminal kinase pathway during eye development. PLoS ONE 2014, 9, e92250. [Google Scholar]
- Liu, Y.; Wang, W.M.; Zou, L.Y.; Li, L.; Feng, L.; Pan, M.Z.; Lv, M.Y.; Cao, Y.; Wang, H.; Kung, H.F. Ubiquitin specific peptidase 5 mediates Histidine-rich protein Hpn induced cell apoptosis in hepatocellular carcinoma through P14-P53 signaling. Proteomics 2017, 17, 1600350. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.-M.; Lu, Y.-F.; Feng, L.; Li, L.; Pan, M.-Z.; Sun, Y.; Suen, C.-W.; Guo, W.; Pang, J.-X. Usp5 functions as an oncogene for stimulating tumorigenesis in hepatocellular carcinoma. Oncotarget 2017, 8, 50655. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhang, C.; Chu, M.; Fan, Y.; Wei, L.; Li, Z.; Yao, Y.; Zhuang, W. miR-125a suppresses malignancy of multiple myeloma by reducing the deubiquitinase USP5. J. Cell. Biochem. 2020, 121, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Vashistha, V.; Bhardwaj, S.; Yadav, B.K.; Yadav, A.K. Depleting deubiquitinating enzymes promotes apoptosis in glioma cell line via RNA binding proteins SF2/ASF1. Biochem. Biophys. Rep. 2020, 24, 100846. [Google Scholar] [PubMed]
- Jing, X.; Chen, Y.; Chen, Y.; Shi, G.; Lv, S.; Cheng, N.; Feng, C.; Xin, Z.; Zhang, L.; Wu, J. Down-regulation of USP8 Inhibits Cholangiocarcinoma Cell Proliferation and Invasion. Cancer Manag. Res. 2020, 12, 2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, M.; Lee, E.; Seong, D.; Seo, J.; Kim, J.; Grootjans, S.; Kim, S.; Vandenabeele, P.; Song, J. USP8 suppresses death receptor-mediated apoptosis by enhancing FLIP L stability. Oncogene 2017, 36, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Le Clorennec, C.; Lazrek, Y.; Dubreuil, O.; Sampaio, C.; Larbouret, C.; Lanotte, R.; Poul, M.-A.; Barret, J.-M.; Prost, J.-F.; Pèlegrin, A. ITCH-dependent proteasomal degradation of c-FLIP induced by the anti-HER3 antibody 9F7-F11 promotes DR5/caspase 8-mediated apoptosis of tumor cells. Cell Commun. Signal. 2019, 17, 106. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Zhang, M.; Cui, R.; Tili, E.; Kim, T.; Lee, T.J.; Peng, Y.; Croce, C. A negative feedback regulatory loop between miR-138 and TP53 is mediated by USP10. Oncotarget 2019, 10, 6288. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Shi, Y.; Xue, J.; Miao, R.; Huang, S.; Wang, T.; Wu, J.; Fu, M.; Wu, Z.H. USP10 inhibits genotoxic NF-κB activation by MCPIP1-facilitated deubiquitination of NEMO. EMBO J. 2013, 32, 3206–3219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, C.; Tong, D.; Xiang, S.; Williams, K.; Bai, W.; Li, G.-M.; Bepler, G.; Zhang, X. Ubiquitin-specific peptidase 10 (USP10) deubiquitinates and stabilizes MutS homolog 2 (MSH2) to regulate cellular sensitivity to DNA damage. J. Biol. Chem. 2016, 291, 10783–10791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Kitaura, H.; Kakita, A.; Kakihana, T.; Katsuragi, Y.; Nameta, M.; Zhang, L.; Iwakura, Y.; Nawa, H.; Higuchi, M. USP10 is a driver of ubiquitinated protein aggregation and aggresome formation to inhibit apoptosis. IScience 2018, 9, 433–450. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Higuchi, M.; Matsuki, H.; Yoshita, M.; Ohsawa, T.; Oie, M.; Fujii, M. Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol. Cell. Biol. 2013, 33, 815–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Higuchi, M.; Makokha, G.N.; Matsuki, H.; Yoshita, M.; Tanaka, Y.; Fujii, M. HTLV-1 Tax oncoprotein stimulates ROS production and apoptosis in T cells by interacting with USP10. Blood J. Am. Soc. Hematol. 2013, 122, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wu, D.; Xu, Z. USP10 protects against cerebral ischemia injury by suppressing inflammation and apoptosis through the inhibition of TAK1 signaling. Biochem. Biophys. Res. Commun. 2019, 516, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Jiangqiao, Z.; Tianyu, W.; Zhongbao, C.; Long, Z.; Jilin, Z.; Xiaoxiong, M.; Tao, Q. Ubiquitin-specific peptidase 10 protects against hepatic ischaemic/reperfusion injury via TAK1 signalling. Front. Immunol. 2020, 11, 506275. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Hang, Y.; Zhang, T.; Tan, L.; Li, S.; Jin, Y. USP10 promotes proliferation and migration and inhibits apoptosis of endometrial stromal cells in endometriosis through activating the Raf-1/MEK/ERK pathway. Am. J. Physiol. Cell Physiol. 2018, 315, C863–C872. [Google Scholar] [CrossRef]
- Higuchi, M.; Kawamura, H.; Matsuki, H.; Hara, T.; Takahashi, M.; Saito, S.; Saito, K.; Jiang, S.; Naito, M.; Kiyonari, H. USP10 is an essential deubiquitinase for hematopoiesis and inhibits apoptosis of long-term hematopoietic stem cells. Stem Cell Rep. 2016, 7, 1116–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Lee, D.M.; Woo, H.G.; Kim, K.D.; Lee, H.J.; Kwon, Y.-J.; Choi, K.S. RNAi screening-based identification of USP10 as a novel regulator of paraptosis. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Yan, G.; Song, X.; Xie, L.; Zhou, Y.; Li, J.; Hu, X.; Li, Z.; Hu, J.; Zhang, Y. Deubiquitylation and stabilization of p21 by USP11 is critical for cell-cycle progression and DNA damage responses. Proc. Natl. Acad. Sci. USA 2018, 115, 4678–4683. [Google Scholar] [CrossRef] [Green Version]
- Ting, X.; Xia, L.; Yang, J.; He, L.; Si, W.; Shang, Y.; Sun, L. USP11 acts as a histone deubiquitinase functioning in chromatin reorganization during DNA repair. Nucleic Acids Res. 2019, 47, 9721–9740. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, A.R.; Apgar, S.; Dolios, G.; Wang, R.; Aaronson, S.A. BRCA2 is ubiquitinated in vivo and interacts with USP11, a deubiquitinating enzyme that exhibits prosurvival function in the cellular response to DNA damage. Mol. Cell. Biol. 2004, 24, 7444–7455. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Luo, A.; Shrivastava, I.; He, M.; Huang, Y.; Bahar, I.; Liu, Z.; Wan, Y. Regulation of XIAP turnover reveals a role for USP11 in promotion of tumorigenesis. EBioMedicine 2017, 15, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Seong, D.; Seo, J.; Jeong, M.; Lee, H.; Song, J. USP11-dependent selective cIAP2 deubiquitylation and stabilization determine sensitivity to Smac mimetics. Cell Death Differ. 2015, 22, 1463–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Li, X.; Chen, J.; Zhao, J.; Wang, J.; Ji, Y.; Shen, Y.; Han, L.; Shi, J.; Zhang, D. USP11, deubiquitinating enzyme, associated with neuronal apoptosis following intracerebral hemorrhage. J. Mol. Neurosci. 2016, 58, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hong, X.; Wei, Z.; Xie, M.; Li, W.; Liu, G.; Guo, H.; Yang, J.; Wei, W.; Zhang, S. Ubiquitination of the HPV oncoprotein E6 is critical for E6/E6AP-mediated p53 degradation. Front. Microbiol. 2019, 10, 2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.S.; Coppens, I.; Saorin, A.; Brady, N.R.; Hamacher-Brady, A. Endolysosomal Targeting of Mitochondria Is Integral to BAX-Mediated Mitochondrial Permeabilization during Apoptosis Signaling. Dev. Cell 2020, 53, 627–645. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.-Y.; Yao, M.; Yang, Z.; Yang, L.; Liu, X.-J.; Yu, J.; Ma, Y.; Zhang, N.; Zhang, X.-Y.; Liu, M.-H. De-regulated STAT5A/miR-202-5p/USP15/Caspase-6 regulatory axis suppresses CML cell apoptosis and contributes to Imatinib resistance. J. Exp. Clin. Cancer Res. 2020, 39, 1–14. [Google Scholar] [CrossRef]
- Zou, Q.; Jin, J.; Hu, H.; Li, H.S.; Romano, S.; Xiao, Y.; Nakaya, M.; Zhou, X.; Cheng, X.; Yang, P. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol. 2014, 15, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Jiang, H.; Du, J.; Li, L.; Li, R.; Lu, J.; Fu, W.; Hou, J. USP15 inhibits multiple myeloma cell apoptosis through activating a feedback loop with the transcription factor NF-κBp65. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Bao, G.; Liu, F. Inhibition of USP15 Prevent Glutamate-Induced Oxidative Damage by Activating Nrf2/HO-1 Signaling Pathway in HT22 Cells. Cell. Mol. Neurobiol. 2020, 40, 999–1020. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Fernandez, A.; Salio, M.; Partridge, T.; Chen, J.; Vere, G.; Greenwood, H.; Olie, C.S.; Damianou, A.; Scott, H.C.; Diaz-Saez, L. Deep analysis of the USP18-dependent ISGylome and proteome unveils important roles for USP18 in tumour cell antigenicity and radiosensitivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Santin, I.; Moore, F.; Grieco, F.A.; Marchetti, P.; Brancolini, C.; Eizirik, D.L. USP18 is a key regulator of the interferon-driven gene network modulating pancreatic beta cell inflammation and apoptosis. Cell Death Dis. 2012, 3, e419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potu, H.; Sgorbissa, A.; Brancolini, C. Identification of USP18 as an Important Regulator of the Susceptibility to IFN-α and Drug-Induced Apoptosis. Cancer Res. 2010, 70, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Manini, I.; Sgorbissa, A.; Potu, H.; Tomasella, A.; Brancolini, C. The DeISGylase USP18 limits TRAIL-induced apoptosis through the regulation of TRAIL levels: Cellular levels of TRAIL influences responsiveness to TRAIL-induced apoptosis. Cancer Biol. Ther. 2013, 14, 1158–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagenais-Lussier, X.; Loucif, H.; Cadorel, H.; Blumberger, J.; Isnard, S.; Bego, M.G.; Cohen, É.A.; Routy, J.-P.; van Grevenynghe, J.; Montreal Primary Infection Study Group. USP18 is a significant driver of memory CD4 T-cell reduced viability caused by type I IFN signaling during primary HIV-1 infection. PLoS Pathog. 2019, 15, e1008060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, W.; Guo, Q.; Zhu, C.; Song, Y.; Feng, H.; Cao, Y.; Du, M.; Chen, H. USP18 promotes cell proliferation and suppressed apoptosis in cervical cancer cells via activating AKT signaling pathway. BMC Cancer 2020, 20, 741. [Google Scholar] [CrossRef] [PubMed]
- Duex, J.E.; Comeau, L.; Sorkin, A.; Purow, B.; Kefas, B. Usp18 regulates epidermal growth factor (EGF) receptor expression and cancer cell survival via microRNA-7. J. Biol. Chem. 2011, 286, 25377–25386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Liu, T.; Jiang, X.; Guo, C.; Liu, A.; Xiao, X. Downregulation of USP18 inhibits growth and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma cells by suppressing BCL2L1. Exp. Cell Res. 2017, 358, 315–322. [Google Scholar] [CrossRef]
- Lai, K.P.; Cheung, A.H.Y.; Tse, W.K.F. Deubiquitinase Usp18 prevents cellular apoptosis from oxidative stress in liver cells. Cell Biol. Int. 2017, 41, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wang, K.; Tang, P.; Chen, S.; Liu, T.; Lei, J.; Yuan, R.; Hu, Z.; Li, W.; Yu, X. Deubiquitinase USP18 promotes the progression of pancreatic cancer via enhancing the Notch1-c-Myc axis. Aging 2020, 12, 19273. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Kim, M.; Seo, D.; Park, J.S.; Lee, J.; Lee, J.; Park, S.H. The Deubiquitinating Enzyme USP20 Regulates the TNFα-Induced NF-κB Signaling Pathway through Stabilization of p62. Int. J. Mol. Sci. 2020, 21, 3116. [Google Scholar] [CrossRef]
- Kim, J.H.; Seo, D.; Kim, S.J.; Choi, D.W.; Park, J.S.; Ha, J.; Choi, J.; Lee, J.H.; Jung, S.M.; Seo, K.W. The deubiquitinating enzyme USP20 stabilizes ULK1 and promotes autophagy initiation. EMBO Rep. 2018, 19, e44378. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, N.; Zhao, Y.; Zhu, X.; Wang, C.; Liu, Q.; Gao, C.; Zhao, X.; Li, J. Oncogenic USP22 supports gastric cancer growth and metastasis by activating c-Myc/NAMPT/SIRT1-dependent FOXO1 and YAP signaling. Aging 2019, 11, 9643. [Google Scholar] [CrossRef]
- Lin, Z.; Yang, H.; Kong, Q.; Li, J.; Lee, S.-M.; Gao, B.; Dong, H.; Wei, J.; Song, J.; Zhang, D.D. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell 2012, 46, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Liu, P.; Sun, D.; Chen, Z.; Hu, J.; Peng, S.; Liu, Y. USP22 down-regulation facilitates human retinoblastoma cell aging and apoptosis via inhibiting TERT/P53 pathway. Eur. Rev. Med. Pharm. Sci. 2017, 21, 2785–2792. [Google Scholar]
- Ling, S.; Li, J.; Shan, Q.; Dai, H.; Lu, D.; Wen, X.; Song, P.; Xie, H.; Zhou, L.; Liu, J. USP22 mediates the multidrug resistance of hepatocellular carcinoma via the SIRT1/AKT/MRP1 signaling pathway. Mol. Oncol. 2017, 11, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Ning, Z.; Lu, C.; Gao, W.; Liang, J.; Yan, Q.; Tan, G.; Liu, J. USP22 induces cisplatin resistance in lung adenocarcinoma by regulating γH2AX-mediated DNA damage repair and Ku70/bax-mediated apoptosis. Front. Pharmacol. 2017, 8, 274. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.X.; Ning, Z.; Gao, W.; Ling, J.; Wang, A.; Luo, H.F.; Liang, Y.; Yan, Q.; Wang, Z.Y. Ubiquitin-specific protease 22-induced autophagy is correlated with poor prognosis of pancreatic cancer. Oncol. Rep. 2014, 32, 2726–2734. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.H.; Yu, Y.; Du, C.; Fu, H.; Wang, J.; Tian, Y. RNA interference-mediated USP22 gene silencing promotes human brain glioma apoptosis and induces cell cycle arrest. Oncol. Lett. 2013, 5, 1290–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosinsky, R.L.; Helms, M.; Zerche, M.; Wohn, L.; Dyas, A.; Prokakis, E.; Kazerouni, Z.B.; Bedi, U.; Wegwitz, F.; Johnsen, S.A. USP22-dependent HSP90AB1 expression promotes resistance to HSP90 inhibition in mammary and colorectal cancer. Cell Death Dis. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Roedig, J.; Kowald, L.; Juretschke, T.; Karlowitz, R.; Ahangarian Abhari, B.; Roedig, H.; Fulda, S.; Beli, P.; van Wijk, S.J. USP22 controls necroptosis by regulating receptor-interacting protein kinase 3 ubiquitination. EMBO Rep. 2020, 22, e50163. [Google Scholar] [PubMed]
- Shi, J.-x.; Wang, Q.-j.; Li, H.; Huang, Q. Silencing of USP22 suppresses high glucose-induced apoptosis, ROS production and inflammation in podocytes. Mol. Biosyst. 2016, 12, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israël, A.; Wallach, D.; Courtois, G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Brummelkamp, T.R.; Nijman, S.M.; Dirac, A.M.; Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 2003, 424, 797–801. [Google Scholar] [CrossRef]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef]
- Schlicher, L.; Wissler, M.; Preiss, F.; Brauns-Schubert, P.; Jakob, C.; Dumit, V.; Borner, C.; Dengjel, J.; Maurer, U. SPATA 2 promotes CYLD activity and regulates TNF-induced NF-κB signaling and cell death. EMBO Rep. 2016, 17, 1485–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, R.L.; Sundberg, J.P.; Sun, S.-C.; Gillespie, V.L.; Heeger, P.; Xiong, H.; Lira, S.A.; Ting, A.T. Immune dysregulation in SHARPIN-deficient mice is dependent on CYLD-mediated cell death. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Moquin, D.M.; McQuade, T.; Chan, F.K.-M. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE 2013, 8, e76841. [Google Scholar] [CrossRef]
- Legarda, D.; Justus, S.J.; Ang, R.L.; Rikhi, N.; Li, W.; Moran, T.M.; Zhang, J.; Mizoguchi, E.; Zelic, M.; Kelliher, M.A. CYLD proteolysis protects macrophages from TNF-mediated auto-necroptosis induced by LPS and licensed by type I IFN. Cell Rep. 2016, 15, 2449–2461. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Liu, J.; Pan, X.; Peng, C.; Xiong, B.; Feng, M.; Yang, X. miR-454 promotes survival and induces oxaliplatin resistance in gastric carcinoma cells by targeting CYLD. Exp. Ther. Med. 2020, 19, 3604–3610. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, L.; Luo, W.; Zhou, X.; Chen, Y.; Yang, K.; Liao, J.; Wu, D.; Cai, L. CYLD Promotes Apoptosis of Nasopharyngeal Carcinoma Cells by Regulating NDRG1. Cancer Manag. Res. 2020, 12, 10639. [Google Scholar] [CrossRef]
- Qi, L.; Zang, H.; Wu, W.; Nagarkatti, P.; Nagarkatti, M.; Liu, Q.; Robbins, J.; Wang, X.; Cui, T. CYLD exaggerates pressure overload-induced cardiomyopathy via suppressing autolysosome efflux in cardiomyocytes. J. Mol. Cell. Cardiol. 2020, 145, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Priem, D.; Devos, M.; Druwé, S.; Martens, A.; Slowicka, K.; Ting, A.T.; Pasparakis, M.; Declerq, W.; Vandenabeele, P.; van Loo, G. A20 protects cells from TNF-induced apoptosis through linear ubiquitin-dependent and-independent mechanisms. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Makarov, R.; Brenji, S.; Schneider, A.T.; Hooiveld, G.J.; Luedde, T.; Leverkus, M.; Yazdi, A.S.; Panayotova-Dimitrova, D. A20 Promotes Ripoptosome Formation and TNF-Induced Apoptosis via cIAPs Regulation and NIK Stabilization in Keratinocytes. Cells 2020, 9, 351. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Mooney, E.C.; Xia, X.-J.; Gupta, N.; Sahingur, S.E. A20 Restricts Inflammatory Response and Desensitizes Gingival Keratinocytes to Apoptosis. Front. Immunol. 2020, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, B.; Kong, X.; Hao, J.; Ye, Y.; Yang, K.; Xu, T.; Zhang, F. A20 protects neuronal apoptosis stimulated by lipopolysaccharide-induced microglial exosomes. Neurosci. Lett. 2019, 712, 134480. [Google Scholar] [CrossRef] [PubMed]
- Bellail, A.C.; Olson, J.J.; Yang, X.; Chen, Z.J.; Hao, C. A20 ubiquitin ligase–mediated polyubiquitination of RIP1 inhibits caspase-8 cleavage and TRAIL-induced apoptosis in glioblastoma. Cancer Discov. 2012, 2, 140–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Jing, N.; Shen, A.; Guo, F.; Song, Y.; Pan, M.; Ma, X.; Zhao, L.; Zhang, H.; Wu, L. MiR-21-5p in macrophage-derived extracellular vesicles affects podocyte pyroptosis in diabetic nephropathy by regulating A20. J. Endocrinol. Investig. 2020. [Google Scholar] [CrossRef]
- Slowicka, K.; Serramito-Gómez, I.; Boada-Romero, E.; Martens, A.; Sze, M.; Petta, I.; Vikkula, H.K.; De Rycke, R.; Parthoens, E.; Lippens, S. Physical and functional interaction between A20 and ATG16L1-WD40 domain in the control of intestinal homeostasis. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiil, B.K.; Damgaard, R.B.; Wagner, S.A.; Keusekotten, K.; Fritsch, M.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Komander, D.; Gyrd-Hansen, M. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol. Cell 2013, 50, 818–830. [Google Scholar] [CrossRef] [Green Version]
- Fiil, B.K.; Gyrd-Hansen, M. OTULIN deficiency causes auto-inflammatory syndrome. Cell Res. 2016, 26, 1176–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damgaard, R.B.; Elliott, P.R.; Swatek, K.N.; Maher, E.R.; Stepensky, P.; Elpeleg, O.; Komander, D.; Berkun, Y. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med. 2019, 11, e9324. [Google Scholar] [CrossRef] [PubMed]
- Heger, K.; Wickliffe, K.E.; Ndoja, A.; Zhang, J.; Murthy, A.; Dugger, D.L.; Maltzman, A.; e Melo, F.D.S.; Hung, J.; Zeng, Y. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 2018, 559, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.; Saleh, M. Post-translational modification of OTULIN regulates ubiquitin dynamics and cell death. Cell Rep. 2019, 29, 3652–3663.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damgaard, R.B.; Jolin, H.E.; Allison, M.E.; Davies, S.E.; Titheradge, H.L.; McKenzie, A.N.; Komander, D. OTULIN protects the liver against cell death, inflammation, fibrosis, and cancer. Cell Death Differ. 2020, 27, 1457–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verboom, L.; Martens, A.; Priem, D.; Hoste, E.; Sze, M.; Vikkula, H.; Van Hove, L.; Voet, S.; Roels, J.; Maelfait, J. OTULIN prevents liver inflammation and hepatocellular carcinoma by inhibiting FADD-and RIPK1 kinase-mediated hepatocyte apoptosis. Cell Rep. 2020, 30, 2237–2247.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.X.; Challagundla, K.B.; Dai, M.S. Positive regulation of p53 stability and activity by the deubiquitinating enzyme Otubain 1. EMBO J. 2012, 31, 576–592. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.-G.; Li, Y.; Sun, X.-X.; Dai, M.-S. Otub1 stabilizes MDMX and promotes its proapoptotic function at the mitochondria. Oncotarget 2017, 8, 11053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunarathna, U.; Kongsema, M.; Zona, S.; Gong, C.; Cabrera, E.; Gomes, A.R.; Man, E.P.; Khongkow, P.; Tsang, J.W.; Khoo, U.-S. OTUB1 inhibits the ubiquitination and degradation of FOXM1 in breast cancer and epirubicin resistance. Oncogene 2016, 35, 1433–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Q.; Chen, J.; Li, X.; Xu, X.; Zhang, N.; Zhou, A.; Zhou, B.; Lu, Q.; Chen, Z. Expression of OTUB1 in hepatocellular carcinoma and its effects on HCC cell migration and invasion. Acta Biochim. Biophys. Sin. 2017, 49, 680–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Li, A.; Shen, J.; Cao, M.; Ning, X.; Yuan, D.; Ji, Y.; Wang, H.; Ke, K. OTUB1 attenuates neuronal apoptosis after intracerebral hemorrhage. Mol. Cell. Biochem. 2016, 422, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, T.; Niessen, K.; De Almagro, M.C.; Izrael-Tomasevic, A.; Fedorova, A.V.; Varfolomeev, E.; Arnott, D.; Deshayes, K.; Kirkpatrick, D.S.; Vucic, D. OTUB1 modulates c-IAP1 stability to regulate signalling pathways. EMBO J. 2013, 32, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Jiang, L.; Tavana, O.; Gu, W. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. 2019, 79, 1913–1924. [Google Scholar] [CrossRef]
- Tian, Z.; D’Arcy, P.; Wang, X.; Ray, A.; Tai, Y.-T.; Hu, Y.; Carrasco, R.D.; Richardson, P.; Linder, S.; Chauhan, D. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood J. Am. Soc. Hematol. 2014, 123, 706–716. [Google Scholar] [CrossRef]
- Wang, X.; Mazurkiewicz, M.; Hillert, E.-K.; Olofsson, M.H.; Pierrou, S.; Hillertz, P.; Gullbo, J.; Selvaraju, K.; Paulus, A.; Akhtar, S. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Paulus, A.; Akhtar, S.; Caulfield, T.; Samuel, K.; Yousaf, H.; Bashir, Y.; Paulus, S.; Tran, D.; Hudec, R.; Cogen, D. Coinhibition of the deubiquitinating enzymes, USP14 and UCHL5, with VLX1570 is lethal to ibrutinib-or bortezomib-resistant Waldenstrom macroglobulinemia tumor cells. Blood Cancer J. 2016, 6, e492. [Google Scholar] [CrossRef]
- Chitta, K.; Paulus, A.; Akhtar, S.; Blake, M.K.K.; Caulfield, T.R.; Novak, A.J.; Ansell, S.M.; Advani, P.; Ailawadhi, S.; Sher, T. Targeted inhibition of the deubiquitinating enzymes, USP 14 and UCHL 5, induces proteotoxic stress and apoptosis in W aldenström macroglobulinaemia tumour cells. Br. J. Haematol. 2015, 169, 377–390. [Google Scholar] [CrossRef]
- Jiang, L.; Sun, Y.; Wang, J.; He, Q.; Chen, X.; Lan, X.; Chen, J.; Dou, Q.P.; Shi, X.; Liu, J. Proteasomal cysteine deubiquitinase inhibitor b-AP15 suppresses migration and induces apoptosis in diffuse large B cell lymphoma. J. Exp. Clin. Cancer Res. 2019, 38, 1–14. [Google Scholar] [CrossRef]
- Sha, B.; Chen, X.; Wu, H.; Li, M.; Shi, J.; Wang, L.; Liu, X.; Chen, P.; Hu, T.; Li, P. Deubiquitylatinase inhibitor b-AP15 induces c-Myc-Noxa-mediated apoptosis in esophageal squamous cell carcinoma. Apoptosis 2019, 24, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, X.; Xu, D.; Yang, L.; Yang, Z.; Yang, Q.; Yan, D.; Zhang, P.; Feng, D.; Liu, J. Autophagy Induced by Proteasomal DUB inhibitor NiPT Restricts NiPT-mediated cancer cell death. Front. Oncol. 2020, 10, 348. [Google Scholar] [CrossRef] [Green Version]
- Fukui, S.; Nagasaka, K.; Miyagawa, Y.; Kikuchi-Koike, R.; Kawata, Y.; Kanda, R.; Ichinose, T.; Sugihara, T.; Hiraike, H.; Wada-Hiraike, O. The proteasome deubiquitinase inhibitor bAP15 downregulates TGF-β/Smad signaling and induces apoptosis via UCHL5 inhibition in ovarian cancer. Oncotarget 2019, 10, 5932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Liu, Z.S.; Wang, X.; Ouyang, L. Ubiquitin C-Terminal Hydrolase L5 (UCHL5) Accelerates the Growth of Endometrial Cancer via Activating the Wnt/β-Catenin Signaling Pathway. Front. Oncol. 2020, 10, 865. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Zhou, J.; Zhou, Y.; Xie, Y.; Jiang, Y.; Wu, J.; Luo, Z.; Liu, G.; Yin, L.; Zhang, X.-L. Mycobacterial EST12 activates a RACK1–NLRP3–gasdermin D pyroptosis–IL-1β immune pathway. Sci. Adv. 2020, 6, eaba4733. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-S.; Wang, X.-F.; Zhang, Y.-J.; Luo, P.; Long, H.-D.; Li, L.; Yang, H.-Q.; Xie, R.-T.; Jia, C.-Y.; Lu, G.-X. Inhibition of USP14 Deubiquitinating Activity as a Potential Therapy for Tumors with p53 Deficiency. Mol. Ther. Oncolytics 2020, 16, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wang, J.; Yuan, X.; Yang, S.; Xu, X.; Li, K.; He, Y.; Wei, L.; Zhang, J.; Tian, Y. IU1 suppresses proliferation of cervical cancer cells through MDM2 degradation. Int. J. Biol. Sci. 2020, 16, 2951. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Huang, C.; Liao, Y.; Liu, Y.; He, J.; Guo, Z.; Jiang, L.; Wang, X.; Liu, J.; Huang, H. Inhibition of USP14 enhances the sensitivity of breast cancer to enzalutamide. J. Exp. Clin. Cancer Res. 2019, 38, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Xia, X.; Liu, N.; Cai, J.; Guo, Z.; Li, Y.; Jiang, L.; Dou, Q.P.; Tang, D.; Huang, H. Growth arrest and apoptosis induction in androgen receptor-positive human breast cancer cells by inhibition of USP14-mediated androgen receptor deubiquitination. Oncogene 2018, 37, 1896–1910. [Google Scholar] [CrossRef] [PubMed]
- Didier, R.; Mallavialle, A.; Jouira, R.B.; Domdom, M.A.; Tichet, M.; Auberger, P.; Luciano, F.; Ohanna, M.; Tartare-Deckert, S.; Deckert, M. Targeting the proteasome-associated deubiquitinating enzyme USP14 impairs melanoma cell survival and overcomes resistance to MAPK-targeting therapies. Mol. Cancer Ther. 2018, 17, 1416–1429. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Ma, G.; Liu, G.; Li, B.; Li, H.; Hao, X.; Liu, L. USP 14 as a novel prognostic marker promotes cisplatin resistance via Akt/ERK signaling pathways in gastric cancer. Cancer Med. 2018, 7, 5577–5588. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Zhang, C.; Bai, C.; Han, Y.P.; Li, Q. MiR-4782-3p inhibited non-small cell lung cancer growth via USP14. Cell. Physiol. Biochem. 2014, 33, 457–467. [Google Scholar] [CrossRef]
- Xu, J.; Deng, Y.; Wang, Y.; Sun, X.; Chen, S.; Fu, G. SPAG5-AS1 inhibited autophagy and aggravated apoptosis of podocytes via SPAG5/AKT/mTOR pathway. Cell Prolif. 2020, 53, e12738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.H.; Kwak, M.; Lee, T.H.; Park, M.-S.; Jeong, I.-H.; Kim, M.J.; Jin, J.-O.; Lee, P.C.-W. USP14 inhibition regulates tumorigenesis by inducing autophagy in lung cancer in vitro. Int. J. Mol. Sci. 2019, 20, 5300. [Google Scholar] [CrossRef] [Green Version]
- Moghadami, A.A.; Aboutalebi Vand Beilankouhi, E.; Kalantary-Charvadeh, A.; Hamzavi, M.; Mosayyebi, B.; Sedghi, H.; Ghorbani Haghjo, A.; Nazari Soltan Ahmad, S. Inhibition of USP14 induces ER stress-mediated autophagy without apoptosis in lung cancer cell line A549. Cell Stress Chaperones 2020, 25, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Ma, R.; Yang, X.; Pang, S. The deubiquitinating enzyme USP14 regulates leukemic chemotherapy drugs-induced cell apoptosis by suppressing ubiquitination of Aurora kinase B. Cell. Physiol. Biochem. 2017, 42, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, R.I.; Pulver, T.; Heilmann, W.; Mooneyham, A.; Mullany, S.; Zhao, X.; Shahi, M.; Richter, J.; Klein, M.; Chen, L. USP14 is a predictor of recurrence in endometrial cancer and a molecular target for endometrial cancer treatment. Oncotarget 2016, 7, 30962. [Google Scholar] [CrossRef]
- Dong, Y.; Hakimi, M.-A.; Chen, X.; Kumaraswamy, E.; Cooch, N.S.; Godwin, A.K.; Shiekhattar, R. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol. Cell 2003, 12, 1087–1099. [Google Scholar] [CrossRef]
- Chen, X.; Arciero, C.A.; Wang, C.; Broccoli, D.; Godwin, A.K. BRCC36 is essential for ionizing radiation–induced BRCA1 phosphorylation and nuclear foci formation. Cancer Res. 2006, 66, 5039–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Kumari, B.; Yadav, U.C. Regulation of oxidized LDL-induced inflammatory process through NLRP3 inflammasome activation by the deubiquitinating enzyme BRCC36. Inflamm. Res. 2019, 68, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, T.; Huang, B.; Luo, M.; Chen, Z.; Zhao, Z.; Wang, J.; Leung, D.; Yang, X.; Chan, K.W. Excessive deubiquitination of NLRP3-R779C variant contributes to very-early-onset inflammatory bowel disease development. J. Allergy Clin. Immunol. 2020, 147, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Rao, Z.; Chen, X.; Wu, J.X.; Xiao, M.; Zhang, J.; Zhang, H.; Yang, S.; Wang, X.; Fang, L.; Wang, B. Vitamin D receptor inhibits NLRP3 activation by impeding its deubiquitination mediated by BRCC3. Front. Immunol. 2019, 10, 2783. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, H.; Bi, X.; Li, Y.; Huang, Z. Targeting the deubiquitinase STAMBPL1 triggers apoptosis in prostate cancer cells by promoting XIAP degradation. Cancer Lett. 2019, 456, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Shahriyar, S.A.; Woo, S.M.; Seo, S.U.; Min, K.-J.; Kwon, T.K. Cepharanthine enhances TRAIL-mediated apoptosis through STAMBPL1-mediated downregulation of survivin expression in renal carcinoma cells. Int. J. Mol. Sci. 2018, 19, 3280. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.J.; Qian, J.; Jin, X.; Li, J.; Guo, C.X.; Yue, X.C. STAMBPL1 knockdown has antitumour effects on gastric cancer biological activities. Oncol. Lett. 2019, 18, 4421–4428. [Google Scholar] [CrossRef] [Green Version]
- Ning, F.; Xin, H.; Liu, J.; Lv, C.; Xin, X.; Wang, M.; Wang, Y.; Zhang, W.; Zhang, X. Structure and function of USP5: Insight into physiological and pathophysiological roles. Pharmacol. Res. 2019, 157, 104557. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Chakraborty, D.; Basu, M.; Ghosh, M.K. Emerging insights into HAUSP (USP7) in physiology, cancer and other diseases. Signal Transduct. Target. Ther. 2018, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Kon, N.; Kobayashi, Y.; Li, M.; Brooks, C.; Ludwig, T.; Gu, W. Inactivation of HAUSP in vivo modulates p53 function. Oncogene 2010, 29, 1270–1279. [Google Scholar] [CrossRef] [Green Version]
- Dufner, A.; Knobeloch, K.-P. Ubiquitin-specific protease 8 (USP8/UBPy): A prototypic multidomain deubiquitinating enzyme with pleiotropic functions. Biochem. Soc. Trans. 2019, 47, 1867–1879. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, U.; Neizer-Ashun, F.; Mukherjee, P.; Bhattacharya, R. When the chains do not break: The role of USP10 in physiology and pathology. Cell Death Dis. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-K.; Chang, Y.-T.; Korinek, M.; Chen, Y.-T.; Yang, Y.-T.; Leu, S.; Lin, I.; Tang, C.-J.; Chiu, C.-C. The regulations of deubiquitinase USP15 and its pathophysiological mechanisms in diseases. Int. J. Mol. Sci. 2017, 18, 483. [Google Scholar] [CrossRef]
- Kang, J.A.; Jeon, Y.J. Emerging roles of USP18: From biology to pathophysiology. Int. J. Mol. Sci. 2020, 21, 6825. [Google Scholar] [CrossRef]
- Wu, C.; Luo, K.; Zhao, F.; Yin, P.; Song, Y.; Deng, M.; Huang, J.; Chen, Y.; Li, L.; Lee, S. USP20 positively regulates tumorigenesis and chemoresistance through β-catenin stabilization. Cell Death Differ. 2018, 25, 1855–1869. [Google Scholar] [CrossRef]
- Shanmugam, I.; Abbas, M.; Ayoub, F.; Mirabal, S.; Bsaili, M.; Caulder, E.K.; Weinstock, D.M.; Tomkinson, A.E.; Hromas, R.; Shaheen, M. Ubiquitin-specific peptidase 20 regulates Rad17 stability, checkpoint kinase 1 phosphorylation and DNA repair by homologous recombination. J. Biol. Chem. 2014, 289, 22739–22748. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.-Y.; Shi, X.-J.; Hu, A.; Wang, J.-Q.; Ding, Y.; Jiang, W.; Sun, M.; Zhao, X.; Luo, J.; Qi, W. Feeding induces cholesterol biosynthesis via the mTORC1–USP20–HMGCR axis. Nature 2020, 588, 479–484. [Google Scholar] [CrossRef]
- Melo-Cardenas, J.; Zhang, Y.; Zhang, D.D.; Fang, D. Ubiquitin-specific peptidase 22 functions and its involvement in disease. Oncotarget 2016, 7, 44848. [Google Scholar] [CrossRef] [Green Version]
- Suenaga, N.; Kuramitsu, M.; Komure, K.; Kanemaru, A.; Takano, K.; Ozeki, K.; Nishimura, Y.; Yoshida, R.; Nakayama, H.; Shinriki, S. Loss of tumor suppressor CYLD expression triggers cisplatin resistance in oral squamous cell carcinoma. Int. J. Mol. Sci. 2019, 20, 5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Ran, Q.; Zhou, Y.; Liu, S.; Zhao, C.; Yu, X.; Zhu, F.; Ji, Y.; Du, Q.; Yang, T. Doxorubicin sensitizes cancer cells to Smac mimetic via synergistic activation of the CYLD/RIPK1/FADD/caspase-8-dependent apoptosis. Apoptosis 2020, 25, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Massoumi, R.; Chmielarska, K.; Hennecke, K.; Pfeifer, A.; Fässler, R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-κB signaling. Cell 2006, 125, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-κ B signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Reyes-Turcu, F.; Licchesi, J.D.; Odenwaelder, P.; Wilkinson, K.D.; Barford, D. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 2009, 10, 466–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Du, F.; Wang, X. TNF-α induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; de Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef] [Green Version]
- Reiley, W.; Zhang, M.; Wu, X.; Granger, E.; Sun, S.-C. Regulation of the deubiquitinating enzyme CYLD by IκB kinase gamma-dependent phosphorylation. Mol. Cell. Biol. 2005, 25, 3886–3895. [Google Scholar] [CrossRef] [Green Version]
- Hutti, J.E.; Shen, R.R.; Abbott, D.W.; Zhou, A.Y.; Sprott, K.M.; Asara, J.M.; Hahn, W.C.; Cantley, L.C. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKε promotes cell transformation. Mol. Cell 2009, 34, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Kalac, M.; Markson, M.; Chan, M.; Brody, J.D.; Bhagat, G.; Ang, R.L.; Legarda, D.; Justus, S.J.; Liu, F. Reversal of CYLD phosphorylation as a novel therapeutic approach for adult T-cell leukemia/lymphoma (ATLL). Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008, 135, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; He, X.; Huang, N.; Yu, J.; Shao, B. A20: A master regulator of arthritis. Arthritis Res. Ther. 2020, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Weinelt, N.; van Wijk, S.J. Ubiquitin-dependent and-independent functions of OTULIN in cell fate control and beyond. Cell Death Differ. 2020, 28, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.R.; McKenzie, A.N.; Komander, D. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell 2016, 166, 1215–1230.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldana, M.; VanderVorst, K.; Berg, A.L.; Lee, H.; Carraway, K.L. Otubain 1: A non-canonical deubiquitinase with an emerging role in cancer. Endocr. Relat. Cancer 2019, 26, R1–R14. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.; Harbour, J.W.; Brugarolas, J.; Bononi, A.; Pagano, I.; Dey, A.; Krausz, T.; Pass, H.I.; Yang, H.; Gaudino, G. Biological mechanisms and clinical significance of BAP1 mutations in human cancer. Cancer Discov. 2020, 10, 1103–1120. [Google Scholar] [CrossRef]
- Luo, H.; Jing, B.; Xia, Y.; Zhang, Y.; Hu, M.; Cai, H.; Tong, Y.; Zhou, L.; Yang, L.; Yang, J. WP1130 reveals USP24 as a novel target in T-cell acute lymphoblastic leukemia. Cancer Cell Int. 2019, 19, 1–14. [Google Scholar] [CrossRef]
- Patel, K.; Ahmed, Z.S.; Huang, X.; Yang, Q.; Ekinci, E.; Neslund-Dudas, C.M.; Mitra, B.; Elnady, F.A.; Ahn, Y.-H.; Yang, H. Discovering proteasomal deubiquitinating enzyme inhibitors for cancer therapy: Lessons from rational design, nature and old drug reposition. Future Med. Chem. 2018, 10, 2087–2108. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.M.; Seo, S.U.; Kubatka, P.; Min, K.-J.; Kwon, T.K. Honokiol Enhances TRAIL-Mediated Apoptosis through STAMBPL1-Induced Survivin and c-FLIP Degradation. Biomolecules 2019, 9, 838. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DUB | Stimulus | RCD | Inhibitory Compound of DUB | Substrate and Molecular Pathway | Associated Physiology and Pathology | Ref. |
|---|---|---|---|---|---|---|
| USP5 | ↓Intrinsic apoptosis | WP1130 | ↑c-Maf | ↑Multiple myeloma | [71] | |
| ↓Intrinsic apoptosis | ↓DNA damage | ↑Pancreatic cancer | [72] | |||
| TRAIL, FasL | ↓Extrinsic apoptosis | G9(+) | *USP5 is cleaved in TRAIL-sensitive cells | ↑Pancreatic cancer, breast cancer | [73] | |
| ↓Intrinsic apoptosis | ↓JNK | ↑Drosophila photoreceptor development | [74] | |||
| Helicobacter pylori | ↓Intrinsic apoptosis | ↓P14ARF, p53 | ↑Hepatocellular carcinoma | [75,76] | ||
| ↓Intrinsic apoptosis | *miR-125a suppresses USP5 expression | ↑Multiple myeloma | [77] | |||
| Bortezomib | ↓Intrinsic apoptosis | ↑hnRNPA1, ↓SF2/ASF1 *SF2/ASF1 suppresses apoptosis in USP5-depleted cells. | ↑Glioma | [78] | ||
| USP7 | Nutlin | ↓Intrinsic apoptosis | HBX41108, P5091, C1, C2 | ↓p53, N-Myc | ↑Colorectal cancer | [36] |
| ↑Intrinsic apoptosis | ↑p53 | ↓Lung cancer | [30] | |||
| Etoposide | ↓Intrinsic apoptosis | HBX41108 | ↑SUV39H1, H3K9me3 | ↑Colorectal cancer | [40] | |
| Doxorubicin, etoposide | ↓Intrinsic apoptosis | P22077 | ↓p53 | ↑Neuroblastoma | [43] | |
| Doxorubicin | ↓Intrinsic apoptosis | P22077 | ↓BAX | ↑Hepatocellular carcinoma | [39] | |
| ↓Intrinsic apoptosis/necrosis | P5091 | ↑Autophagy | ↑Ovarian cancer | [42] | ||
| Senescence | ↓Intrinsic apoptosis | P5091/P22077 | ↓p53 | ↑Senescence-associated secretory phenotype | [33] | |
| ↓Intrinsic apoptosis | P5091/P22077 | ↓ER/ROS stress | ↑Colorectal cancer | [35] | ||
| ↓Intrinsic apoptosis | P5091 | ↑c-Maf, MafB | ↑Multiple myeloma | [29] | ||
| Paclitaxel, docetaxel | ↓Intrinsic apoptosis | P5091 | ↓PLK1 | ↑Prostate cancer | [44] | |
| ↓Intrinsic apoptosis | P217564 | ↑Foxp3, Tip60, UHRF1, DNMT1 | ↑Colorectal cancer, prostate cancer, leukemia | [28] | ||
| Irradiation | ↓Intrinsic apoptosis | HBX19818 | ↑Homologous recombination repair | ↑Leukemia | [41] | |
| ↓Intrinsic apoptosis | P5091/P22077 | ↑GATA1 | ↑Erythroblast differentiation | [31] | ||
| Dexamethasone, irradiation | * Activated caspase-3 cleaves USP7 | Thymocyte development | [38] | |||
| USP7 | TNFα, cycloheximide | ↑Extrinsic apoptosis | ↑RIPK1 | ↑Liver injury | [37] | |
| Erastin | ↑Ferroptosis | ↓H2Bub1 | ↓Lung cancer | [32] | ||
| USP8 | ↓Intrinsic apoptosis | ↑AKT | ↑Cholangiocarcinoma | [79] | ||
| αFas, TRAIL, cycloheximide | ↓Extrinsic apoptosis | ↑c-FLIP | ↑Cervical cancer, melanoma | [80] | ||
| 9F7-F11 | ↑Extrinsic apoptosis | ↓ITCH, c-FLIP | ↓Pancreatic cancer, breast cancer, prostate cancer | [81] | ||
| USP10 | Irradiation | ↑Intrinsic apoptosis | ↑p53 | ↓Colorectal cancer, prostate cancer | [82] | |
| ↑Intrinsic apoptosis | ↑p53 *miR-138 suppresses USP10 expression | ↓Colorectal cancer, cervical cancer, lung carcinoma, non-small cell lung cancer cell line | [83] | |||
| Etoposide, camptothecin, irradiation | ↑Intrinsic apoptosis | ↓NEMO | ↓Fibrosarcoma | [84] | ||
| MNNG, 6-TG | ↓Intrinsic apoptosis | ↑MSH2 | ↑Lung cancer | [85] | ||
| MG-132, bortezomib | ↓Intrinsic apoptosis | ↑p62 | ↓Parkinson’s disease | [86] | ||
| Arsenite | ↓Intrinsic apoptosis | ↓ROS-induced stress | ↑Osteosarcoma, adrenal cortex adenocarcinoma, cervical cancer | [87] | ||
| Arsenite | ↓Intrinsic apoptosis | ↓ROS-induced stress *Tax suppresses the activity of USP10 | ↑Adult T-cell leukemia | [88] | ||
| Ischemic injury | ↓Intrinsic apoptosis | ↓TAK1 | ↓Ischemia-reperfusion | [89,90] | ||
| ↓Intrinsic apoptosis | ↑Raf-1 | ↑Endometriosis | [91] | |||
| Cytokine deprivation | ↓Intrinsic apoptosis | ↑Hematopoiesis | [92] | |||
| Curcumin | ↑Paraptosis | Spoutin-1 | ↑ERK, JNK, ER stress, mitochondrial dilation | ↓Breast cancer | [93] | |
| USP11 | Etoposide, doxorubicin | ↓Intrinsic apoptosis | ↑p21 | ↑Lung cancer, colorectal cancer | [94] | |
| Camptothecin, VP-16 | ↓Intrinsic apoptosis | ↓Histone ubiquitination | ↑Cervical cancer, osteosarcoma | [95] | ||
| Mitomycin C | ↓Intrinsic apoptosis | ↑BRCA2 | ↑Breast cancer, pancreatic cancer | [96] | ||
| Loss of adhesion, cisplatin | ↓Intrinsic apoptosis, anoikis | Mitoxantrone | ↑XIAP | ↑Breast cancer | [97] | |
| TRAIL, TNFα, SMAC mimetics | ↓Intrinsic apoptosis | Mitoxantrone | ↑c-IAP-2 | ↑Colorectal cancer, melanoma | [98] | |
| Hemin | ↑Extrinsic apoptosis | ↑Fas, FasL, caspase-3 | ↑Intracerebral hemorrhage | [99] | ||
| USP15 | Papilloma virus | ↑Intrinsic apoptosis | ↑p53 | ↓Papilloma virus infection | [100] | |
| TNFα, actinomycin D, staurosporine | ↑Intrinsic, extrinsic apoptosis | ↑Cytochrome c release | ↑Breast cancer, cervical cancer | [101] | ||
| Imatinib | ↑intrinsic apoptosis | ↑Caspase-6 *SPATA5A/miR-202-5p suppresses USP15 expression | ↓Chronic myeloid leukemia | [102] | ||
| ↓Intrinsic apoptosis | ↑MDM2 *USP15 promotes T-cell activity against cancer cell | ↑Colorectal cancer, melanoma | [103] | |||
| ↓Intrinsic apoptosis | ↑p65 | ↑Multiple myeloma | [104] | |||
| Glutamate induced oxidative stress | ↓Intrinsic apoptosis | ↑ROS, caspase, ↓Bcl-2 | ↓Epilepsy | [105] | ||
| USP18 | IFN, irradiation | ↓Intrinsic apoptosis | ↑Acute myeloid leukemia | [106] | ||
| IFN | ↓Intrinsic apoptosis | ↓STAT/DP5, PUMA, Bim | ↓Type 1 diabetes | [107] | ||
| IFN, bortezomib | ↓Extrinsic apoptosis | ↓TRAIL | ↓Breast cancer | [108,109] | ||
| HIV, IFN | ↑Intrinsic apoptosis | ↑PTEN, ↓AKT | ↑HIV infection | [110] | ||
| ↓Intrinsic apoptosis | ↓AKT | ↑Cervical cancer | [111] | |||
| ↓Intrinsic apoptosis | ↓miR-7, ↑EGFR | ↑Glioma, cervical cancer | [112] | |||
| ↓Intrinsic apoptosis | ↑Bcl-2 | ↑HBV-associated hepatocellular carcinoma | [113] | |||
| Oxidative stress | ↓Intrinsic apoptosis | ↓p53, caspase | ↓Oxidative liver injury | [114] | ||
| ↓Intrinsic apoptosis | ↑Notch1, c-MYC | ↑Pancreatic cancer | [115] | |||
| USP20 | TNFα, cycloheximide | ↓Extrinsic apoptosis | ↑p62 | ↑Cervical cancer | [116] | |
| HBSS | ↓Intrinsic apoptosis | ↑ULK1 | ↑Cervical cancer | [117] | ||
| USP22 | ↓Intrinsic apoptosis | ↑c-MYC | ↑Gastric cancer | [118] | ||
| Etoposide | ↓Intrinsic apoptosis | ↑SIRT1, ↓p53 | ↑Embryonic development | [119] | ||
| ↓Intrinsic apoptosis | ↓TERT, p53 | ↑Retinoblastoma | [120] | |||
| 5-FU | ↓Intrinsic apoptosis | ↑SIRT1/AKT/MRP1 | ↑Hepatocellular carcinoma | [121] | ||
| Cisplatin | ↓Intrinsic apoptosis | ↓H2A ubiquitination, ↑SIRT1 | ↑Lung adenocarcinoma | [122] | ||
| ↓Intrinsic apoptosis | ↑ERK1/2, Autophagy | ↑Pancreatic cancer | [123] | |||
| ↓Intrinsic apoptosis | ↑Glioma | [124] | ||||
| Ganetespib | ↓Intrinsic apoptosis | ↑HSP90 | ↑Colorectal cancer, breast cancer | [125] | ||
| TNFα, BV6, zVAD-FMK | ↑Necroptosis | ↑RIPK3 | ↓Colorectal cancer | [126] | ||
| High glucose stress | ↑Intrinsic apoptosis | ↑Caspase-3, Bax, inflammation | ↑Diabetic nephropathy | [127] | ||
| CYLD | TNFα, cycloheximide | ↑Extrinsic apoptosis | ↓TNFR/NF-κB | [128,129,130,131] | ||
| TNFα, cycloheximide, zVAD-FMK | ↑Extrinsic apoptosis, necroptosis | ↑RIPK1 | ↑Autoinflammation, immunodeficiency | [132] | ||
| TNFα, LPS, LBW242, zVAD-FMK | ↑Necroptosis | ↑RIPK1/necrosome *Caspase-8 cleaves CYLD | [133,134] | |||
| Oxaliplatin | ↑Intrinsic apoptosis | * miR-454 suppresses CYLD | ↓Gastric cancer | [135] | ||
| ↑Intrinsic apoptosis | ↑NDRG1 | ↓Nasopharyngeal carcinoma | [136] | |||
| Transverse aortic arch constriction | ↑Intrinsic apoptosis | ↓mTOR/autolysosomal clearance | ↑Cardiomyopathy | [137] | ||
| A20 | TNFα | ↓Extrinsic apoptosis | ↓TNFR/NF-κB | [138] | ||
| TNFα | ↑Extrinsic apoptosis | ↑TNFR/NF-κB/NIK/ripoptosome | ↑Psoriasis | [139] | ||
| Porphyromonas gingivalis, TNFα, cycloheximide | ↓Intrinsic, extrinsic apoptosis | ↓Caspase-3 | ↓Periodontal disease | [140] | ||
| LPS-induced microglial exosome | ↓Intrinsic apoptosis | ↓Caspase-3, Bax, ↑Bcl-2 | ↓Brain injury | [141] | ||
| TRAIL | ↓Extrinsic apoptosis | ↓RIPK1/caspase-8 | ↑Glioblastoma | [142] | ||
| TRAIL | ↓Extrinsic apoptosis | ↓Caspase-8 | ↑Lung cancer, colorectal cancer | [143] | ||
| ↓Pyroptosis | ↓Caspase-1 | [144] | ||||
| High glucose | ↓Pyroptosis | ↓NLRP3 *miR-21-5p suppresses A20 | ↑Diabetic nephropathy | [145] | ||
| ↓Unclassified cell death | ↓ATGL16L1 | ↓IBD-like pathology | [146] | |||
| OTULIN | TNFα, L18-MDP | ↑Extrinsic apoptosis | ↓LUBAC, NF-κB | ↓Cervical cancer, osteosarcoma | [147,148] | |
| TNFα, cycloheximide, zVAD-FMK | ↓Extrinsic apoptosis, necroptosis | ↑LUBAC, ↓caspase *DUSP14 dephosphorylates OTULIN | ↓OTULIN-related autoinflammatory syndrome (ORAS) | [149,150,151,152] | ||
| ↓Extrinsic apoptosis | ↓mTOR, IFN | ↓ORAS-associated liver diseases, hepatocellular carcinoma | [153,154] | |||
| OTUB1 | Neocarzinostatin, etoposide, UV | ↑Intrinsic apoptosis | ↑p53 | ↓Osteosarcoma | [155] | |
| UV | ↑Intrinsic apoptosis | ↑MDMX | ↓Osteosarcoma | [156] | ||
| Epirubicin | ↓Intrinsic apoptosis | ↑FOXM1 | ↑Breast cancer | [157] | ||
| ↓Intrinsic apoptosis | ↓Bax, caspase | ↑Hepatocellular carcinoma | [158] | |||
| Intracerebral hemorrhage, hemin | ↓Intrinsic apoptosis | ↓Bax, ↑Bcl-2 | ↓Brain injury | [159] | ||
| TWEAK, BV6 | ↓Extrinsic apoptosis | ↑c-IAP-1 | ↑Ovarian carcinoma, melanoma, fibrosarcoma, breast cancer | [160] | ||
| tert-butyl hydroperoxide | ↓Ferroptosis | ↑SLC7A11 | ↑Non-small cell lung cancer, neuroblastoma, osteosarcoma | [161] | ||
| BAP1 | Gemcitabine | ↑Intrinsic apoptosis | ↓Malignant mesothelioma | [51] | ||
| Erastin | ↑Ferroptosis | ↓H2A ubiquitination | ↓Renal cell carcinoma | [49,54] | ||
| H2O2, C2-ceramide, menadione, 5-FU, irradiation, UV | ↑Intrinsic apoptosis | ↑IP3R3 | ↓Pleural mesothelioma, | [45] | ||
| ↑Intrinsic apoptosis | ↑Bax | ↓Neuroblastoma | [47] | |||
| Reduced serum | ↓Intrinsic apoptosis | ↑Survivin | ↑Cutaneous melanoma | [46] | ||
| Glucose deprivation | ↓Intrinsic apoptosis | ↓ATF3, CHOP | ↑Renal cell carcinoma | [48] | ||
| TRAIL | ↓Extrinsic apoptosis | ↓DR4/5 | ↑Pleural mesothelioma | [45] | ||
| ↓Intrinsic apoptosis | ↓Histone ubiquitination of promoters of Bcl2 and Mcl2 | ↑Survival of RNF2-active cancer | [53] | |||
| BAP1 | ↓Intrinsic apoptosis | ↓DNA repair *EZH2 protects cancer cells without active BAP1 | ↑Clear cell renal cell carcinoma | [52] | ||
| UCHL1 | ↑Intrinsic apoptosis | ↑p53 | ↓Nasopharyngeal carcinoma, breast cancer | [62,67] | ||
| ↑Intrinsic apoptosis | ↑p53, Bax, caspase-3 | ↑Spermatogenesis | [63,64] | |||
| Doxorubicin, cisplatin, irradiation | ↑Intrinsic apoptosis | LDN-57444 | ↑NOXA | ↓Melanoma, colorectal cancer | [65] | |
| UCHL1 | Cisplatin | ↑Intrinsic apoptosis | ↑Bax, ↓BCL2, BCL11A, AEN, XIAP, AKT | ↓Ovarian cancer | [69] | |
| IFN, TNFα | ↑Extrinsic apoptosis | LDN-57444 | ↓Bcl-2 | ↓Multipotent mesenchymal stromal cell survival | [57] | |
| Deep hypothermic circulatory arrest | ↑Intrinsic apoptosis | ↑Neuronal injury by cardiac dysfunction | [61] | |||
| Hypoxia | ↑Intrinsic apoptosis | ↑Hypoxic-ischemic encephalopathy | [70] | |||
| Transverse aortic constriction | ↑Intrinsic apoptosis | LDN-57444 | ↑EGFR | ↓Cardiac hypertrophy | [56] | |
| High glucose | ↑Apoptosis, necroptosis | ↑RIPK1, RIPK3 | ↑Diabetic nephropathy | [59] | ||
| IAPP accumulation | ↓Intrinsic apoptosis | ↑Lysosomal degradation | ↓Type 2 diabetes | [68] | ||
| Oxygen-glucose deprivation | ↓Intrinsic apoptosis | ↓p27 | ↓Embryonal carcinoma, neuroblastoma | [60] | ||
| Ischemic injury | ↓Intrinsic apoptosis | ↓BACE1 | ↓Alzheimer’s disease | [55] | ||
| Gentamicin | ↓Intrinsic apoptosis | ↑Autophagosomal clearance | ↓Ototoxicity of antibiotics | [58] | ||
| Loss of adhesion | ↓Anoikis | ↑FAK | ↑Cervical cancer | [66] | ||
| UCHL5/ USP14 | SAHA, lenalidomide, deexamethasone | ↓Intrinsic, extrinsic apoptosis | b-AP15 | ↓ER stress | ↑Multiple myeloma | [162] |
| ↓Intrinsic apoptosis | b-AP15, VLX1570 | ↓Proteasome-associated polyubiquitin | ↑Multiple myeloma | [163] | ||
| Ibrutinib | ↓Intrinsic apoptosis | b-AP15, VLX1570 | ↑NF-κB, BCR-associated genes, ↓Cellular stress | ↑Waldenström macroglobulinemia | [164,165] | |
| ↓Intrinsic apoptosis | b-AP15 | ↑Wnt, TGFβ | ↑Diffuse large B-cell lymphoma | [166] | ||
| ↓Intrinsic apoptosis | b-AP15 | ↑c-MYC/NOXA | ↑Esophageal squamous cell carcinoma | [167] | ||
| ↓Intrinsic apoptosis | NiPT | ↑Lung cancer | [168] | |||
| UCHL5 | ↓Intrinsic apoptosis | b-AP15 | ↑TGFβ | ↑Ovarian cancer | [169] | |
| ↓Intrinsic apoptosis | ↑Wnt | ↑Endometrial cancer | [170] | |||
| Mycobacterium tuberculosis | ↑Pyroptosis | ↑NLRP3 | ↓Mycobacterium tuberculosis infection | [171] | ||
| USP14 | ↓Intrinsic apoptosis | IU1 | ↑COPS6 | ↑Osteosarcoma, B cell lymphoma | [172] | |
| ↓Intrinsic apoptosis | IU1 | ↓MDM2 | ↑Cervical cancer | [173] | ||
| Enzalutamide | ↓Intrinsic apoptosis | IU1 | ↑Androgen receptor | ↑Breast cancer | [174,175] | |
| Staurosporine | ↓Intrinsic apoptosis | ↑Mitochondria homeostasis ↓ER stress, ROS | ↑Melanoma | [176] | ||
| Cisplatin | ↓Intrinsic apoptosis | ↑AKT/ERK | ↑Gastric cancer | [177] | ||
| ↓Intrinsic apoptosis | *miR-4782-3p suppresses USP14 | ↑Non-small cell lung cancer | [178] | |||
| High glucose | ↑Intrinsic apoptosis | ↑SPAG5 | ↑Diabetic nephropathy | [179] | ||
| ↓Autophagic cell death | IU1 | ↓ER stress-mediated autophagy, JNK | ↑Lung cancer | [180,181] | ||
| ↓Intrinsic apoptosis | b-AP15 | ↑Aurora B | ↑Leukemia | [182] | ||
| ↓Intrinsic apoptosis | VLX1570 | ↑Endometrial cancer | [183] | |||
| BRCC36 | Irradiation | ↓Intrinsic apoptosis | ↑BRCC | ↑Breast cancer, cervical cancer | [184,185] | |
| LPS, HLLOMe, silica, oxLDL | ↑Pyroptosis | G5 | ↑NLRP3 | ↑Pyrogenic response | [11,186,187] | |
| LPS | ↑Pyroptosis | ↑NLRP3 *Vdr suppresses BRCC36 | ↑Pyrogenic response | [188] | ||
| STAMBPL1 | ↓Intrinsic apoptosis | ↑XIAP | ↑Prostate cancer | [189] | ||
| TRAIL | ↓Extrinsic apoptosis | Honokiol, cepharanthine | ↑Survivin, c-FLIP | ↑Renal cell carcinoma, lung cancer, kidney carcinoma | [189,190] | |
| ↓Intrinsic apoptosis | ↓NF-κB | ↑Gastric cancer | [191] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-S.; Kim, S.; Hwang, G.; Song, J. Deubiquitinases: Modulators of Different Types of Regulated Cell Death. Int. J. Mol. Sci. 2021, 22, 4352. https://doi.org/10.3390/ijms22094352
Lee C-S, Kim S, Hwang G, Song J. Deubiquitinases: Modulators of Different Types of Regulated Cell Death. International Journal of Molecular Sciences. 2021; 22(9):4352. https://doi.org/10.3390/ijms22094352
Chicago/Turabian StyleLee, Choong-Sil, Seungyeon Kim, Gyuho Hwang, and Jaewhan Song. 2021. "Deubiquitinases: Modulators of Different Types of Regulated Cell Death" International Journal of Molecular Sciences 22, no. 9: 4352. https://doi.org/10.3390/ijms22094352
APA StyleLee, C. -S., Kim, S., Hwang, G., & Song, J. (2021). Deubiquitinases: Modulators of Different Types of Regulated Cell Death. International Journal of Molecular Sciences, 22(9), 4352. https://doi.org/10.3390/ijms22094352