
Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
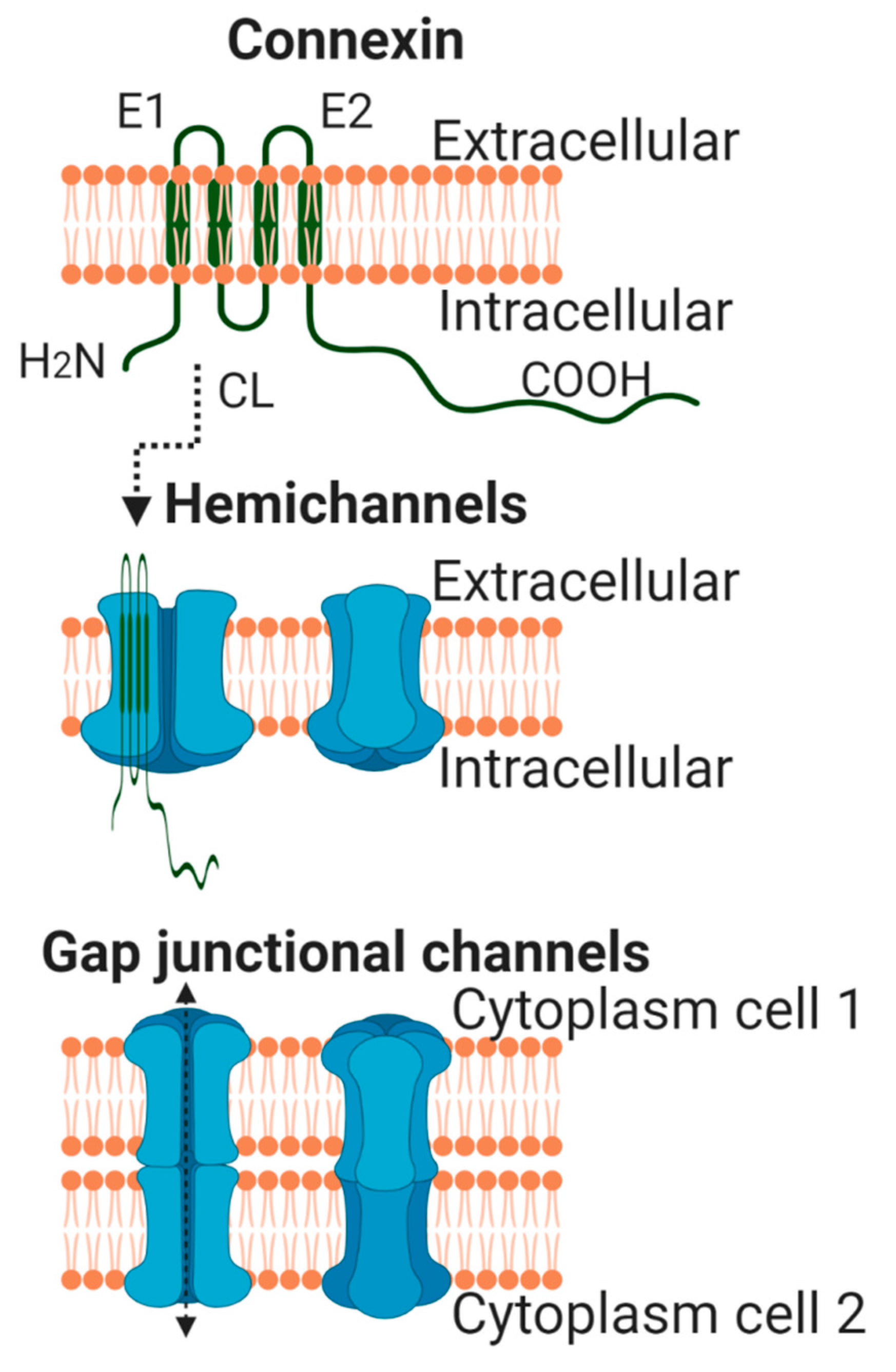
2. General Aspects of Connexin Biology
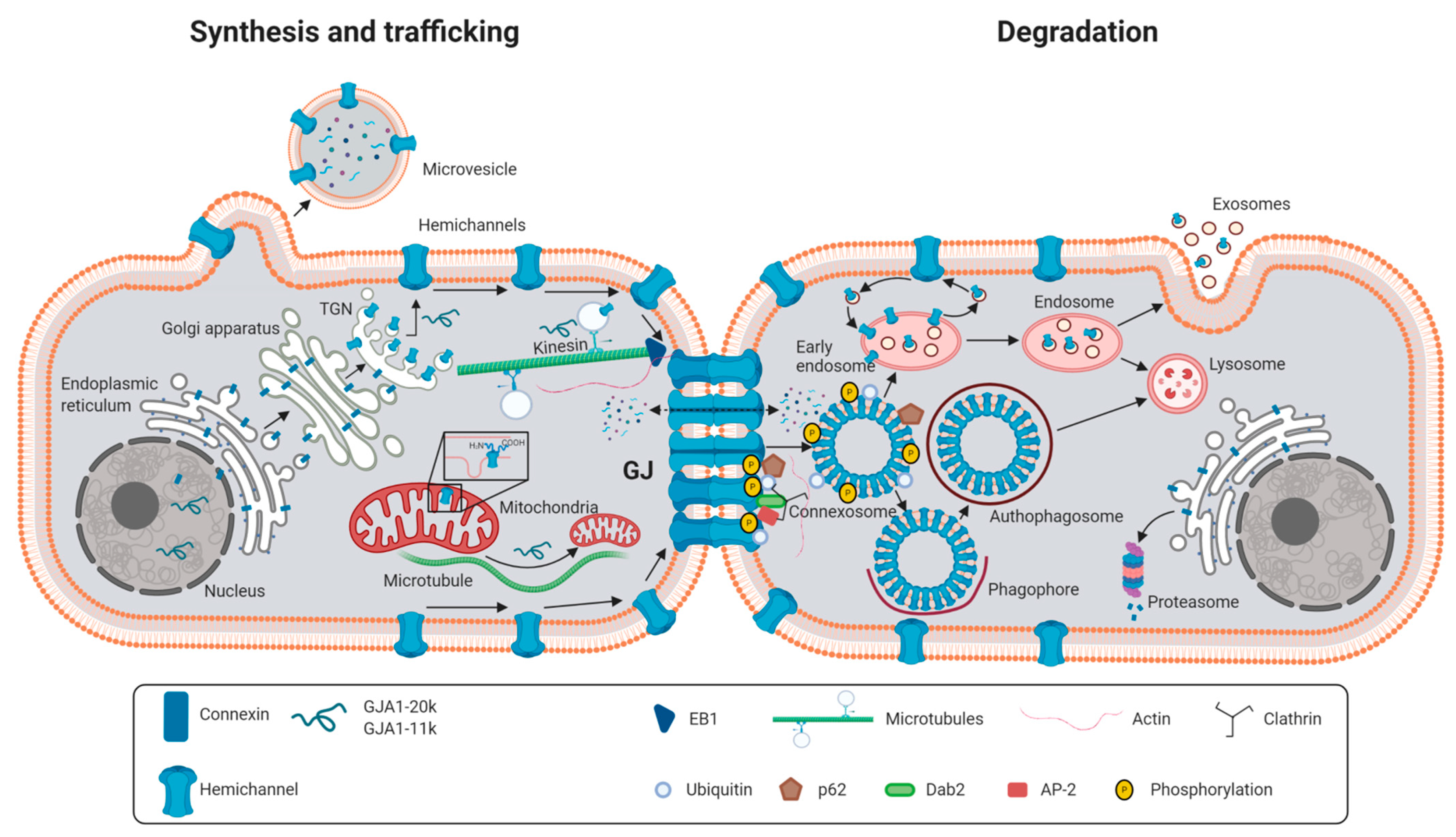
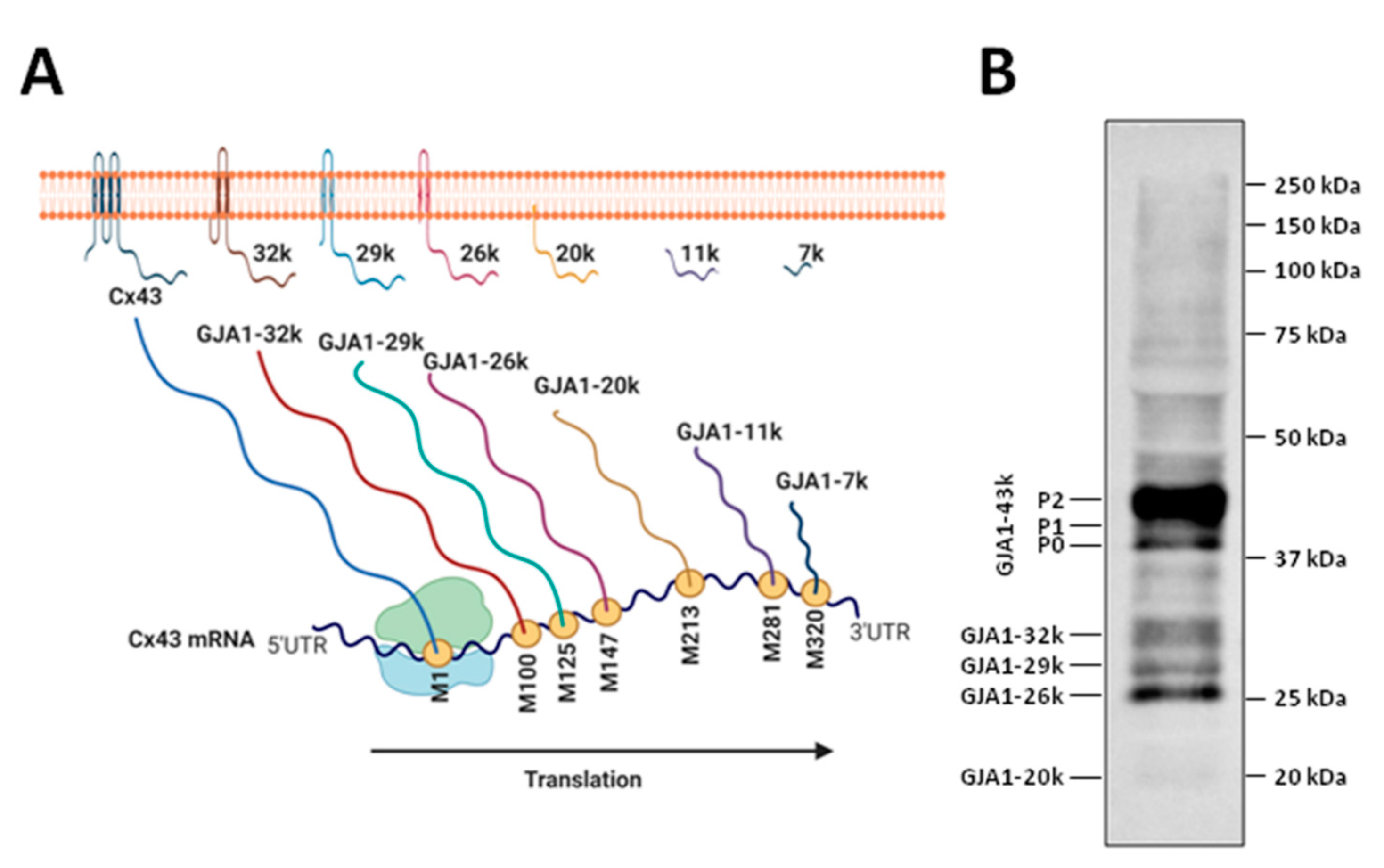
2.1. Synthesis and Degradation of Connexins
2.2. Permeability and Conductance of Connexin Channels
2.3. Gating Regulation of Connexin Channels
2.3.1. Gating by Transjunctional Voltage (Vj) and Transmembrane Potential (Vm)
2.3.2. Gating by Intracellular pH
2.3.3. Calcium- and Calmodulin-Dependent Gating
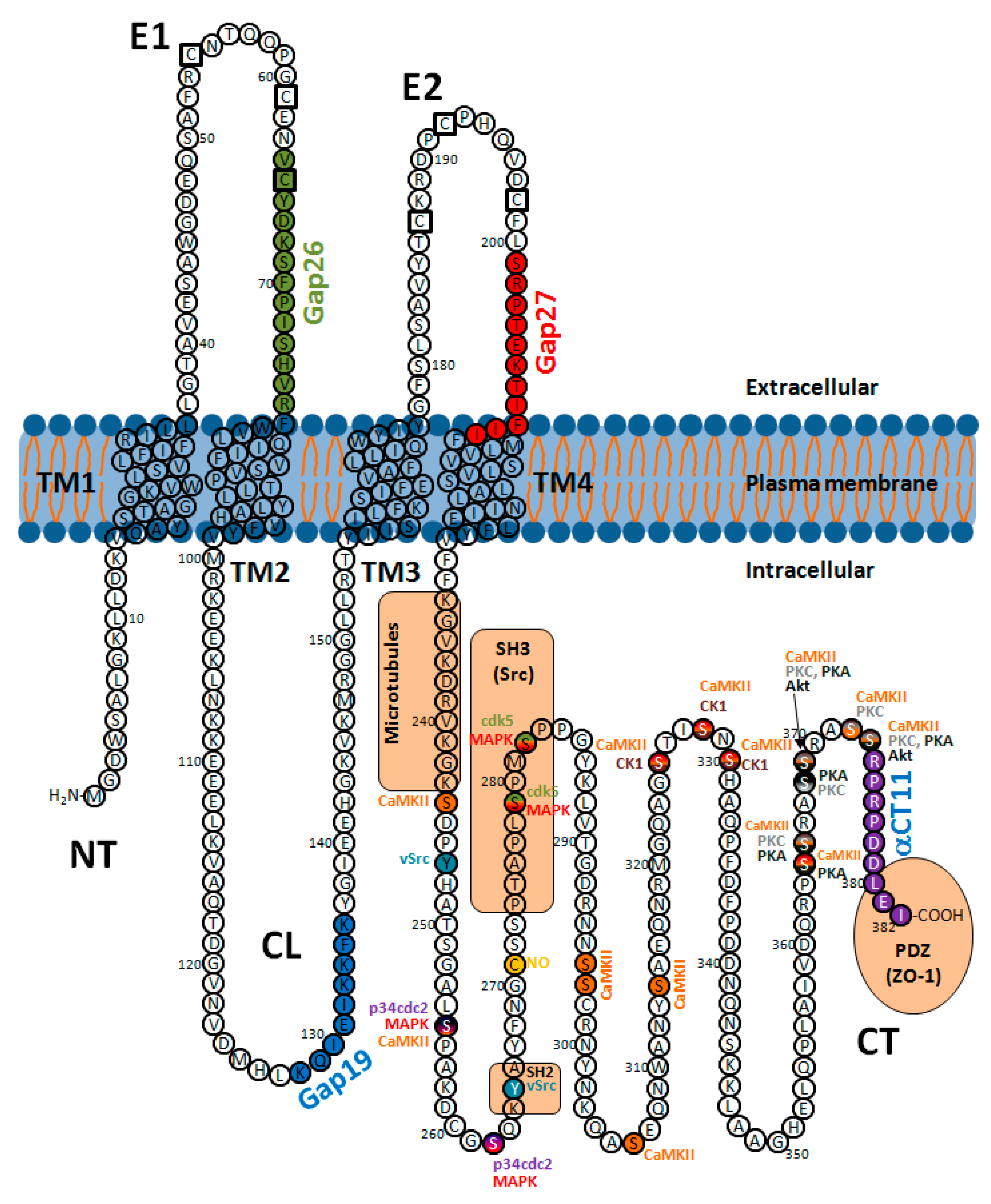
2.4. Regulation by Post-Translational Modifications
2.4.1. Phosphorylation
Cx43 Channels
Cx40 Channels
Cx45 Channels
2.4.2. Redox Regulation: S-Nitrosylation and Carbonylation
2.4.3. Acetylation
2.4.4. SUMOylation
2.4.5. Ubiquitination
2.5. The Connexin Interactome or Connexome
2.5.1. Interactions of Sarcolemmal Connexins with Cytoskeletal Proteins
2.5.2. Interactions of Sarcolemmal Connexins with Other Junctional Proteins
2.5.3. Interactions of Sarcolemmal Connexins with Receptors and Ion Channels
2.5.4. Other Interactors of Sarcolemmal Connexins
2.5.5. Interactions of Mitochondrial Connexins
3. Cardiac Connexins
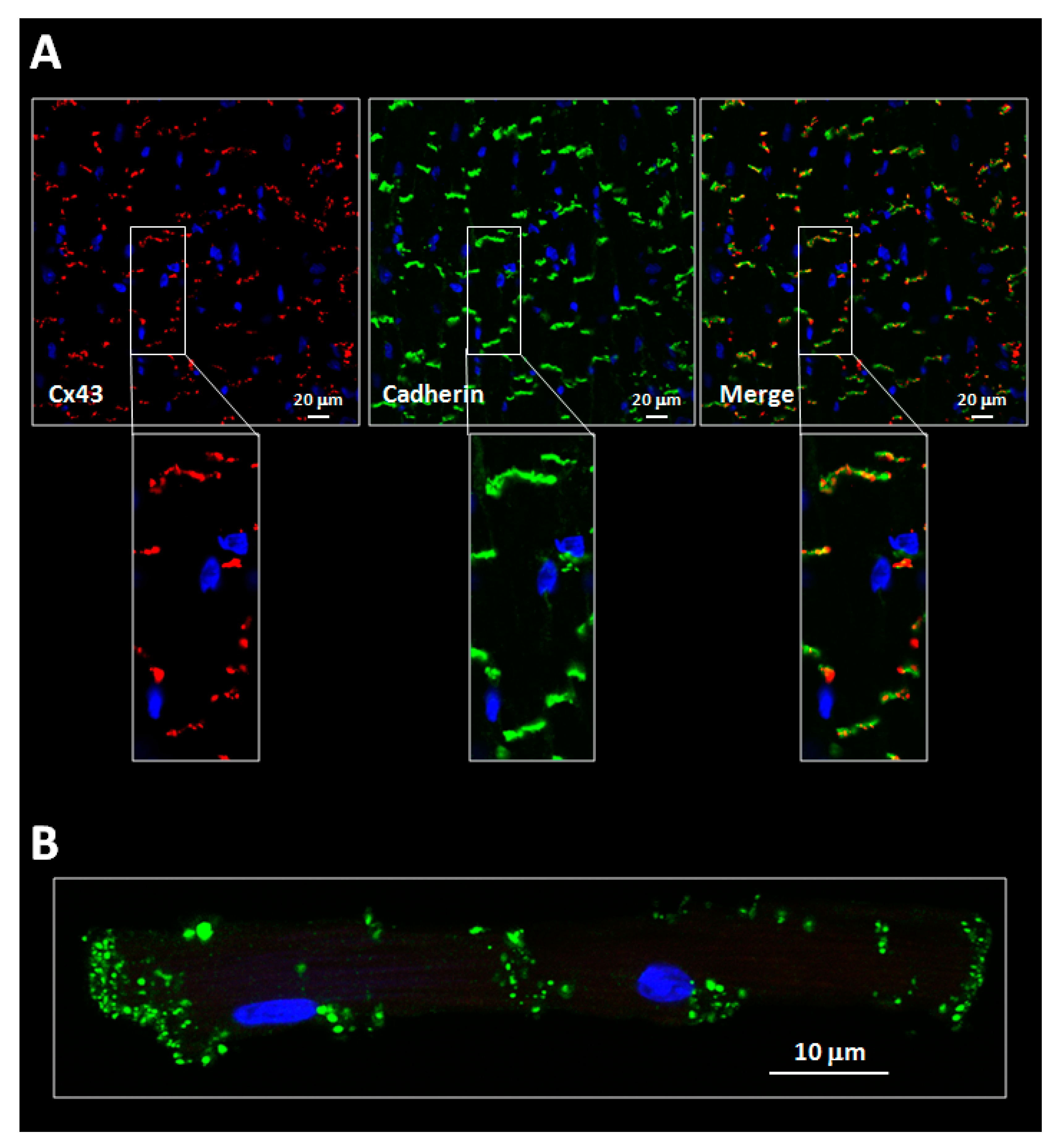
3.1. Alterations in Connexin Distribution and Phosphorylation under Pathological Conditions
3.2. Functions of Cardiac Connexins
3.2.1. Sarcolemmal Connexins: Gap Junction-Dependent Role in Cardiac Electrical Coupling
The Cable Theory of Electrical Conduction and the Influence of Tissue Anisotropy
Gap Junctions and Electrical Coupling in the Supraventricular Conduction System
Gap Junctions and Electrical Coupling in the Ventricular Myocardium
Gap Junctions and Ischemic Ib Ventricular Arrhythmias
3.2.2. Sarcolemmal Connexins: Gap Junction-Dependent Role in Chemical Coupling
Involvement in Myocardial Ischemia/Reperfusion Injury
Studies in Transgenic Mice Models
The “Good Samaritan” Effect
Chemical Coupling through Other Cardiac Connexin Isoforms
Chemical Coupling and Regulation of Cell Growth, Migration and Differentiation
3.2.3. Gap Junction-Independent Functions of Unopposed Sarcolemmal Hemichannels: Involvement in Paracrine Communication and Dysregulation of Cell Homeostasis
Flux of Intracellular Metabolites through Opened Hemichannels
Calcium Influx and Cell Edema
Involvement of Unopposed Hemichannels in Myocardial Ischemia/Reperfusion Injury
3.2.4. Involvement of Sarcolemmal Connexins in Long-Distance Communication through Tunneling Nanotubes and Extracellular Vesicles
3.2.5. Mitochondrial Connexins
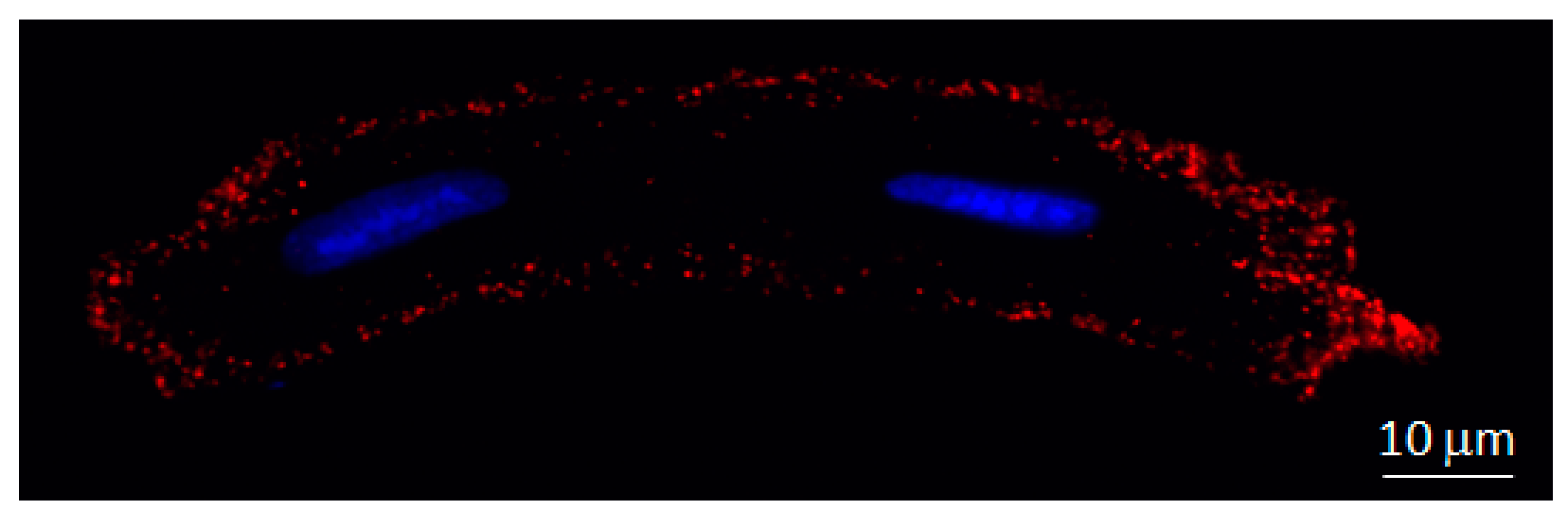
Presence of Cx43 at Cardiomyocyte Mitochondria
Functions of Mitochondrial Cx43
Involvement in Preconditioning Protection
Involvement in Chemotherapy-Induced Cardiotoxicity
Other Mitochondrial Connexins
3.2.6. Nuclear Connexins
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dobrowolski, R.; Willecke, K. Connexin-caused genetic diseases and corresponding mouse models. Antioxid. Redox Signal. 2009, 11, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Sohl, G.; Willecke, K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun. Adhes. 2003, 10, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.C.; Berthoud, V.M. Gap junction gene and protein families: Connexins, innexins, and pannexins. Biochim. Biophys. Acta 2018, 1860, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Sosinsky, G.E.; Nicholson, B.J. Structural organization of gap junction channels. Biochim. Biophys. Acta 2005, 1711, 99–125. [Google Scholar] [CrossRef] [Green Version]
- Van Veen, A.A.; van Rijen, H.V.; Opthof, T. Cardiac gap junction channels: Modulation of expression and channel properties. Cardiovasc. Res. 2001, 51, 217–229. [Google Scholar] [CrossRef]
- Laird, D.W. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim. Biophys. Acta 2005, 1711, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, A.P. Connexin phosphorylation as a regulatory event linked to channel gating. Biochim. Biophys. Acta 2005, 1711, 164–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta 2018, 1860, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Procida, K.; Jorgensen, L.; Schmitt, N.; Delmar, M.; Taffet, S.M.; Holstein-Rathlou, N.H.; Nielsen, M.S.; Braunstein, T.H. Phosphorylation of connexin43 on serine 306 regulates electrical coupling. Heart Rhythm 2009, 6, 1632–1638. [Google Scholar] [CrossRef] [Green Version]
- Fishman, G.I.; Eddy, R.L.; Shows, T.B.; Rosenthal, L.; Leinwand, L.A. The human connexin gene family of gap junction proteins: Distinct chromosomal locations but similar structures. Genomics 1991, 10, 250–256. [Google Scholar] [CrossRef]
- Axelsen, L.N.; Calloe, K.; Holstein-Rathlou, N.H.; Nielsen, M.S. Managing the complexity of communication: Regulation of gap junctions by post-translational modification. Front. Pharmacol. 2013, 4, 130. [Google Scholar] [CrossRef] [Green Version]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.S.; Nygaard, A.L.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar] [PubMed] [Green Version]
- Shaw, R.M.; Fay, A.J.; Puthenveedu, M.A.; von Zastrow, M.; Jan, Y.N.; Jan, L.Y. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 2007, 128, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Severs, N.J. The cardiac gap junction and intercalated disc. Int. J. Cardiol. 1990, 26, 137–173. [Google Scholar] [CrossRef]
- Harris, A.L. Emerging issues of connexin channels: Biophysics fills the gap. Q. Rev. Biophys. 2001, 34, 325–472. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Chen, Y.; Reuss, L.; Altenberg, G.A. Functional expression in Xenopus oocytes of gap-junctional hemichannels formed by a cysteine-less connexin 43. J. Biol. Chem. 2004, 279, 9689–9692. [Google Scholar] [CrossRef] [Green Version]
- Koval, M.; Molina, S.A.; Burt, J.M. Mix and match: Investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 2014, 588, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Beyer, E.C. Are these connexins compatible and does it matter? Channels 2015, 9, 63–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Xu, Q.; Veenstra, R.D. Functional formation of heterotypic gap junction channels by connexins-40 and -43. Channels 2014, 8, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Rackauskas, M.; Kreuzberg, M.M.; Pranevicius, M.; Willecke, K.; Verselis, V.K.; Bukauskas, F.F. Gating properties of heterotypic gap junction channels formed of connexins 40, 43, and 45. Biophys. J. 2007, 92, 1952–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, X.; Doble, B.W.; Kardami, E. The carboxy-tail of connexin-43 localizes to the nucleus and inhibits cell growth. Mol. Cell Biochem. 2003, 242, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Dodoni, G.; Rodriguez-Sinovas, A.; Cabestrero, A.; Ruiz-Meana, M.; Gres, P.; Konietzka, I.; Lopez-Iglesias, C.; Garcia-Dorado, D.; Di Lisa, F.; et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Sinovas, A.; Boengler, K.; Cabestrero, A.; Gres, P.; Morente, M.; Ruiz-Meana, M.; Konietzka, I.; Miro, E.; Totzeck, A.; Heusch, G.; et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ. Res. 2006, 99, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sinovas, A.; Ruiz-Meana, M.; Denuc, A.; Garcia-Dorado, D. Mitochondrial Cx43, an important component of cardiac preconditioning. Biochim. Biophys. Acta 2018, 1860, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.S.; Koval, M. Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease. Int. J. Mol. Sci. 2018, 19, 1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epifantseva, I.; Shaw, R.M. Intracellular trafficking pathways of Cx43 gap junction channels. Biochim. Biophys. Acta 2018, 1860, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Musil, L.S.; Goodenough, D.A. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 1993, 74, 1065–1077. [Google Scholar] [CrossRef]
- Smyth, J.W.; Vogan, J.M.; Buch, P.J.; Zhang, S.S.; Fong, T.S.; Hong, T.T.; Shaw, R.M. Actin Cytoskeleton Rest Stops Regulate Anterograde Traffic of Connexin 43 Vesicles to the Plasma Membrane. Circ. Res. 2012, 110, 978–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fort, A.G.; Murray, J.W.; Dandachi, N.; Davidson, M.W.; Dermietzel, R.; Wolkoff, A.W.; Spray, D.C. In vitro motility of liver connexin vesicles along microtubules utilizes kinesin motors. J. Biol. Chem. 2011, 286, 22875–22885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.L.; Ali, M.Y.; Warshaw, D.M. Cargo transport: Molecular motors navigate a complex cytoskeleton. Curr. Opin. Cell Biol. 2008, 20, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.M.; Dubash, A.D.; Kreitzer, G.; Green, K.J. Disease mutations in desmoplakin inhibit Cx43 membrane targeting mediated by desmoplakin-EB1 interactions. J. Cell Biol. 2014, 206, 779–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauf, U.; Giepmans, B.N.; Lopez, P.; Braconnot, S.; Chen, S.C.; Falk, M.M. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 10446–10451. [Google Scholar] [CrossRef] [Green Version]
- Gaietta, G.; Deerinck, T.J.; Adams, S.R.; Bouwer, J.; Tour, O.; Laird, D.W.; Sosinsky, G.E.; Tsien, R.Y.; Ellisman, M.H. Multicolor and electron microscopic imaging of connexin trafficking. Science 2002, 296, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Rhett, J.M.; Veeraraghavan, R.; Poelzing, S.; Gourdie, R.G. The perinexus: Sign-post on the path to a new model of cardiac conduction? Trends Cardiovasc. Med. 2013, 23, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Ongstad, E.L.; Jourdan, J.; Gourdie, R.G. Cx43 associates with Na(v)1.5 in the cardiomyocyte perinexus. J. Membr. Biol. 2012, 245, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo-Macias, A.; Agullo-Pascual, E.; Delmar, M. The cardiac connexome: Non-canonical functions of connexin43 and their role in cardiac arrhythmias. Semin. Cell Dev. Biol. 2016, 50, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.S.; Shaw, R.M. Trafficking highways to the intercalated disc: New insights unlocking the specificity of connexin 43 localization. Cell Commun. Adhes. 2014, 21, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.G.; Reynhout, J.K.; TenBroek, E.M.; Quade, B.J.; Yasumura, T.; Davidson, K.G.; Sheridan, J.D.; Rash, J.E. Gap junction assembly: Roles for the formation plaque and regulation by the C-terminus of connexin43. Mol. Biol. Cell 2012, 23, 71–86. [Google Scholar] [CrossRef]
- Johnson, R.; Hammer, M.; Sheridan, J.; Revel, J.P. Gap junction formation between reaggregated Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 4536–4540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solan, J.L.; Lampe, P.D. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta 2018, 1860, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Revel, J.P.; Karnovsky, M.J. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J. Cell Biol. 1967, 33, C7–C12. [Google Scholar] [CrossRef]
- Bukauskas, F.F.; Jordan, K.; Bukauskiene, A.; Bennett, M.V.; Lampe, P.D.; Laird, D.W.; Verselis, V.K. Clustering of connexin 43-enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2556–2561. [Google Scholar] [CrossRef] [Green Version]
- Windoffer, R.; Beile, B.; Leibold, A.; Thomas, S.; Wilhelm, U.; Leube, R.E. Visualization of gap junction mobility in living cells. Cell Tissue Res. 2000, 299, 347–362. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Fallon, R.F.; Goodenough, D.A. Five-hour half-life of mouse liver gap-junction protein. J. Cell Biol. 1981, 90, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Leithe, E.; Kjenseth, A.; Sirnes, S.; Stenmark, H.; Brech, A.; Rivedal, E. Ubiquitylation of the gap junction protein connexin-43 signals its trafficking from early endosomes to lysosomes in a process mediated by Hrs and Tsg101. J. Cell Sci. 2009, 122, 3883–3893. [Google Scholar] [CrossRef] [Green Version]
- Kjenseth, A.; Fykerud, T.; Rivedal, E.; Leithe, E. Regulation of gap junction intercellular communication by the ubiquitin system. Cell Signal. 2010, 22, 1267–1273. [Google Scholar] [CrossRef]
- Falk, M.M.; Kells, R.M.; Berthoud, V.M. Degradation of connexins and gap junctions. FEBS Lett. 2014, 588, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Laing, J.G.; Beyer, E.C. The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J. Biol. Chem. 1995, 270, 26399–26403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Shao, Q.; Igdoura, S.A.; Alaoui-Jamali, M.A.; Laird, D.W. Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J. Biol. Chem. 2003, 278, 30005–30014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanSlyke, J.K.; Deschenes, S.M.; Musil, L.S. Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol. Biol. Cell 2000, 11, 1933–1946. [Google Scholar] [CrossRef] [Green Version]
- Su, V.; Hoang, C.; Geerts, D.; Lau, A.F. CIP75 (connexin43-interacting protein of 75 kDa) mediates the endoplasmic reticulum dislocation of connexin43. Biochem. J. 2014, 458, 57–67. [Google Scholar] [CrossRef] [PubMed]
- VanSlyke, J.K.; Musil, L.S. Dislocation and degradation from the ER are regulated by cytosolic stress. J. Cell Biol. 2002, 157, 381–394. [Google Scholar] [CrossRef]
- Leithe, E.; Rivedal, E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J. Cell Sci. 2004, 117, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leithe, E.; Rivedal, E. Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J. Biol. Chem. 2004, 279, 50089–50096. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.C.; Kristensen, A.R.; Foster, L.J.; Naus, C.C. Association of connexin43 with E3 ubiquitin ligase TRIM21 reveals a mechanism for gap junction phosphodegron control. J. Proteome Res. 2012, 11, 6134–6146. [Google Scholar] [CrossRef]
- Girao, H.; Catarino, S.; Pereira, P. Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp. Cell Res. 2009, 315, 3587–3597. [Google Scholar] [CrossRef]
- Bejarano, E.; Girao, H.; Yuste, A.; Patel, B.; Marques, C.; Spray, D.C.; Pereira, P.; Cuervo, A.M. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol. Biol. Cell 2012, 23, 2156–2169. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.; Chodock, R.; Hand, A.R.; Laird, D.W. The origin of annular junctions: A mechanism of gap junction internalization. J. Cell Sci. 2001, 114, 763–773. [Google Scholar] [PubMed]
- Archard, H.O.; Denys, F.R. Development of annular gap junctions in guinea pig epithelia. J. Oral Pathol. Med. 1979, 8, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Auth, T.; Schlüter, S.; Urschel, S.; Kussmann, P.; Sonntag, S.; Höher, T.; Kreuzberg, M.M.; Dobrowolski, R.; Willecke, K. The TSG101 protein binds to connexins and is involved in connexin degradation. Exp. Cell Res. 2009, 315, 1053–1062. [Google Scholar] [CrossRef]
- Musil, L.S.; Le, A.C.; VanSlyke, J.K.; Roberts, L.M. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J. Biol. Chem. 2000, 275, 25207–25215. [Google Scholar] [CrossRef] [Green Version]
- Dunn, C.A.; Su, V.; Lau, A.F.; Lampe, P.D. Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J. Biol. Chem. 2012, 287, 2600–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukauskas, F.F.; Peracchia, C. Distinct behaviors of chemical and voltage sensitive gates of gap junction channel. Curr. Top. Membr. 2000, 49, 207–221. [Google Scholar]
- Veenstra, R.D. Ion permeation through connexin gap junction channels: Effects on conductance and selectivity. Curr. Top. Membr. 2000, 49, 95–129. [Google Scholar]
- Skerrett, I.M.; Smith, J.F.; Nicholson, B.J. Mechanistic differences between chemical and electrical gating of gap junctions. Curr. Top. Membr. 2000, 49, 249–269. [Google Scholar]
- Moreno, A.P.; Chanson, M.; Elenes, S.; Anumonwo, J.; Scerri, I.; Gu, H.; Taffet, S.M.; Delmar, M. Role of the carboxyl terminal of connexin43 in transjunctional fast voltage gating. Circ. Res. 2002, 90, 450–457. [Google Scholar] [CrossRef]
- Bukauskas, F.F.; Bukauskiene, A.; Verselis, V.K. Conductance and permeability of the residual state of connexin43 gap junction channels. J. Gen. Physiol. 2002, 119, 171–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukauskas, F.F.; Peracchia, C. Two distinct gating mechanisms in gap junction channels: CO2-sensitive and voltage-sensitive. Biophys. J. 1997, 72, 2137–2142. [Google Scholar] [CrossRef] [Green Version]
- Bevans, C.G.; Kordel, M.; Rhee, S.K.; Harris, A.L. Isoform composition of connexin channels determines selectivity among second messengers and uncharged molecules. J. Biol. Chem. 1998, 273, 2808–2816. [Google Scholar] [CrossRef] [Green Version]
- Valiunas, V.; Beyer, E.C.; Brink, P.R. Cardiac gap junction channels show quantitative differences in selectivity. Circ. Res. 2002, 91, 104–111. [Google Scholar] [CrossRef]
- Beblo, D.A.; Veenstra, R.D. Monovalent cation permeation through the connexin40 gap junction channel. Cs, Rb, K, Na, Li, TEA, TMA, TBA, and effects of anions Br, Cl, F, acetate, aspartate, glutamate, and NO3. J. Gen. Physiol. 1997, 109, 509–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veenstra, R.D.; Wang, H.Z.; Beyer, E.C.; Brink, P.R. Selective dye and ionic permeability of gap junction channels formed by connexin45. Circ. Res. 1994, 75, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Valiunas, V.; Cohen, I.S.; Brink, P.R. Defining the factors that affect solute permeation of gap junction channels. Biochim. Biophys. Acta 2018, 1860, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Valiunas, V. Cyclic nucleotide permeability through unopposed connexin hemichannels. Front. Pharmacol. 2013, 4, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 2008, 28, 4702–4711. [Google Scholar] [CrossRef] [PubMed]
- Kanaporis, G.; Brink, P.R.; Valiunas, V. Gap junction permeability: Selectivity for anionic and cationic probes. Am. J. Physiol. Cell Physiol. 2011, 300, C600–C609. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.L. Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 2007, 94, 120–143. [Google Scholar] [CrossRef] [Green Version]
- Valiunas, V.; White, T.W. Connexin43 and connexin50 channels exhibit different permeability to the second messenger inositol triphosphate. Sci. Rep. 2020, 10, 8744. [Google Scholar] [CrossRef]
- De Mello, W.C. Cell-to-cell diffusion of glucose in the mammalian heart is disrupted by high glucose. Implications for the diabetic heart. Exp. Cell Res. 2015, 334, 239–245. [Google Scholar] [CrossRef]
- Goldberg, G.S.; Moreno, A.P.; Lampe, P.D. Gap junctions between cells expressing connexin 43 or 32 show inverse permselectivity to adenosine and ATP. J. Biol. Chem. 2002, 277, 36725–36730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Dorado, D.; Rodriguez-Sinovas, A.; Ruiz-Meana, M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovasc. Res. 2004, 61, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Rackauskas, M.; Neverauskas, V.; Skeberdis, V.A. Diversity and properties of connexin gap junction channels. Medicina 2010, 46, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saez, J.C.; Retamal, M.A.; Basilio, D.; Bukauskas, F.F.; Bennett, M.V. Connexin-based gap junction hemichannels: Gating mechanisms. Biochim. Biophys. Acta 2005, 1711, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, A.P.; Lau, A.F. Gap junction channel gating modulated through protein phosphorylation. Prog. Biophys. Mol. Biol. 2007, 94, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Dhein, S. Gap junction channels in the cardiovascular system: Pharmacological and physiological modulation. Trends Pharmacol. Sci. 1998, 19, 229–241. [Google Scholar] [CrossRef]
- Kwak, B.R.; van Veen, T.A.; Analbers, L.J.; Jongsma, H.J. TPA increases conductance but decreases permeability in neonatal rat cardiomyocyte gap junction channels. Exp. Cell Res. 1995, 220, 456–463. [Google Scholar] [CrossRef]
- Bargiello, T.A.; Oh, S.; Tang, Q.; Bargiello, N.K.; Dowd, T.L.; Kwon, T. Gating of Connexin Channels by transjunctional-voltage: Conformations and models of open and closed states. Biochim. Biophys. Acta Biomembr. 2018, 1860, 22–39. [Google Scholar] [CrossRef]
- Gonzalez, D.; Gomez-Hernandez, J.M.; Barrio, L.C. Molecular basis of voltage dependence of connexin channels: An integrative appraisal. Prog. Biophys. Mol. Biol. 2007, 94, 66–106. [Google Scholar] [CrossRef] [PubMed]
- Revilla, A.; Castro, C.; Barrio, L.C. Molecular dissection of transjunctional voltage dependence in the connexin-32 and connexin-43 junctions. Biophys. J. 1999, 77, 1374–1383. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Cassara, C.; Lin, X.; Veenstra, R.D. Calcium-calmodulin gating of a pH-insensitive isoform of connexin43 gap junctions. Biochem. J. 2019, 476, 1137–1148. [Google Scholar] [CrossRef]
- Bukauskas, F.F.; Bukauskiene, A.; Bennett, M.V.; Verselis, V.K. Gating properties of gap junction channels assembled from connexin43 and connexin43 fused with green fluorescent protein. Biophys. J. 2001, 81, 137–152. [Google Scholar] [CrossRef] [Green Version]
- Shibayama, J.; Gutierrez, C.; Gonzalez, D.; Kieken, F.; Seki, A.; Carrion, J.R.; Sorgen, P.L.; Taffet, S.M.; Barrio, L.C.; Delmar, M. Effect of charge substitutions at residue his-142 on voltage gating of connexin43 channels. Biophys. J. 2006, 91, 4054–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revilla, A.; Bennett, M.V.; Barrio, L.C. Molecular determinants of membrane potential dependence in vertebrate gap junction channels. Proc. Natl. Acad. Sci. USA 2000, 97, 14760–14765. [Google Scholar] [CrossRef] [Green Version]
- Delmar, M.; Coombs, W.; Sorgen, P.; Duffy, H.S.; Taffet, S.M. Structural bases for the chemical regulation of Connexin43 channels. Cardiovasc. Res. 2004, 62, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Turin, L.; Warner, A. Carbon dioxide reversibly abolishes ionic communication between cells of early amphibian embryo. Nature 1977, 270, 56–57. [Google Scholar] [CrossRef] [PubMed]
- Turin, L.; Warner, A.E. Intracellular pH in early Xenopus embryos: Its effect on current flow between blastomeres. J. Physiol. 1980, 300, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Reber, W.R.; Weingart, R. Ungulate cardiac purkinje fibres: The influence of intracellular pH on the electrical cell-to-cell coupling. J. Physiol. 1982, 328, 87–104. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Taffet, S.; Stoner, L.; Delmar, M.; Vallano, M.L.; Jalife, J. A structural basis for the unequal sensitivity of the major cardiac and liver gap junctions to intracellular acidification: The carboxyl tail length. Biophys. J. 1993, 64, 1422–1433. [Google Scholar] [CrossRef] [Green Version]
- Morley, G.E.; Taffet, S.M.; Delmar, M. Intramolecular interactions mediate pH regulation of connexin43 channels. Biophys. J. 1996, 70, 1294–1302. [Google Scholar] [CrossRef] [Green Version]
- Ek-Vitorin, J.F.; Calero, G.; Morley, G.E.; Coombs, W.; Taffet, S.M.; Delmar, M. pH regulation of connexin43: Molecular analysis of the gating particle. Biophys. J. 1996, 71, 1273–1284. [Google Scholar] [CrossRef] [Green Version]
- Stergiopoulos, K.; Alvarado, J.L.; Mastroianni, M.; Ek-Vitorin, J.F.; Taffet, S.M.; Delmar, M. Hetero-domain interactions as a mechanism for the regulation of connexin channels. Circ. Res. 1999, 84, 1144–1155. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.K.; Jagielnicki, M.; McIntire, W.E.; Purdy, M.D.; Dharmarajan, V.; Griffin, P.R.; Yeager, M. A Steric “Ball-and-Chain” Mechanism for pH-Mediated Regulation of Gap Junction Channels. Cell Rep. 2020, 31, 107482. [Google Scholar] [CrossRef]
- Bouvier, D.; Spagnol, G.; Chenavas, S.; Kieken, F.; Vitrac, H.; Brownell, S.; Kellezi, A.; Forge, V.; Sorgen, P.L. Characterization of the structure and intermolecular interactions between the connexin40 and connexin43 carboxyl-terminal and cytoplasmic loop domains. J. Biol. Chem. 2009, 284, 34257–34271. [Google Scholar] [CrossRef] [Green Version]
- Ek, J.F.; Delmar, M.; Perzova, R.; Taffet, S.M. Role of histidine 95 on pH gating of the cardiac gap junction protein connexin43. Circ. Res. 1994, 74, 1058–1064. [Google Scholar] [CrossRef] [Green Version]
- Trexler, E.B.; Bukauskas, F.F.; Bennett, M.V.; Bargiello, T.A.; Verselis, V.K. Rapid and direct effects of pH on connexins revealed by the connexin46 hemichannel preparation. J. Gen. Physiol. 1999, 113, 721–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vuyst, E.; Decrock, E.; De Bock, M.; Yamasaki, H.; Naus, C.C.; Evans, W.H.; Leybaert, L. Connexin hemichannels and gap junction channels are differentially influenced by lipopolysaccharide and basic fibroblast growth factor. Mol. Biol. Cell 2007, 18, 34–46. [Google Scholar] [CrossRef]
- Sahu, G.; Bera, A.K. Contribution of intracellular calcium and pH in ischemic uncoupling of cardiac gap junction channels formed of connexins 43, 40, and 45: A critical function of C-terminal domain. PLoS ONE 2013, 8, e60506. [Google Scholar] [CrossRef]
- Peracchia, C. Chemical gating of gap junction channels; roles of calcium, pH and calmodulin. Biochim. Biophys. Acta 2004, 1662, 61–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lurtz, M.M.; Louis, C.F. Intracellular calcium regulation of connexin43. Am. J. Physiol. Cell Physiol. 2007, 293, C1806–C1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazrak, A.; Peracchia, C. Gap junction gating sensitivity to physiological internal calcium regardless of pH in Novikoff hepatoma cells. Biophys. J. 1993, 65, 2002–2012. [Google Scholar] [CrossRef] [Green Version]
- Rose, B.; Loewenstein, W.R. Permeability of cell junction depends on local cytoplasmic calcium activity. Nature 1975, 254, 250–252. [Google Scholar] [CrossRef] [PubMed]
- De Mello, W.C. Effect of intracellular injection of calcium and strontium on cell communication in heart. J. Physiol. 1975, 250, 231–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deleze, J. The recovery of resting potential and input resistance in sheep heart injured by knife or laser. J. Physiol. 1970, 208, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Peracchia, C.; Wang, X.G.; Peracchia, L.L. Slow gating of gap junction channels and calmodulin. J. Membr. Biol. 2000, 178, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Peracchia, C.; Sotkis, A.; Wang, X.G.; Peracchia, L.L.; Persechini, A. Calmodulin directly gates gap junction channels. J. Biol. Chem. 2000, 275, 26220–26224. [Google Scholar] [CrossRef] [Green Version]
- Peracchia, C. Calmodulin-Mediated Regulation of Gap Junction Channels. Int. J. Mol. Sci. 2020, 21, 485. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Salarian, M.; Chen, Y.; Zhuo, Y.; Brown, N.E.; Hepler, J.R.; Yang, J.J. Direct visualization of interaction between calmodulin and connexin45. Biochem. J. 2017, 474, 4035–4051. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, W.; Lurtz, M.M.; Ye, Y.; Huang, Y.; Lee, H.W.; Chen, Y.; Louis, C.F.; Yang, J.J. Identification of the calmodulin binding domain of connexin43. J. Biol. Chem. 2007, 282, 35005–35017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noma, A.; Tsuboi, N. Dependence of junctional conductance on proton, calcium and magnesium ions in cardiac paired cells of guinea-pig. J. Physiol. 1987, 382, 193–211. [Google Scholar] [CrossRef]
- White, R.L.; Doeller, J.E.; Verselis, V.K.; Wittenberg, B.A. Gap junctional conductance between pairs of ventricular myocytes is modulated synergistically by H+ and Ca++. J. Gen. Physiol. 1990, 95, 1061–1075. [Google Scholar] [CrossRef] [Green Version]
- Orellana, J.A.; Sanchez, H.A.; Schalper, K.A.; Figueroa, V.; Saez, J.C. Regulation of intercellular calcium signaling through calcium interactions with connexin-based channels. Adv. Exp. Med. Biol. 2012, 740, 777–794. [Google Scholar] [PubMed]
- DeVries, S.H.; Schwartz, E.A. Hemi-gap-junction channels in solitary horizontal cells of the catfish retina. J. Physiol. 1992, 445, 201–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, W.; Ramachandran, J.; Alsamarah, A.; Luo, Y.; Harris, A.L.; Contreras, J.E. Mechanism of gating by calcium in connexin hemichannels. Proc. Natl. Acad. Sci. USA 2016, 113, E7986–E7995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalper, K.A.; Palacios-Prado, N.; Retamal, M.A.; Shoji, K.F.; Martinez, A.D.; Saez, J.C. Connexin hemichannel composition determines the FGF-1-induced membrane permeability and free [Ca2+]i responses. Mol. Biol. Cell 2008, 19, 3501–3513. [Google Scholar] [CrossRef] [Green Version]
- John, S.A.; Kondo, R.; Wang, S.Y.; Goldhaber, J.I.; Weiss, J.N. Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 1999, 274, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Kondo, R.P.; Wang, S.Y.; John, S.A.; Weiss, J.N.; Goldhaber, J.I. Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J. Mol. Cell Cardiol. 2000, 32, 1859–1872. [Google Scholar] [CrossRef]
- Thimm, J.; Mechler, A.; Lin, H.; Rhee, S.; Lal, R. Calcium-dependent open/closed conformations and interfacial energy maps of reconstituted hemichannels. J. Biol. Chem. 2005, 280, 10646–10654. [Google Scholar] [CrossRef] [Green Version]
- De Vuyst, E.; Decrock, E.; Cabooter, L.; Dubyak, G.R.; Naus, C.C.; Evans, W.H.; Leybaert, L. Intracellular calcium changes trigger connexin 32 hemichannel opening. EMBO J. 2006, 25, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vuyst, E.; Wang, N.; Decrock, E.; De Bock, M.; Vinken, M.; Van Moorhem, M.; Lai, C.; Culot, M.; Rogiers, V.; Cecchelli, R.; et al. Ca(2+) regulation of connexin 43 hemichannels in C6 glioma and glial cells. Cell Calcium 2009, 46, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, T.W. Vergleichende Untersuchungen zur Lehre von der Muskel und Nervenelektricität. Pflugers Arch. 1877, 15, 116–148. [Google Scholar] [CrossRef]
- Jalife, J.; Morley, G.E.; Vaidya, D. Connexins and impulse propagation in the mouse heart. J. Cardiovasc. Electrophysiol. 1999, 10, 1649–1663. [Google Scholar] [CrossRef]
- Thevenin, A.F.; Kowal, T.J.; Fong, J.T.; Kells, R.M.; Fisher, C.G.; Falk, M.M. Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology 2013, 28, 93–116. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, S.R.; Billaud, M.; Lohman, A.W.; Taddeo, E.P.; Isakson, B.E. Posttranslational modifications in connexins and pannexins. J. Membr. Biol. 2012, 245, 319–332. [Google Scholar] [CrossRef] [Green Version]
- D’hondt, C.; Iyyathurai, J.; Vinken, M.; Rogiers, V.; Leybaert, L.; Himpens, B.; Bultynck, G. Regulation of connexin- and pannexin-based channels by post-translational modifications. Biol. Cell 2013, 105, 373–398. [Google Scholar] [CrossRef] [Green Version]
- Pogoda, K.; Kameritsch, P.; Retamal, M.A.; Vega, J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: A revision. BMC Cell Biol. 2016, 17 (Suppl. S1), 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampe, P.D.; Lau, A.F. Regulation of gap junctions by phosphorylation of connexins. Arch. Biochem. Biophys. 2000, 384, 205–215. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.C.; Gouw, J.W.; Naus, C.C.; Foster, L.J. Connexin multi-site phosphorylation: Mass spectrometry-based proteomics fills the gap. Biochim. Biophys. Acta 2013, 1828, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.D.; Solan, J.L.; Dolejsi, M.K.; Lampe, P.D. Analysis of connexin phosphorylation sites. Methods 2000, 20, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Abudara, V.; Bechberger, J.; Freitas-Andrade, M.; De Bock, M.; Wang, N.; Bultynck, G.; Naus, C.C.; Leybaert, L.; Giaume, C. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giepmans, B.N. Gap junctions and connexin-interacting proteins. Cardiovasc. Res. 2004, 62, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, D.S.; Beyer, E.C.; Paul, D.L.; Kobe, S.S.; Lau, A.F. Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol. Cell. Biol. 1990, 10, 1754–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquez-Rosado, L.; Solan, J.L.; Dunn, C.A.; Norris, R.P.; Lampe, P.D. Connexin43 phosphorylation in brain, cardiac, endothelial and epithelial tissues. Biochim. Biophys. Acta 2012, 1818, 1985–1992. [Google Scholar] [CrossRef] [Green Version]
- Musil, L.S.; Cunningham, B.A.; Edelman, G.M.; Goodenough, D.A. Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J. Cell Biol. 1990, 111, 2077–2088. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.D.; Lampe, P.D. Casein kinase 1 regulates connexin-43 gap junction assembly. J. Biol. Chem. 2002, 277, 44962–44968. [Google Scholar] [CrossRef] [Green Version]
- Lampe, P.D.; Cooper, C.D.; King, T.J.; Burt, J.M. Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J. Cell Sci. 2006, 119, 3435–3442. [Google Scholar] [CrossRef] [Green Version]
- Van Veen, T.A.; van Rijen, H.V.; Jongsma, H.J. Electrical conductance of mouse connexin45 gap junction channels is modulated by phosphorylation. Cardiovasc. Res. 2000, 46, 496–510. [Google Scholar] [CrossRef] [Green Version]
- Van Rijen, H.V.; van Veen, T.A.; Hermans, M.M.; Jongsma, H.J. Human connexin40 gap junction channels are modulated by cAMP. Cardiovasc. Res. 2000, 45, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Saez, J.C.; Nairn, A.C.; Czernik, A.J.; Fishman, G.I.; Spray, D.C.; Hertzberg, E.L. Phosphorylation of connexin43 and the regulation of neonatal rat cardiac myocyte gap junctions. J. Mol. Cell Cardiol. 1997, 29, 2131–2145. [Google Scholar] [CrossRef]
- Bao, X.; Altenberg, G.A.; Reuss, L. Mechanism of regulation of the gap junction protein connexin 43 by protein kinase C-mediated phosphorylation. Am. J. Physiol. Cell Physiol. 2004, 286, C647–C654. [Google Scholar] [CrossRef]
- Zou, J.; Yue, X.Y.; Zheng, S.C.; Zhang, G.; Chang, H.; Liao, Y.C.; Zhang, Y.; Xue, M.Q.; Qi, Z. Cholesterol modulates function of connexin 43 gap junction channel via PKC pathway in H9c2 cells. Biochim. Biophys. Acta 2014, 1838, 2019–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampe, P.D.; Lau, A.F. The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.K.; Cheng, H.H.; Wang, S.D.; Yeih, D.F.; Wang, S.M. PKCvarepsilon mediates serine phosphorylation of connexin43 induced by lysophosphatidylcholine in neonatal rat cardiomyocytes. Toxicology 2013, 314, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fu, Y.; Peng, J.; Wu, D.; Yu, M.; Xu, C.; Wang, Q.; Tao, L. Simvastatin-induced up-regulation of gap junctions composed of connexin 43 sensitize Leydig tumor cells to etoposide: An involvement of PKC pathway. Toxicology 2013, 312, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; TenBroek, E.M.; Burt, J.M.; Kurata, W.E.; Johnson, R.G.; Lau, A.F. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 2000, 149, 1503–1512. [Google Scholar] [CrossRef]
- Bao, X.; Lee, S.C.; Reuss, L.; Altenberg, G.A. Change in permeant size selectivity by phosphorylation of connexin 43 gap-junctional hemichannels by PKC. Proc. Natl. Acad. Sci. USA 2007, 104, 4919–4924. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Reuss, L.; Altenberg, G.A. Regulation of purified and reconstituted connexin 43 hemichannels by protein kinase C-mediated phosphorylation of Serine 368. J. Biol. Chem. 2004, 279, 20058–20066. [Google Scholar] [CrossRef] [Green Version]
- Hawat, G.; Baroudi, G. Differential modulation of unapposed connexin 43 hemichannel electrical conductance by protein kinase C isoforms. Pflugers Arch. 2008, 456, 519–527. [Google Scholar] [CrossRef]
- Shah, M.M.; Martinez, A.M.; Fletcher, W.H. The connexin43 gap junction protein is phosphorylated by protein kinase A and protein kinase C: In vivo and in vitro studies. Mol. Cell Biochem. 2002, 238, 57–68. [Google Scholar] [CrossRef]
- TenBroek, E.M.; Lampe, P.D.; Solan, J.L.; Reynhout, J.K.; Johnson, R.G. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J. Cell Biol. 2001, 155, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Paulson, A.F.; Lampe, P.D.; Meyer, R.A.; TenBroek, E.; Atkinson, M.M.; Walseth, T.F.; Johnson, R.G. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J. Cell Sci. 2000, 113, 3037–3049. [Google Scholar]
- Burghardt, R.C.; Barhoumi, R.; Sewall, T.C.; Bowen, J.A. Cyclic AMP induces rapid increases in gap junction permeability and changes in the cellular distribution of connexin43. J. Membr. Biol. 1995, 148, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Burt, J.M.; Spray, D.C. Inotropic agents modulate gap junctional conductance between cardiac myocytes. Am. J. Physiol. 1988, 254, H1206–H1210. [Google Scholar] [CrossRef]
- Shah, K.; Gupta, S.; Ghosh, J.; Bajpai, J.; Maheshwari, A. Acute non-ST elevation myocardial infarction following paclitaxel administration for ovarian carcinoma: A case report and review of literature. J. Cancer Res. Ther. 2012, 8, 442–444. [Google Scholar] [PubMed]
- Yogo, K.; Ogawa, T.; Akiyama, M.; Ishida, N.; Takeya, T. Identification and functional analysis of novel phosphorylation sites in Cx43 in rat primary granulosa cells. FEBS Lett. 2002, 531, 132–136. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Robayna, I.J.; Falender, A.E.; Ochsner, S.; Firestone, G.L.; Richards, J.S. Follicle-Stimulating hormone (FSH) stimulates phosphorylation and activation of protein kinase B (PKB/Akt) and serum and glucocorticoid-lnduced kinase (Sgk): Evidence for A kinase-independent signaling by FSH in granulosa cells. Mol. Endocrinol. 2000, 14, 1283–1300. [Google Scholar] [CrossRef] [PubMed]
- Ock, S.; Lee, W.S.; Kim, H.M.; Park, K.S.; Kim, Y.K.; Kook, H.; Park, W.J.; Lee, T.J.; Abel, E.D.; Kim, J. Connexin43 and zonula occludens-1 are targets of Akt in cardiomyocytes that correlate with cardiac contractile dysfunction in Akt deficient hearts. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1183–1191. [Google Scholar] [CrossRef]
- Park, D.J.; Wallick, C.J.; Martyn, K.D.; Lau, A.F.; Jin, C.; Warn-Cramer, B.J. Akt phosphorylates Connexin43 on Ser373, a “mode-1” binding site for 14-3-3. Cell Commun. Adhes. 2007, 14, 211–226. [Google Scholar] [CrossRef] [Green Version]
- Batra, N.; Riquelme, M.A.; Burra, S.; Kar, R.; Gu, S.; Jiang, J.X. Direct regulation of osteocytic connexin 43 hemichannels through AKT kinase activated by mechanical stimulation. J. Biol. Chem. 2014, 289, 10582–10591. [Google Scholar] [CrossRef] [Green Version]
- Dunn, C.A.; Lampe, P.D. Injury-triggered Akt phosphorylation of Cx43: A ZO-1-driven molecular switch that regulates gap junction size. J. Cell Sci. 2014, 127, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Takens-Kwak, B.R.; Jongsma, H.J. Cardiac gap junctions: Three distinct single channel conductances and their modulation by phosphorylating treatments. Pflugers Arch. 1992, 422, 198–200. [Google Scholar] [CrossRef]
- Kwak, B.R.; Saez, J.C.; Wilders, R.; Chanson, M.; Fishman, G.I.; Hertzberg, E.L.; Spray, D.C.; Jongsma, H.J. Effects of cGMP-dependent phosphorylation on rat and human connexin43 gap junction channels. Pflugers Arch. 1995, 430, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.R.; Jongsma, H.J. Regulation of cardiac gap junction channel permeability and conductance by several phosphorylating conditions. Mol. Cell Biochem. 1996, 157, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Laing, J.G.; Kanter, E.M.; Berthoud, V.M.; Bao, M.; Rohrs, H.W.; Townsend, R.R.; Yamada, K.A. Identification of CaMKII phosphorylation sites in Connexin43 by high-resolution mass spectrometry. J. Proteome Res. 2011, 10, 1098–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hund, T.J.; Decker, K.F.; Kanter, E.; Mohler, P.J.; Boyden, P.A.; Schuessler, R.B.; Yamada, K.A.; Rudy, Y. Role of activated CaMKII in abnormal calcium homeostasis and I(Na) remodeling after myocardial infarction: Insights from mathematical modeling. J. Mol. Cell Cardiol. 2008, 45, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelsen, L.N.; Stahlhut, M.; Mohammed, S.; Larsen, B.D.; Nielsen, M.S.; Holstein-Rathlou, N.H.; Andersen, S.; Jensen, O.N.; Hennan, J.K.; Kjolbye, A.L. Identification of ischemia-regulated phosphorylation sites in connexin43: A possible target for the antiarrhythmic peptide analogue rotigaptide (ZP123). J. Mol. Cell Cardiol. 2006, 40, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Warn-Cramer, B.J.; Cottrell, G.T.; Burt, J.M.; Lau, A.F. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, 9188–9196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warn-Cramer, B.J.; Lampe, P.D.; Kurata, W.E.; Kanemitsu, M.Y.; Loo, L.W.; Eckhart, W.; Lau, A.F. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J. Biol. Chem. 1996, 271, 3779–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimlamool, W.; Andrews, R.M.; Falk, M.M. Connexin43 phosphorylation by PKC and MAPK signals VEGF-mediated gap junction internalization. Mol. Biol. Cell 2015, 26, 2755–2768. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.E.; Mitra, S.; Katoch, P.; Kelsey, L.S.; Johnson, K.R.; Mehta, P.P. Phosphorylation on Ser-279 and Ser-282 of connexin43 regulates endocytosis and gap junction assembly in pancreatic cancer cells. Mol. Biol. Cell 2013, 24, 715–733. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, G.T.; Lin, R.; Warn-Cramer, B.J.; Lau, A.F.; Burt, J.M. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am. J. Physiol. Cell Physiol. 2003, 284, C511–C520. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kam, Y.; Koo, S.K.; Joe, C.O. Gating connexin 43 channels reconstituted in lipid vesicles by mitogen-activated protein kinase phosphorylation. J. Biol. Chem. 1999, 274, 5581–5587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, G.J.; Chen, Q.; Chen, L.J.; Shu, Y.; Bu, L.L.; Shao, X.Y.; Zhang, P.; Jiao, F.J.; Shi, J.; Tian, B. Phosphorylation of Connexin 43 by Cdk5 Modulates Neuronal Migration During Embryonic Brain Development. Mol. Neurobiol. 2016, 53, 2969–2982. [Google Scholar] [CrossRef] [PubMed]
- Kanemitsu, M.Y.; Jiang, W.; Eckhart, W. Cdc2-mediated phosphorylation of the gap junction protein, connexin43, during mitosis. Cell Growth Differ. 1998, 9, 13–21. [Google Scholar]
- Lampe, P.D.; Kurata, W.E.; Warn-Cramer, B.J.; Lau, A.F. Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J. Cell Sci. 1998, 111, 833–841. [Google Scholar]
- Solan, J.L.; Lampe, P.D. Src Regulation of Cx43 Phosphorylation and Gap Junction Turnover. Biomolecules 2020, 10, 1596. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun. Adhes. 2008, 15, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warn-Cramer, B.J.; Lau, A.F. Regulation of gap junctions by tyrosine protein kinases. Biochim. Biophys. Acta 2004, 1662, 81–95. [Google Scholar] [CrossRef] [Green Version]
- Loo, L.W.; Berestecky, J.M.; Kanemitsu, M.Y.; Lau, A.F. pp60src-mediated phosphorylation of connexin 43, a gap junction protein. J. Biol. Chem. 1995, 270, 12751–12761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanemitsu, M.Y.; Loo, L.W.; Simon, S.; Lau, A.F.; Eckhart, W. Tyrosine phosphorylation of connexin 43 by v-Src is mediated by SH2 and SH3 domain interactions. J. Biol. Chem. 1997, 272, 22824–22831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, L.W.; Kanemitsu, M.Y.; Lau, A.F. In vivo association of pp60v-src and the gap-junction protein connexin 43 in v-src-transformed fibroblasts. Mol Carcinog. 1999, 25, 187–195. [Google Scholar] [CrossRef]
- Lin, R.; Warn-Cramer, B.J.; Kurata, W.E.; Lau, A.F. v-Src phosphorylation of connexin 43 on Tyr247 and Tyr265 disrupts gap junctional communication. J. Cell Biol. 2001, 154, 815–827. [Google Scholar] [CrossRef]
- Zhou, L.; Kasperek, E.M.; Nicholson, B.J. Dissection of the molecular basis of pp60(v-src) induced gating of connexin 43 gap junction channels. J. Cell Biol. 1999, 144, 1033–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolon, M.L.; Peng, T.; Kidder, G.M.; Tyml, K. Lipopolysaccharide plus hypoxia and reoxygenation synergistically reduce electrical coupling between microvascular endothelial cells by dephosphorylating connexin40. J. Cell Physiol. 2008, 217, 350–359. [Google Scholar] [CrossRef]
- Bao, M.; Kanter, E.M.; Huang, R.Y.; Maxeiner, S.; Frank, M.; Zhang, Y.; Schuessler, R.B.; Smith, T.W.; Townsend, R.R.; Rohrs, H.W.; et al. Residual Cx45 and its relationship to Cx43 in murine ventricular myocardium. Channels 2011, 5, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, I.E.; Sanchez, H.A.; Martinez, A.D.; Retamal, M.A. Redox-mediated regulation of connexin proteins; focus on nitric oxide. Biochim. Biophys. Acta 2018, 1860, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A. Connexin and Pannexin hemichannels are regulated by redox potential. Front. Physiol. 2014, 5, 80. [Google Scholar] [CrossRef] [Green Version]
- Retamal, M.A.; Garcia, I.E.; Pinto, B.I.; Pupo, A.; Baez, D.; Stehberg, J.; Del Rio, R.; Gonzalez, C. Extracellular Cysteine in Connexins: Role as Redox Sensors. Front. Physiol. 2016, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, A.C.; Billaud, M.; Johnstone, S.R.; Best, A.K.; Yemen, S.; Dwyer, S.T.; Looft-Wilson, R.; Lysiak, J.J.; Gaston, B.; Palmer, L.; et al. Compartmentalized connexin 43 s-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Miskolczi, G.; Gonczi, M.; Kovacs, M.; Vegh, A. Examination of the effect of sodium nitrite on gap junction function during ischaemia and reperfusion in anaesthetized dogs. Acta Biol. Hung. 2017, 68, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Contreras, J.E.; Sanchez, H.A.; Eugenin, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.; Saez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar] [CrossRef] [Green Version]
- Retamal, M.A.; Cortes, C.J.; Reuss, L.; Bennett, M.V.; Saez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef] [Green Version]
- Retamal, M.A.; Schalper, K.A.; Shoji, K.F.; Bennett, M.V.; Saez, J.C. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential. Proc. Natl. Acad. Sci. USA 2007, 104, 8322–8327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirca, M.; Kleinbongard, P.; Soetkamp, D.; Heger, J.; Csonka, C.; Ferdinandy, P.; Schulz, R. Interaction between connexin 43 and nitric oxide synthase in mice heart mitochondria. J. Cell Mol. Med. 2015, 19, 815–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon-Paravic, C.G.; Figueroa, V.A.; Guzman, D.J.; Valderrama, C.F.; Vallejos, A.A.; Fiori, M.C.; Altenberg, G.A.; Reuss, L.; Retamal, M.A. Carbon monoxide (CO) is a novel inhibitor of connexin hemichannels. J. Biol. Chem. 2014, 289, 36150–36157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimann, F.J.; Dröse, S.; Bonke, E.; Schneider, L.; Tybl, E.; Wittig, I.; Heidler, J.; Heide, H.; Josipovic, I.; Leisegang, M.; et al. TLR2-Dependent Reversible Oxidation of Connexin 43 at Cys260 Modifies Electrical Coupling After Experimental Myocardial Ischemia/Reperfusion. J. Cardiovasc. Transl. Res. 2019, 12, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Sakata, Y.; Dong, J.W.; Vallejo, J.G.; Huang, C.H.; Baker, J.S.; Tracey, K.J.; Tacheuchi, O.; Akira, S.; Mann, D.L. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H503–H509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, S.; Xie, L.H.; John, S.A.; Subramaniam, S.; Lal, R. A novel role for connexin hemichannel in oxidative stress and smoking-induced cell injury. PLoS ONE 2007, 2, e712. [Google Scholar] [CrossRef] [Green Version]
- Colussi, C.; Rosati, J.; Straino, S.; Spallotta, F.; Berni, R.; Stilli, D.; Rossi, S.; Musso, E.; Macchi, E.; Mai, A.; et al. Nε-lysyne acetylation determines dissociation from GAP junctions and lateralization of connexin 43 in normal and dystrophic heart. Proc. Natl. Acad. Sci. USA 2011, 108, 2795–2800. [Google Scholar] [CrossRef] [Green Version]
- Meraviglia, V.; Azzimato, V.; Colussi, C.; Florio, M.C.; Binda, A.; Panariti, A.; Qanud, K.; Suffredini, S.; Gennaccaro, L.; Miragoli, M.; et al. Acetylation mediates Cx43 reduction caused by electrical stimulation. J. Mol. Cell Cardiol. 2015, 87, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjenseth, A.; Fykerud, T.A.; Sirnes, S.; Bruun, J.; Yohannes, Z.; Kolberg, M.; Omori, Y.; Rivedal, E.; Leithe, E. The Gap Junction Channel Protein Connexin 43 Is Covalently Modified and Regulated by SUMOylation. J. Biol. Chem. 2012, 287, 15851–15861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Scholz, C.; Kelstrup, C.D.; Young, C.; Nielsen, M.L.; Olsen, J.V.; Brakebusch, C.; Choudhary, C. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol. Cell Proteom. 2012, 11, 1578–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basheer, W.A.; Harris, B.S.; Mentrup, H.L.; Abreha, M.; Thames, E.L.; Lea, J.B.; Swing, D.A.; Copeland, N.G.; Jenkins, N.A.; Price, R.L.; et al. Cardiomyocyte-specific overexpression of the ubiquitin ligase Wwp1 contributes to reduction in Connexin 43 and arrhythmogenesis. J. Mol. Cell Cardiol. 2015, 88, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fykerud, T.A.; Kjenseth, A.; Schink, K.O.; Sirnes, S.; Bruun, J.; Omori, Y.; Brech, A.; Rivedal, E.; Leithe, E. Smad ubiquitination regulatory factor-2 controls gap junction intercellular communication by modulating endocytosis and degradation of connexin43. J. Cell Sci. 2012, 125, 3966–3976. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.L.; Lai, S.Y.; Lai, W.A.; Lee, M.T.; Liao, C.F.; Ke, F.C.; Hwang, J.J. CRTC2 and Nedd4 ligase involvement in FSH and TGFβ1 upregulation of connexin43 gap junction. J. Mol. Endocrinol. 2015, 55, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Catarino, S.; Ramalho, J.S.; Marques, C.; Pereira, P.; Girao, H. Ubiquitin-mediated internalization of connexin43 is independent of the canonical endocytic tyrosine-sorting signal. Biochem. J. 2011, 437, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010, 20, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Decrock, E.; Leybaert, L.; Bultynck, G.; Himpens, B.; Vanhaecke, T.; Rogiers, V. Non-channel functions of connexins in cell growth and cell death. Biochim. Biophys. Acta 2012, 1818, 2002–2008. [Google Scholar] [CrossRef] [PubMed]
- Herve, J.C.; Derangeon, M.; Sarrouilhe, D.; Giepmans, B.N.; Bourmeyster, N. Gap junctional channels are parts of multiprotein complexes. Biochim. Biophys. Acta 2012, 1818, 1844–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins-Marques, T.; Anjo, S.I.; Pereira, P.; Manadas, B.; Girao, H. Interacting Network of the Gap Junction (GJ) Protein Connexin43 (Cx43) is Modulated by Ischemia and Reperfusion in the Heart. Mol. Cell Proteom. 2015, 14, 3040–3055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giepmans, B.N.; Verlaan, I.; Hengeveld, T.; Janssen, H.; Calafat, J.; Falk, M.M.; Moolenaar, W.H. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 2001, 11, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Saidi Brikci-Nigassa, A.; Clement, M.J.; Ha-Duong, T.; Adjadj, E.; Ziani, L.; Pastre, D.; Curmi, P.A.; Savarin, P. Phosphorylation controls the interaction of the connexin43 C-terminal domain with tubulin and microtubules. Biochemistry 2012, 51, 4331–4342. [Google Scholar] [CrossRef]
- Giepmans, B.N.; Verlaan, I.; Moolenaar, W.H. Connexin-43 interactions with ZO-1 and alpha- and beta-tubulin. Cell Commun. Adhes. 2001, 8, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Rhee, D.Y.; Zhao, X.Q.; Francis, R.J.; Huang, G.Y.; Mably, J.D.; Lo, C.W. Connexin 43 regulates epicardial cell polarity and migration in coronary vascular development. Development 2009, 136, 3185–3193. [Google Scholar] [CrossRef] [Green Version]
- Giepmans, B.N.; Moolenaar, W.H. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr. Biol. 1998, 8, 931–934. [Google Scholar] [CrossRef] [Green Version]
- Toyofuku, T.; Yabuki, M.; Otsu, K.; Kuzuya, T.; Hori, M.; Tada, M. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J. Biol. Chem. 1998, 273, 12725–12731. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Barker, R.J.; Hunter, A.W.; Zhang, Y.; Jourdan, J.; Gourdie, R.G. Quantitative analysis of ZO-1 colocalization with Cx43 gap junction plaques in cultures of rat neonatal cardiomyocytes. Microsc. Microanal. 2005, 11, 244–248. [Google Scholar] [CrossRef]
- Hunter, A.W.; Barker, R.J.; Zhu, C.; Gourdie, R.G. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol. Biol. Cell 2005, 16, 5686–5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maass, K.; Shibayama, J.; Chase, S.E.; Willecke, K.; Delmar, M. C-terminal truncation of connexin43 changes number, size, and localization of cardiac gap junction plaques. Circ. Res. 2007, 101, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Wayakanon, P.; Bhattacharjee, R.; Nakahama, K.; Morita, I. The role of the Cx43 C-terminus in GJ plaque formation and internalization. Biochem. Biophys. Res. Commun. 2012, 420, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Butkevich, E.; Hulsmann, S.; Wenzel, D.; Shirao, T.; Duden, R.; Majoul, I. Drebrin is a novel connexin-43 binding partner that links gap junctions to the submembrane cytoskeleton. Curr. Biol. 2004, 14, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosi, C.; Ren, C.; Spagnol, G.; Cavin, G.; Cone, A.; Grintsevich, E.E.; Sosinsky, G.E.; Sorgen, P.L. Connexin43 Forms Supramolecular Complexes through Non-Overlapping Binding Sites for Drebrin, Tubulin, and ZO-1. PLoS ONE 2016, 11, e0157073. [Google Scholar] [CrossRef]
- Guillemot, L.; Paschoud, S.; Pulimeno, P.; Foglia, A.; Citi, S. The cytoplasmic plaque of tight junctions: A scaffolding and signalling center. Biochim. Biophys. Acta 2008, 1778, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, K.; Chiba, H.; Fujita, H.; Kojima, T.; Saito, T.; Endo, T.; Sawada, N. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J. Cell Physiol. 2006, 208, 123–132. [Google Scholar] [CrossRef]
- Niessen, C.M.; Gottardi, C.J. Molecular components of the adherens junction. Biochim. Biophys. Acta 2008, 1778, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.J.; Francis, R.; Xu, X.; Lo, C.W. Connexin43 associated with an N-cadherin-containing multiprotein complex is required for gap junction formation in NIH3T3 cells. J. Biol. Chem. 2005, 280, 19925–19936. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.C.; Tsai, R.Y.; Chung, T.H. Role of catenins in the development of gap junctions in rat cardiomyocytes. J. Cell Biochem 2003, 88, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Sato, P.Y.; Coombs, W.; Lin, X.; Nekrasova, O.; Green, K.J.; Isom, L.L.; Taffet, S.M.; Delmar, M. Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ. Res. 2011, 109, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agullo-Pascual, E.; Reid, D.A.; Keegan, S.; Sidhu, M.; Fenyo, D.; Rothenberg, E.; Delmar, M. Super-resolution fluorescence microscopy of the cardiac connexome reveals plakophilin-2 inside the connexin43 plaque. Cardiovasc. Res. 2013, 100, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Cruz, F.M.; Sanz-Rosa, D.; Roche-Molina, M.; Garcia-Prieto, J.; Garcia-Ruiz, J.M.; Pizarro, G.; Jimenez-Borreguero, L.J.; Torres, M.; Bernad, A.; Ruiz-Cabello, J.; et al. Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J. Am. Coll Cardiol. 2015, 65, 1438–1450. [Google Scholar] [CrossRef]
- Batra, N.; Burra, S.; Siller-Jackson, A.J.; Gu, S.; Xia, X.; Weber, G.F.; DeSimone, D.; Bonewald, L.F.; Lafer, E.M.; Sprague, E.; et al. Mechanical stress-activated integrin α5β1 induces opening of connexin 43 hemichannels. Proc. Natl. Acad. Sci. USA 2012, 109, 3359–3364. [Google Scholar] [CrossRef] [Green Version]
- Zemljic-Harpf, A.E.; Godoy, J.C.; Platoshyn, O.; Asfaw, E.K.; Busija, A.R.; Domenighetti, A.A.; Ross, R.S. Vinculin directly binds zonula occludens-1 and is essential for stabilizing connexin-43-containing gap junctions in cardiac myocytes. J. Cell Sci. 2014, 127, 1104–1116. [Google Scholar] [CrossRef] [Green Version]
- Agullo-Pascual, E.; Delmar, M. The noncanonical functions of Cx43 in the heart. J. Membr. Biol. 2012, 245, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Delmar, M. Connexin43 regulates sodium current; ankyrin-G modulates gap junctions: The intercalated disc exchanger. Cardiovasc. Res. 2012, 93, 220–222. [Google Scholar] [CrossRef]
- Petitprez, S.; Zmoos, A.F.; Ogrodnik, J.; Balse, E.; Raad, N.; El Haou, S.; Albesa, M.; Bittihn, P.; Luther, S.; Lehnart, S.E.; et al. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ. Res. 2011, 108, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Liu, N.; Lu, J.; Zhang, J.; Anumonwo, J.M.; Isom, L.L.; Fishman, G.I.; Delmar, M. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm 2011, 8, 1923–1930. [Google Scholar] [CrossRef] [Green Version]
- Oxford, E.M.; Musa, H.; Maass, K.; Coombs, W.; Taffet, S.M.; Delmar, M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ. Res. 2007, 101, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Sato, P.Y.; Musa, H.; Coombs, W.; Guerrero-Serna, G.; Patiño, G.A.; Taffet, S.M.; Isom, L.L.; Delmar, M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 2009, 105, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Jansen, J.A.; Noorman, M.; Musa, H.; Stein, M.; de Jong, S.; van der, N.R.; Hund, T.J.; Mohler, P.J.; Vos, M.A.; van Veen, T.A.; et al. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm 2012, 9, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Morley, G.E.; Vaidya, D.; Samie, F.H.; Lo, C.; Delmar, M.; Jalife, J. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J. Cardiovasc. Electrophysiol. 1999, 10, 1361–1375. [Google Scholar] [CrossRef]
- Danik, S.B.; Liu, F.; Zhang, J.; Suk, H.J.; Morley, G.E.; Fishman, G.I.; Gutstein, D.E. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ. Res. 2004, 95, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.A.; Rodriguez-Sinovas, A.; Fernandez-Sanz, C.; Ruiz-Meana, M.; Garcia-Dorado, D. Effects of a reduction in the number of gap junction channels or in their conductance on ischemia-reperfusion arrhythmias in isolated mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2442–H2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongsma, H.J.; Wilders, R. Gap junctions in cardiovascular disease. Circ. Res. 2000, 86, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Sperelakis, N. Electric field model: An alternative mechanism for cell-to-cell propagation in cardiac muscle and smooth muscle. J. Gastrointest. Mot. 1991, 3, 64–83. [Google Scholar] [CrossRef]
- Sperelakis, N. An electric field mechanism for transmission of excitation between myocardial cells. Circ. Res. 2002, 91, 985–987. [Google Scholar] [CrossRef]
- Kleber, A.G.; Saffitz, J.E. Role of the intercalated disc in cardiac propagation and arrhythmogenesis. Front. Physiol. 2014, 5, 404. [Google Scholar] [CrossRef] [Green Version]
- Veeraraghavan, R.; Poelzing, S.; Gourdie, R.G. Intercellular electrical communication in the heart: A new, active role for the intercalated disk. Cell Commun. Adhes. 2014, 21, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Kucera, J.P.; Rohr, S.; Rudy, Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ. Res. 2002, 91, 1176–1182. [Google Scholar] [CrossRef] [Green Version]
- Schubert, A.L.; Schubert, W.; Spray, D.C.; Lisanti, M.P. Connexin family members target to lipid raft domains and interact with caveolin-1. Biochemistry 2002, 41, 5754–5764. [Google Scholar] [CrossRef]
- Langlois, S.; Cowan, K.N.; Shao, Q.; Cowan, B.J.; Laird, D.W. Caveolin-1 and -2 interact with connexin43 and regulate gap junctional intercellular communication in keratinocytes. Mol. Biol. Cell 2008, 19, 912–928. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Zhou, J.; Zelenka, P.S.; Takemoto, D.J. Protein kinase Cgamma regulation of gap junction activity through caveolin-1-containing lipid rafts. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5259–5268. [Google Scholar] [CrossRef]
- Rinaldi, F.; Hartfield, E.M.; Crompton, L.A.; Badger, J.L.; Glover, C.P.; Kelly, C.M.; Rosser, A.E.; Uney, J.B.; Caldwell, M.A. Cross-regulation of Connexin43 and β-catenin influences differentiation of human neural progenitor cells. Cell Death Dis. 2014, 5, e1017. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.T.; Bechberger, J.F.; Ozog, M.A.; Perbal, B.; Naus, C.C. CCN3 (NOV) interacts with connexin43 in C6 glioma cells: Possible mechanism of connexin-mediated growth suppression. J. Biol. Chem. 2004, 279, 36943–36950. [Google Scholar] [CrossRef] [Green Version]
- Sin, W.C.; Tse, M.; Planque, N.; Perbal, B.; Lampe, P.D.; Naus, C.C. Matricellular protein CCN3 (NOV) regulates actin cytoskeleton reorganization. J. Biol. Chem. 2009, 284, 29935–29944. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, S.R.; Kroncke, B.M.; Straub, A.C.; Best, A.K.; Dunn, C.A.; Mitchell, L.A.; Peskova, Y.; Nakamoto, R.K.; Koval, M.; Lo, C.W.; et al. MAPK phosphorylation of connexin 43 promotes binding of cyclin E and smooth muscle cell proliferation. Circ. Res. 2012, 111, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, T.; Dai, P.; Harada, Y.; Hino, H.; Tsukahara, F.; Maru, Y.; Otsuji, E.; Takamatsu, T. Connexin43 functions as a novel interacting partner of heat shock cognate protein 70. Sci. Rep. 2013, 3, 2719. [Google Scholar] [CrossRef] [Green Version]
- Bivi, N.; Lezcano, V.; Romanello, M.; Bellido, T.; Plotkin, L.I. Connexin43 interacts with β-arrestin: A pre-requisite for osteoblast survival induced by parathyroid hormone. J. Cell Biochem. 2011, 112, 2920–2930. [Google Scholar] [CrossRef] [Green Version]
- Denuc, A.; Nunez, E.; Calvo, E.; Loureiro, M.; Miro-Casas, E.; Guaras, A.; Vazquez, J.; Garcia-Dorado, D. New protein-protein interactions of mitochondrial connexin 43 in mouse heart. J. Cell Mol. Med. 2016, 20, 794–803. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhao, X.; Yao, Y.; Qi, X.; Yuan, Y.; Hu, Y. Connexin 43 interacts with Bax to regulate apoptosis of pancreatic cancer through a gap junction-independent pathway. Int. J. Oncol. 2012, 41, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Waza, A.A.; Andrabi, K.; Hussain, M.U. Protein kinase C (PKC) mediated interaction between conexin43 (Cx43) and K(+)(ATP) channel subunit (Kir6.1) in cardiomyocyte mitochondria: Implications in cytoprotection against hypoxia induced cell apoptosis. Cell Signal. 2014, 26, 1909–1917. [Google Scholar] [CrossRef]
- Schmidtmann, M. Über die intracellulaire Wasserstoffionenkonzentration unter physiologischen und einigen pathologischen Bedingungen. Z Gesamte Exp. Med. 1925, 45, 714–742. [Google Scholar] [CrossRef]
- Sjostrand, F.S.; Andersson-Cedergren, E.; Dewey, M.M. The ultrastructure of the intercalated discs of frog, mouse and guinea pig cardiac muscle. J. Ultrastruct. Res. 1958, 1, 271–287. [Google Scholar] [CrossRef]
- Gilula, N.B.; Reeves, O.R.; Steinbach, A. Metabolic coupling, ionic coupling and cell contacts. Nature 1972, 235, 262–265. [Google Scholar] [CrossRef]
- Ongstad, E.; Kohl, P. Fibroblast-myocyte coupling in the heart: Potential relevance for therapeutic interventions. J. Mol. Cell Cardiol. 2016, 91, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Lambiase, P.D.; Tinker, A. Connexins in the heart. Cell Tissue Res. 2015, 360, 675–684. [Google Scholar] [CrossRef]
- Zhou, P.; Pu, W.T. Recounting Cardiac Cellular Composition. Circ. Res. 2016, 118, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos, R.C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongbloed, M.R.; Mahtab, E.A.; Blom, N.A.; Schalij, M.J.; Gittenberger-de Groot, A.C. Development of the cardiac conduction system and the possible relation to predilection sites of arrhythmogenesis. Sci. World J. 2008, 8, 239–269. [Google Scholar] [CrossRef] [PubMed]
- Van Kempen, M.J.; Velde, I.T.; Wessels, A.; Oosthoek, P.W.; Gros, D.; Jongsma, H.J.; Moorman, A.F.; Lamers, W.H. Differential connexin distribution accommodates cardiac function in different species. Microsc. Res. Tech. 1995, 31, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Gros, D.B.; Jongsma, H.J. Connexins in mammalian heart function. Bioessays 1996, 18, 719–730. [Google Scholar] [CrossRef]
- Van Kempen, M.J.; Fromaget, C.; Gros, D.; Moorman, A.F.; Lamers, W.H. Spatial distribution of connexin43, the major cardiac gap junction protein, in the developing and adult rat heart. Circ. Res. 1991, 68, 1638–1651. [Google Scholar] [CrossRef] [Green Version]
- Vozzi, C.; Dupont, E.; Coppen, S.R.; Yeh, H.I.; Severs, N.J. Chamber-related differences in connexin expression in the human heart. J. Mol. Cell Cardiol. 1999, 31, 991–1003. [Google Scholar] [CrossRef]
- Coppen, S.R.; Dupont, E.; Rothery, S.; Severs, N.J. Connexin45 expression is preferentially associated with the ventricular conduction system in mouse and rat heart. Circ. Res. 1998, 82, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Kreuzberg, M.M.; Sohl, G.; Kim, J.S.; Verselis, V.K.; Willecke, K.; Bukauskas, F.F. Functional properties of mouse connexin30.2 expressed in the conduction system of the heart. Circ. Res. 2005, 96, 1169–1177. [Google Scholar] [CrossRef] [Green Version]
- Kreuzberg, M.M.; Liebermann, M.; Segschneider, S.; Dobrowolski, R.; Dobrzynski, H.; Kaba, R.; Rowlinson, G.; Dupont, E.; Severs, N.J.; Willecke, K. Human connexin31.9, unlike its orthologous protein connexin30.2 in the mouse, is not detectable in the human cardiac conduction system. J. Mol. Cell Cardiol. 2009, 46, 553–559. [Google Scholar] [CrossRef]
- Chi, N.C.; Bussen, M.; Brand-Arzamendi, K.; Ding, C.; Olgin, J.E.; Shaw, R.M.; Martin, G.R.; Stainier, D.Y. Cardiac conduction is required to preserve cardiac chamber morphology. Proc. Natl. Acad. Sci. USA 2010, 107, 14662–14667. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.A.; Rodriguez-Sinovas, A.; Barba, I.; Miro-Casas, E.; Fernandez-Sanz, C.; Ruiz-Meana, M.; Alburquerque-Bejar, J.J.; Garcia-Dorado, D. Activation of RISK and SAFE pathways is not involved in the effects of Cx43 deficiency on tolerance to ischemia-reperfusion injury and preconditioning protection. Basic Res. Cardiol. 2013, 108, 351. [Google Scholar] [CrossRef]
- Remo, B.F.; Giovannone, S.; Fishman, G.I. Connexin43 cardiac gap junction remodeling: Lessons from genetically engineered murine models. J. Membr. Biol. 2012, 245, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Beardslee, M.A.; Lerner, D.L.; Tadros, P.N.; Laing, J.G.; Beyer, E.C.; Yamada, K.A.; Kleber, A.G.; Schuessler, R.B.; Saffitz, J.E. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ. Res. 2000, 87, 656–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, T.; Ohnuma, Y.; Kuno, A.; Tanno, M.; Ichikawa, Y.; Nakamura, Y.; Yano, T.; Miki, T.; Sakamoto, J.; Shimamoto, K. Protective role of gap junctions in preconditioning against myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H214–H221. [Google Scholar] [CrossRef] [PubMed]
- Severs, N.J.; Coppen, S.R.; Dupont, E.; Yeh, H.I.; Ko, Y.S.; Matsushita, T. Gap junction alterations in human cardiac disease. Cardiovasc. Res. 2004, 62, 368–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.H.; Green, C.R.; Peters, N.S.; Rothery, S.; Severs, N.J. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am. J. Pathol. 1991, 139, 801–821. [Google Scholar]
- Peters, N.S.; Coromilas, J.; Severs, N.J.; Wit, A.L. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation 1997, 95, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Tansey, E.E.; Kwaku, K.F.; Hammer, P.E.; Cowan, D.B.; Federman, M.; Levitsky, S.; McCully, J.D. Reduction and redistribution of gap and adherens junction proteins after ischemia and reperfusion. Ann. Thorac. Surg. 2006, 82, 1472–1479. [Google Scholar] [CrossRef] [Green Version]
- Vetterlein, F.; Muhlfeld, C.; Cetegen, C.; Volkmann, R.; Schrader, C.; Hellige, G. Redistribution of connexin43 in regional acute ischemic myocardium: Influence of ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H813–H819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins-Marques, T.; Catarino, S.; Marques, C.; Matafome, P.; Ribeiro-Rodrigues, T.; Baptista, R.; Pereira, P.; Girao, H. Heart ischemia results in connexin43 ubiquitination localized at the intercalated discs. Biochimie 2015, 112, 196–201. [Google Scholar] [CrossRef]
- Cabo, C.; Yao, J.; Boyden, P.A.; Chen, S.; Hussain, W.; Duffy, H.S.; Ciaccio, E.J.; Peters, N.S.; Wit, A.L. Heterogeneous gap junction remodeling in reentrant circuits in the epicardial border zone of the healing canine infarct. Cardiovasc. Res. 2006, 72, 241–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.K.; Schuessler, R.B.; Saffitz, J.E. Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ. Res. 2003, 92, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.M.; Lin, M.S.; Chou, T.F.; Tsai, C.H.; Chang, N.C. Adjunctive 17beta-estradiol administration reduces infarct size by altered expression of canine myocardial connexin43 protein. Cardiovasc. Res. 2004, 63, 109–117. [Google Scholar] [CrossRef]
- Zhang, Y.; Kakinuma, Y.; Ando, M.; Katare, R.G.; Yamasaki, F.; Sugiura, T.; Sato, T. Acetylcholine inhibits the hypoxia-induced reduction of connexin43 protein in rat cardiomyocytes. J. Pharmacol. Sci. 2006, 101, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Shintani-Ishida, K.; Unuma, K.; Yoshida, K. Ischemia enhances translocation of connexin43 and gap junction intercellular communication, thereby propagating contraction band necrosis after reperfusion. Circ. J. 2009, 73, 1661–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srisakuldee, W.; Jeyaraman, M.M.; Nickel, B.E.; Tanguy, S.; Jiang, Z.S.; Kardami, E. Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc. Res. 2009, 83, 672–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, D.; Cruciani, V.; Sundset, R.; Ytrehus, K.; Mikalsen, S.O. Ischemia induces closure of gap junctional channels and opening of hemichannels in heart-derived cells and tissue. Cell Physiol. Biochem. 2011, 28, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Yan, X.; Yang, Y.; Chen, M.; Wu, L.; Gou, Z.; Sun, Z.; Talabieke, S.; Zheng, Y.; Luo, D. Connexin 43 dephosphorylation contributes to arrhythmias and cardiomyocyte apoptosis in ischemia/reperfusion hearts. Basic Res. Cardiol. 2019, 114, 40. [Google Scholar] [CrossRef]
- Luke, R.A.; Saffitz, J.E. Remodeling of ventricular conduction pathways in healed canine infarct border zones. J. Clin. Investig. 1991, 87, 1594–1602. [Google Scholar] [CrossRef]
- Kostin, S.; Rieger, M.; Dammer, S.; Hein, S.; Richter, M.; Klövekorn, W.P.; Bauer, E.P.; Schaper, J. Gap junction remodeling and altered connexin43 expression in the failing human heart. Mol. Cell Biochem. 2003, 242, 135–144. [Google Scholar] [CrossRef]
- Kostin, S.; Dammer, S.; Hein, S.; Klovekorn, W.P.; Bauer, E.P.; Schaper, J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc. Res. 2004, 62, 426–436. [Google Scholar] [CrossRef]
- Dupont, E.; Matsushita, T.; Kaba, R.A.; Vozzi, C.; Coppen, S.R.; Khan, N.; Kaprielian, R.; Yacoub, M.H.; Severs, N.J. Altered connexin expression in human congestive heart failure. J. Mol. Cell Cardiol. 2001, 33, 359–371. [Google Scholar] [CrossRef]
- Kitamura, H.; Ohnishi, Y.; Yoshida, A.; Okajima, K.; Azumi, H.; Ishida, A.; Galeano, E.J.; Kubo, S.; Hayashi, Y.; Itoh, H.; et al. Heterogeneous loss of connexin43 protein in nonischemic dilated cardiomyopathy with ventricular tachycardia. J. Cardiovasc. Electrophysiol. 2002, 13, 865–870. [Google Scholar] [CrossRef]
- Chkourko, H.S.; Guerrero-Serna, G.; Lin, X.; Darwish, N.; Pohlmann, J.R.; Cook, K.E.; Martens, J.R.; Rothenberg, E.; Musa, H.; Delmar, M. Remodeling of mechanical junctions and of microtubule-associated proteins accompany cardiac connexin43 lateralization. Heart Rhythm 2012, 9, 1133–1140. [Google Scholar] [CrossRef] [Green Version]
- Tribulova, N.; Okruhlicova, L.; Imanaga, I.; Hirosawa, N.; Ogawa, K.; Weismann, P. Factors involved in the susceptibility of spontaneously hypertensive rats to low K+-induced arrhythmias. Gen. Physiol. Biophys. 2003, 22, 369–382. [Google Scholar]
- Bruce, A.F.; Rothery, S.; Dupont, E.; Severs, N.J. Gap junction remodelling in human heart failure is associated with increased interaction of connexin43 with ZO-1. Cardiovasc. Res. 2008, 77, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Sasano, C.; Honjo, H.; Takagishi, Y.; Uzzaman, M.; Emdad, L.; Shimizu, A.; Murata, Y.; Kamiya, K.; Kodama, I. Internalization and dephosphorylation of connexin43 in hypertrophied right ventricles of rats with pulmonary hypertension. Circ. J. 2007, 71, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, M.S.; Mihm, M.J.; Cook, A.C.; Schanbacher, B.L.; Bauer, J.A. Alterations in connexin 43 during diabetic cardiomyopathy: Competition of tyrosine nitration versus phosphorylation. J. Diabetes 2015, 7, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Okruhlicova, L.; Tribulova, N.; Misejkova, M.; Kucka, M.; Stetka, R.; Slezak, J.; Manoach, M. Gap junction remodelling is involved in the susceptibility of diabetic rats to hypokalemia-induced ventricular fibrillation. Acta Histochem. 2002, 104, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Stables, C.L.; Musa, H.; Mitra, A.; Bhushal, S.; Deo, M.; Guerrero-Serna, G.; Mironov, S.; Zarzoso, M.; Vikstrom, K.L.; Cawthorn, W.; et al. Reduced Na+ current density underlies impaired propagation in the diabetic rabbit ventricle. J. Mol. Cell Cardiol. 2014, 69, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonda, T.A.; Szynaka, B.; Sokolowska, M.; Dziemidowicz, M.; Winnicka, M.M.; Chyczewski, L.; Kaminski, K.A. Remodeling of the intercalated disc related to aging in the mouse heart. J. Cardiol. 2016, 68, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Flenniken, A.M.; Osborne, L.R.; Anderson, N.; Ciliberti, N.; Fleming, C.; Gittens, J.E.; Gong, X.Q.; Kelsey, L.B.; Lounsbury, C.; Moreno, L.; et al. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development 2005, 132, 4375–4386. [Google Scholar] [CrossRef] [Green Version]
- Kalcheva, N.; Qu, J.; Sandeep, N.; Garcia, L.; Zhang, J.; Wang, Z.; Lampe, P.D.; Suadicani, S.O.; Spray, D.C.; Fishman, G.I. Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc. Natl. Acad. Sci. USA 2007, 104, 20512–20516. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolski, R.; Sasse, P.; Schrickel, J.W.; Watkins, M.; Kim, J.S.; Rackauskas, M.; Troatz, C.; Ghanem, A.; Tiemann, K.; Degen, J.; et al. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum. Mol. Genet. 2008, 17, 539–554. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef]
- Kaplan, S.R.; Gard, J.J.; Protonotarios, N.; Tsatsopoulou, A.; Spiliopoulou, C.; Anastasakis, A.; Squarcioni, C.P.; McKenna, W.J.; Thiene, G.; Basso, C.; et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm 2004, 1, 3–11. [Google Scholar] [CrossRef]
- Kaplan, S.R.; Gard, J.J.; Carvajal-Huerta, L.; Ruiz-Cabezas, J.C.; Thiene, G.; Saffitz, J.E. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc. Pathol. 2004, 13, 26–32. [Google Scholar] [CrossRef]
- Lyon, R.C.; Mezzano, V.; Wright, A.T.; Pfeiffer, E.; Chuang, J.; Banares, K.; Castaneda, A.; Ouyang, K.; Cui, L.; Contu, R.; et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum. Mol. Genet. 2014, 23, 1134–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polontchouk, L.; Haefliger, J.A.; Ebelt, B.; Schaefer, T.; Stuhlmann, D.; Mehlhorn, U.; Kuhn-Regnier, F.; De Vivie, E.R.; Dhein, S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J. Am. Coll. Cardiol. 2001, 38, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Dupont, E.; Ko, Y.; Rothery, S.; Coppen, S.R.; Baghai, M.; Haw, M.; Severs, N.J. The gap-junctional protein connexin40 is elevated in patients susceptible to postoperative atrial fibrillation. Circulation 2001, 103, 842–849. [Google Scholar] [CrossRef]
- Kostin, S.; Klein, G.; Szalay, Z.; Hein, S.; Bauer, E.P.; Schaper, J. Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 2002, 54, 361–379. [Google Scholar] [CrossRef] [Green Version]
- Gemel, J.; Levy, A.E.; Simon, A.R.; Bennett, K.B.; Ai, X.; Akhter, S.; Beyer, E.C. Connexin40 abnormalities and atrial fibrillation in the human heart. J. Mol. Cell Cardiol. 2014, 76, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Lai, Y.J.; Yeh, H.I.; Chen, C.L.; Sun, S.; Wu, S.J.; Lin, F.Y. Atrial gap junctions, NF-kappaB and fibrosis in patients undergoing coronary artery bypass surgery: The relationship with postoperative atrial fibrillation. Cardiology 2009, 112, 81–88. [Google Scholar] [CrossRef]
- van der Velden, H.M.; van Kempen, M.J.; Wijffels, M.C.; van Zijverden, M.; Groenewegen, W.A.; Allessie, M.A.; Jongsma, H.J. Altered pattern of connexin40 distribution in persistent atrial fibrillation in the goat. J. Cardiovasc. Electrophysiol. 1998, 9, 596–607. [Google Scholar] [CrossRef]
- Weidmann, S. The electrical constants of Purkinje fibres. J. Physiol. 1952, 118, 348–360. [Google Scholar] [CrossRef]
- Weidmann, S. The diffusion of radiopotassium across intercalated disks of mammalian cardiac muscle. J. Physiol. 1966, 187, 323–342. [Google Scholar] [CrossRef]
- Lo, C.W. Role of gap junctions in cardiac conduction and development: Insights from the connexin knockout mice. Circ. Res. 2000, 87, 346–348. [Google Scholar] [CrossRef] [Green Version]
- Jansen, J.A.; van Veen, A.A.; de Bakker, J.M.; van Rijen, H.V. Cardiac connexins and impulse propagation. J. Mol. Cell Cardiol. 2010, 48, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Kleber, A.G.; Rudy, Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 2004, 84, 431–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeraraghavan, R.; Gourdie, R.G.; Poelzing, S. Mechanisms of cardiac conduction: A history of revisions. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H619–H627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidmann, S. Electrical constants of trabecular muscle from mammalian heart. J. Physiol. 1970, 210, 1041–1054. [Google Scholar] [CrossRef]
- Draper, M.H.; Mya-Tu, M. A comparison of the conduction velocity in cardiac tissues of various mammals. Q. J. Exp. Physiol. Cogn. Med. Sci. 1959, 44, 91–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, T.; Takayama, N.; Shimamoto, T. Directional difference of conduction velocity in the cardiac ventricular syncytium studied by microelectrodes. Circ. Res. 1959, 7, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Knisley, S.B.; Hill, B.C. Effects of bipolar point and line stimulation in anisotropic rabbit epicardium: Assessment of the critical radius of curvature for longitudinal block. IEEE Trans. Biomed. Eng 1995, 42, 957–966. [Google Scholar] [CrossRef]
- Eloff, B.C.; Lerner, D.L.; Yamada, K.A.; Schuessler, R.B.; Saffitz, J.E.; Rosenbaum, D.S. High resolution optical mapping reveals conduction slowing in connexin43 deficient mice. Cardiovasc. Res. 2001, 51, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Gutstein, D.E.; Morley, G.E.; Tamaddon, H.; Vaidya, D.; Schneider, M.D.; Chen, J.; Chien, K.R.; Stuhlmann, H.; Fishman, G.I. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ. Res. 2001, 88, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Peskoff, A. Electric potential in cylindrical syncytia and muscle fibers. Bull. Math. Biol. 1979, 41, 183–192. [Google Scholar] [CrossRef]
- Peskoff, A. Electric potential in three-dimensional electrically syncytial tissues. Bull. Math. Biol. 1979, 41, 163–181. [Google Scholar] [CrossRef]
- Spach, M.S.; Miller, W.T., III; Dolber, P.C.; Kootsey, J.M.; Sommer, J.R.; Mosher, C.E., Jr. The functional role of structural complexities in the propagation of depolarization in the atrium of the dog. Cardiac conduction disturbances due to discontinuities of effective axial resistivity. Circ. Res. 1982, 50, 175–191. [Google Scholar] [CrossRef] [Green Version]
- Spach, M.S.; Miller, W.T., III; Geselowitz, D.B.; Barr, R.C.; Kootsey, J.M.; Johnson, E.A. The discontinuous nature of propagation in normal canine cardiac muscle. Evidence for recurrent discontinuities of intracellular resistance that affect the membrane currents. Circ. Res. 1981, 48, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Rohr, S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc. Res. 2004, 62, 309–322. [Google Scholar] [CrossRef]
- Shaw, R.M.; Rudy, Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ. Res. 1997, 81, 727–741. [Google Scholar] [CrossRef]
- Cole, W.C.; Picone, J.B.; Sperelakis, N. Gap junction uncoupling and discontinuous propagation in the heart. A comparison of experimental data with computer simulations. Biophys. J. 1988, 53, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Spach, M.S.; Heidlage, J.F.; Dolber, P.C.; Barr, R.C. Electrophysiological effects of remodeling cardiac gap junctions and cell size: Experimental and model studies of normal cardiac growth. Circ. Res. 2000, 86, 302–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilders, R.; Verheijck, E.E.; Kumar, R.; Goolsby, W.N.; van Ginneken, A.C.; Joyner, R.W.; Jongsma, H.J. Model clamp and its application to synchronization of rabbit sinoatrial node cells. Am. J. Physiol. 1996, 271, H2168–H2182. [Google Scholar] [CrossRef] [PubMed]
- Boyett, M.R.; Honjo, H.; Kodama, I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res. 2000, 47, 658–687. [Google Scholar] [CrossRef]
- Verheule, S.; van Batenburg, C.A.; Coenjaerts, F.E.; Kirchhoff, S.; Willecke, K.; Jongsma, H.J. Cardiac conduction abnormalities in mice lacking the gap junction protein connexin40. J. Cardiovasc. Electrophysiol. 1999, 10, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Hagendorff, A.; Schumacher, B.; Kirchhoff, S.; Lüderitz, B.; Willecke, K. Conduction disturbances and increased atrial vulnerability in Connexin40-deficient mice analyzed by transesophageal stimulation. Circulation 1999, 99, 1508–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firouzi, M.; Ramanna, H.; Kok, B.; Jongsma, H.J.; Koeleman, B.P.; Doevendans, P.A.; Groenewegen, W.A.; Hauer, R.N. Association of human connexin40 gene polymorphisms with atrial vulnerability as a risk factor for idiopathic atrial fibrillation. Circ. Res. 2004, 95, e29–e33. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.A.; Schuessler, R.B.; Berul, C.I.; Beardslee, M.A.; Beyer, E.C.; Mendelsohn, M.E.; Saffitz, J.E. Disparate effects of deficient expression of connexin43 on atrial and ventricular conduction: Evidence for chamber-specific molecular determinants of conduction. Circulation 1998, 97, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojodjojo, P.; Kanagaratnam, P.; Segal, O.R.; Hussain, W.; Peters, N.S. The effects of carbenoxolone on human myocardial conduction: A tool to investigate the role of gap junctional uncoupling in human arrhythmogenesis. J. Am. Coll. Cardiol. 2006, 48, 1242–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, G.; Leong-Poi, H.; Mangat, I.; Moe, G.W.; Hu, X.; So, P.P.; Tarulli, E.; Ramadeen, A.; Rossman, E.I.; Hennan, J.K.; et al. Effects of Chronic Gap Junction Conduction-Enhancing Antiarrhythmic Peptide GAP-134 Administration on Experimental Atrial Fibrillation in Dogs. Circ. Arrhythm. Electrophysiol. 2009, 2, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Rossman, E.I.; Liu, K.; Morgan, G.A.; Swillo, R.E.; Krueger, J.A.; Gardell, S.J.; Butera, J.; Gruver, M.; Kantrowitz, J.; Feldman, H.S.; et al. The gap junction modifier, GAP-134 [(2S,4R)-1-(2-aminoacetyl)-4-benzamido-pyrrolidine-2-carboxylic acid], improves conduction and reduces atrial fibrillation/flutter in the canine sterile pericarditis model. J. Pharmacol. Exp. Ther. 2009, 329, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Guerra, J.M.; Everett, T.H.; Lee, K.W.; Wilson, E.; Olgin, J.E. Effects of the gap junction modifier rotigaptide (ZP123) on atrial conduction and vulnerability to atrial fibrillation. Circulation 2006, 114, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Aonuma, S.; Kohama, Y.; Akai, K.; Komiyama, Y.; Nakajima, S.; Wakabayashi, M.; Makino, T. Studies on heart. XIX. Isolation of an atrial peptide that improves the rhythmicity of cultured myocardial cell clusters. Chem. Pharm. Bull. 1980, 28, 3332–3339. [Google Scholar] [CrossRef] [Green Version]
- Aonuma, S.; Kohama, Y.; Makino, T.; Fujisawa, Y. Studies of heart. XXI. Amino acid sequence of antiarrhythmic peptide (AAP) isolated from atria. J. Pharmacobiodyn. 1982, 5, 40–48. [Google Scholar] [CrossRef]
- Axelsen, L.N.; Haugan, K.; Stahlhut, M.; Kjolbye, A.L.; Hennan, J.K.; Holstein-Rathlou, N.H.; Petersen, J.S.; Nielsen, M.S. Increasing gap junctional coupling: A tool for dissecting the role of gap junctions. J. Membr. Biol. 2007, 216, 23–35. [Google Scholar] [CrossRef]
- Dhein, S.; Hagen, A.; Jozwiak, J.; Dietze, A.; Garbade, J.; Barten, M.; Kostelka, M.; Mohr, F.W. Improving cardiac gap junction communication as a new antiarrhythmic mechanism: The action of antiarrhythmic peptides. Naunyn Schmiedebergs Arch. Pharmacol. 2010, 381, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Kruger, O.; Maxeiner, S.; Kim, J.S.; van Rijen, H.V.; de Bakker, J.M.; Eckardt, D.; Tiemann, K.; Lewalter, T.; Ghanem, A.; Luderitz, B.; et al. Cardiac morphogenetic defects and conduction abnormalities in mice homozygously deficient for connexin40 and heterozygously deficient for connexin45. J. Mol. Cell Cardiol. 2006, 41, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Henriquez, A.P.; Vogel, R.; Muller-Borer, B.J.; Henriquez, C.S.; Weingart, R.; Cascio, W.E. Influence of dynamic gap junction resistance on impulse propagation in ventricular myocardium: A computer simulation study. Biophys. J. 2001, 81, 2112–2121. [Google Scholar] [CrossRef] [Green Version]
- Seidel, T.; Salameh, A.; Dhein, S. A simulation study of cellular hypertrophy and connexin lateralization in cardiac tissue. Biophys. J. 2010, 99, 2821–2830. [Google Scholar] [CrossRef] [Green Version]
- Zi, M.; Kimura, T.E.; Liu, W.; Jin, J.; Higham, J.; Kharche, S.; Hao, G.; Shi, Y.; Shen, W.; Prehar, S.; et al. Mitogen-activated protein kinase kinase 4 deficiency in cardiomyocytes causes connexin 43 reduction and couples hypertrophic signals to ventricular arrhythmogenesis. J. Biol. Chem. 2011, 286, 17821–17830. [Google Scholar] [CrossRef] [Green Version]
- Rohr, S.; Kucera, J.P.; Kleber, A.G. Slow conduction in cardiac tissue, I: Effects of a reduction of excitability versus a reduction of electrical coupling on microconduction. Circ. Res. 1998, 83, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Sinovas, A.; Garcia-Dorado, D.; Ruiz-Meana, M.; Soler-Soler, J. Enhanced effect of gap junction uncouplers on macroscopic electrical properties of reperfused myocardium. J. Physiol. 2004, 559, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Akar, F.G.; Roth, B.J.; Rosenbaum, D.S. Optical measurement of cell-to-cell coupling in intact heart using subthreshold electrical stimulation. Am. J. Physiol. Heart Circ. Physiol 2001, 281, H533–H542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keevil, V.L.; Huang, C.L.; Chau, P.L.; Sayeed, R.A.; Vandenberg, J.I. The effect of heptanol on the electrical and contractile function of the isolated, perfused rabbit heart. Pflugers Arch. 2000, 440, 275–282. [Google Scholar] [CrossRef]
- Balke, C.W.; Lesh, M.D.; Spear, J.F.; Kadish, A.; Levine, J.H.; Moore, E.N. Effects of cellular uncoupling on conduction in anisotropic canine ventricular myocardium. Circ. Res. 1988, 63, 879–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmar, M.; Michaels, D.C.; Johnson, T.; Jalife, J. Effects of increasing intercellular resistance on transverse and longitudinal propagation in sheep epicardial muscle. Circ. Res. 1987, 60, 780–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhein, S.; Krusemann, K.; Schaefer, T. Effects of the gap junction uncoupler palmitoleic acid on the activation and repolarization wavefronts in isolated rabbit hearts. Br. J. Pharmacol. 1999, 128, 1375–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, P.S.; Gray, R.; Kojodjojo, P.; Jabr, R.; Chowdhury, R.; Fry, C.H.; Peters, N.S. Relationship between gap-junctional conductance and conduction velocity in mammalian myocardium. Circ. Arrhythm. Electrophysiol. 2013, 6, 1208–1214. [Google Scholar] [CrossRef] [Green Version]
- Tse, G.; Yeo, J.M.; Tse, V.; Kwan, J.; Sun, B. Gap junction inhibition by heptanol increases ventricular arrhythmogenicity by reducing conduction velocity without affecting repolarization properties or myocardial refractoriness in Langendorff-perfused mouse hearts. Mol. Med. Rep. 2016, 14, 4069–4074. [Google Scholar] [CrossRef]
- Dhein, S.; Hammerath, S.B. Aspects of the intercellular communication in aged hearts: Effects of the gap junction uncoupler palmitoleic acid. Naunyn Schmiedebergs Arch. Pharmacol. 2001, 364, 397–408. [Google Scholar]
- Ohara, T.; Qu, Z.; Lee, M.H.; Ohara, K.; Omichi, C.; Mandel, W.J.; Chen, P.S.; Karagueuzian, H.S. Increased vulnerability to inducible atrial fibrillation caused by partial cellular uncoupling with heptanol. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1116–H1122. [Google Scholar] [CrossRef] [Green Version]
- Reaume, A.G.; de Sousa, P.A.; Kulkarni, S.; Langille, B.L.; Zhu, D.; Davies, T.C.; Juneja, S.C.; Kidder, G.M.; Rossant, J. Cardiac malformation in neonatal mice lacking connexin43. Science 1995, 267, 1831–1834. [Google Scholar] [CrossRef]
- Guerrero, P.A.; Schuessler, R.B.; Davis, L.M.; Beyer, E.C.; Johnson, C.M.; Yamada, K.A.; Saffitz, J.E. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J. Clin. Investig. 1997, 99, 1991–1998. [Google Scholar] [CrossRef]
- Eckardt, D.; Kirchhoff, S.; Kim, J.S.; Degen, J.; Theis, M.; Ott, T.; Wiesmann, F.; Doevendans, P.A.; Lamers, W.H.; de Bakker, J.M.; et al. Cardiomyocyte-restricted deletion of connexin43 during mouse development. J. Mol. Cell Cardiol. 2006, 41, 963–971. [Google Scholar] [CrossRef]
- Van Rijen, H.V.; Eckardt, D.; Degen, J.; Theis, M.; Ott, T.; Willecke, K.; Jongsma, H.J.; Opthof, T.; de Bakker, J.M. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 2004, 109, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Stein, M.; van Veen, T.A.; Hauer, R.N.; de Bakker, J.M.; van Rijen, H.V. A 50% reduction of excitability but not of intercellular coupling affects conduction velocity restitution and activation delay in the mouse heart. PLoS ONE 2011, 6, e20310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckardt, D.; Theis, M.; Degen, J.; Ott, T.; van Rijen, H.V.; Kirchhoff, S.; Kim, J.S.; de Bakker, J.M.; Willecke, K. Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J. Mol. Cell Cardiol. 2004, 36, 101–110. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A.; Sanchez, J.A.; Gonzalez-Loyola, A.; Barba, I.; Morente, M.; Aguilar, R.; Agullo, E.; Miro-Casas, E.; Esquerda, N.; Ruiz-Meana, M.; et al. Effects of substitution of Cx43 by Cx32 on myocardial energy metabolism, tolerance to ischemia and preconditioning protection. J. Physiol. 2010, 588, 1139–1151. [Google Scholar] [CrossRef]
- Lerner, D.L.; Yamada, K.A.; Schuessler, R.B.; Saffitz, J.E. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation 2000, 101, 547–552. [Google Scholar] [CrossRef] [Green Version]
- Betsuyaku, T.; Kanno, S.; Lerner, D.L.; Schuessler, R.B.; Saffitz, J.E.; Yamada, K.A. Spontaneous and inducible ventricular arrhythmias after myocardial infarction in mice. Cardiovasc. Pathol. 2004, 13, 156–164. [Google Scholar] [CrossRef]
- Gutstein, D.E.; Danik, S.B.; Sereysky, J.B.; Morley, G.E.; Fishman, G.I. Subdiaphragmatic murine electrophysiological studies: Sequential determination of ventricular refractoriness and arrhythmia induction. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1091–H1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danik, S.B.; Rosner, G.; Lader, J.; Gutstein, D.E.; Fishman, G.I.; Morley, G.E. Electrical remodeling contributes to complex tachyarrhythmias in connexin43-deficient mouse hearts. FASEB J. 2008, 22, 1204–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohr, S.; Kucera, J.P.; Fast, V.G.; Kleber, A.G. Paradoxical improvement of impulse conduction in cardiac tissue by partial cellular uncoupling. Science 1997, 275, 841–844. [Google Scholar] [CrossRef]
- Jozwiak, J.; Dhein, S. Local effects and mechanisms of antiarrhythmic peptide AAP10 in acute regional myocardial ischemia: Electrophysiological and molecular findings. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 378, 459–470. [Google Scholar] [CrossRef]
- Kjolbye, A.L.; Dikshteyn, M.; Eloff, B.C.; Deschenes, I.; Rosenbaum, D.S. Maintenance of intercellular coupling by the antiarrhythmic peptide rotigaptide suppresses arrhythmogenic discordant alternans. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H41–H49. [Google Scholar] [CrossRef]
- Hennan, J.K.; Swillo, R.E.; Morgan, G.A.; Rossman, E.I.; Kantrowitz, J.; Butera, J.; Petersen, J.S.; Gardell, S.J.; Vlasuk, G.P. GAP-134 ([2S,4R]-1-[2-Aminoacetyl]4-Benzamidopyrrolidine-2-Carboxylic Acid) Prevents Spontaneous Ventricular Arrhythmias and Reduces Infarct Size During Myocardial Ischemia/Reperfusion Injury in Open-Chest Dogs. J. Cardiovasc. Pharmacol. Ther. 2009, 14, 207–214. [Google Scholar] [CrossRef]
- Hennan, J.K.; Swillo, R.E.; Morgan, G.A.; Keith, J.C., Jr.; Schaub, R.G.; Smith, R.P.; Feldman, H.S.; Haugan, K.; Kantrowitz, J.; Wang, P.J.; et al. Rotigaptide (ZP123) prevents spontaneous ventricular arrhythmias and reduces infarct size during myocardial ischemia/reperfusion injury in open-chest dogs. J. Pharmacol. Exp. Ther. 2006, 317, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Quan, X.Q.; Bai, R.; Lu, J.G.; Patel, C.; Liu, N.; Ruan, Y.; Chen, B.D.; Ruan, L.; Zhang, C.T. Pharmacological Enhancement of Cardiac Gap Junction Coupling Prevents Arrhythmias in Canine LQT2 Model. Cell Commun. Adhes. 2009, 16, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Quan, X.; Li, L.; Bai, R.; Ni, M.; Xu, R.; Zhang, C. Increasing gap junction coupling suppresses ibutilide-induced torsades de pointes. Exp. Ther. Med. 2014, 7, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Greener, I.D.; Sasano, T.; Wan, X.; Igarashi, T.; Strom, M.; Rosenbaum, D.S.; Donahue, J.K. Connexin43 gene transfer reduces ventricular tachycardia susceptibility after myocardial infarction. J. Am. Coll Cardiol. 2012, 60, 1103–1110. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.S.; Rojas, A.G. The initiation of ventricular fibrillation due to coronary occlusion. Exp. Med. Surg. 1943, 1, 105–122. [Google Scholar]
- Rodriguez-Sinovas, A.; Cinca, J. Sudden death (II). Myocardial ischemia and ventricular arrhythmias in experimental models: Triggering mechanisms. Rev. Esp. Cardiol. 1999, 52, 851–859. [Google Scholar]
- Kaplinsky, E.; Ogawa, S.; Balke, C.W.; Dreifus, L.S. Two periods of early ventricular arrhythmia in the canine acute myocardial infarction model. Circulation 1979, 60, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhard, M.M.; Gersing, E.; Brockhoff, C.J.; Schnabel, P.A.; Bretschneider, H.J. Impedance spectroscopy: A method for surveillance of ischemia tolerance of the heart. Thorac. Cardiovasc. Surg. 1987, 35, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Kleber, A.G.; Riegger, C.B.; Janse, M.J. Electrical uncoupling and increase of extracellular resistance after induction of ischemia in isolated, arterially perfused rabbit papillary muscle. Circ. Res. 1987, 61, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascio, W.E.; Yan, G.X.; Kleber, A.G. Passive electrical properties, mechanical activity, and extracellular potassium in arterially perfused and ischemic rabbit ventricular muscle. Effects of calcium entry blockade or hypocalcemia. Circ. Res. 1990, 66, 1461–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.L.; Mazon, P.; Verberne, H.J.; Sleeswijk, M.E.; Coronel, R.; Opthof, T.; Janse, M.J. Ischaemic preconditioning delays ischaemia induced cellular electrical uncoupling in rabbit myocardium by activation of ATP sensitive potassium channels. Cardiovasc. Res. 1993, 27, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Fleischhauer, J.; Lehmann, L.; Kleber, A.G. Electrical resistances of interstitial and microvascular space as determinants of the extracellular electrical field and velocity of propagation in ventricular myocardium. Circulation 1995, 92, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Dekker, L.R.; Fiolet, J.W.; VanBavel, E.; Coronel, R.; Opthof, T.; Spaan, J.A.; Janse, M.J. Intracellular Ca2+, intercellular electrical coupling, and mechanical activity in ischemic rabbit papillary muscle. Effects of preconditioning and metabolic blockade. Circ. Res. 1996, 79, 237–246. [Google Scholar] [CrossRef]
- Owens, L.M.; Fralix, T.A.; Murphy, E.; Cascio, W.E.; Gettes, L.S. Correlation of ischemia-induced extracellular and intracellular ion changes to cell-to-cell electrical uncoupling in isolated blood-perfused rabbit hearts. Experimental Working Group. Circulation 1996, 94, 10–13. [Google Scholar] [CrossRef]
- Cascio, W.E.; Yang, H.; Johnson, T.A.; Muller-Borer, B.J.; Lemasters, J.J. Electrical properties and conduction in reperfused papillary muscle. Circ. Res. 2001, 89, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Casas, O.; Bragos, R.; Riu, P.J.; Rosell, J.; Tresanchez, M.; Warren, M.; Rodriguez-Sinovas, A.; Carreno, A.; Cinca, J. In vivo and in situ ischemic tissue characterization using electrical impedance spectroscopy. Ann. N. Y. Acad. Sci. 1999, 873, 51–58. [Google Scholar] [CrossRef]
- Ellenby, M.I.; Small, K.W.; Wells, R.M.; Hoyt, D.J.; Lowe, J.E. On-line detection of reversible myocardial ischemic injury by measurement of myocardial electrical impedance. Ann. Thorac. Surg. 1987, 44, 587–597. [Google Scholar] [CrossRef]
- Fallert, M.A.; Mirotznik, M.S.; Downing, S.W.; Savage, E.B.; Foster, K.R.; Josephson, M.E.; Bogen, D.K. Myocardial electrical impedance mapping of ischemic sheep hearts and healing aneurysms. Circulation 1993, 87, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.T.; Fleet, W.F.; Johnson, T.A.; Engle, C.L.; Cascio, W.E. The Ib phase of ventricular arrhythmias in ischemic in situ porcine heart is related to changes in cell-to-cell electrical coupling. Experimental Cardiology Group, University of North Carolina. Circulation 1995, 92, 3051–3060. [Google Scholar] [CrossRef]
- Cinca, J.; Warren, M.; Carreno, A.; Tresanchez, M.; Armadans, L.; Gomez, P.; Soler-Soler, J. Changes in myocardial electrical impedance induced by coronary artery occlusion in pigs with and without preconditioning: Correlation with local ST-segment potential and ventricular arrhythmias. Circulation 1997, 96, 3079–3086. [Google Scholar] [CrossRef]
- Padilla, F.; Garcia-Dorado, D.; Rodriguez-Sinovas, A.; Ruiz-Meana, M.; Inserte, J.; Soler-Soler, J. Protection afforded by ischemic preconditioning is not mediated by effects on cell-to-cell electrical coupling during myocardial ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1909–H1916. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A.; Garcia-Dorado, D.; Padilla, F.; Inserte, J.; Barrabes, J.A.; Ruiz-Meana, M.; Agullo, L.; Soler-Soler, J. Pre-treatment with the Na+/H+ exchange inhibitor cariporide delays cell-to-cell electrical uncoupling during myocardial ischemia. Cardiovasc. Res. 2003, 58, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Meana, M.; Garcia-Dorado, D.; Hofstaetter, B.; Piper, H.M.; Soler-Soler, J. Propagation of cardiomyocyte hypercontracture by passage of Na(+) through gap junctions. Circ. Res. 1999, 85, 280–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, G.S.; Valiunas, V.; Brink, P.R. Selective permeability of gap junction channels. Biochim. Biophys. Acta 2004, 1662, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessen, H.; Harz, H.; Bedner, P.; Kramer, K.; Willecke, K. Selective permeability of different connexin channels to the second messenger inositol 1,4,5-trisphosphate. J. Cell Sci. 2000, 113, 1365–1372. [Google Scholar]
- Bedner, P.; Niessen, H.; Odermatt, B.; Kretz, M.; Willecke, K.; Harz, H. Selective permeability of different connexin channels to the second messenger cyclic AMP. J. Biol. Chem. 2006, 281, 6673–6681. [Google Scholar] [CrossRef] [Green Version]
- Kanaporis, G.; Mese, G.; Valiuniene, L.; White, T.W.; Brink, P.R.; Valiunas, V. Gap junction channels exhibit connexin-specific permeability to cyclic nucleotides. J. Gen. Physiol. 2008, 131, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Frame, M.K.; de Feijter, A.W. Propagation of mechanically induced intercellular calcium waves via gap junctions and ATP receptors in rat liver epithelial cells. Exp. Cell Res. 1997, 230, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, N.R.; Henriksen, Z.; Brot, C.; Eriksen, E.F.; Sorensen, O.H.; Civitelli, R.; Steinberg, T.H. Human osteoblastic cells propagate intercellular calcium signals by two different mechanisms. J. Bone Miner. Res. 2000, 15, 1024–1032. [Google Scholar] [CrossRef]
- Leybaert, L.; Sanderson, M.J. Intercellular Ca(2+) waves: Mechanisms and function. Physiol. Rev. 2012, 92, 1359–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suadicani, S.O.; Flores, C.E.; Urban-Maldonado, M.; Beelitz, M.; Scemes, E. Gap junction channels coordinate the propagation of intercellular Ca2+ signals generated by P2Y receptor activation. Glia 2004, 48, 217–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, P.; Weingart, R. Cell pairs isolated from adult guinea pig and rat hearts: Effects of [Ca2+]i on nexal membrane resistance. Pflugers Arch. 1987, 409, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, H.; Toyama, J.; Tsuboi, N.; Kamiya, K.; Kodama, I. ATP directly affects junctional conductance between paired ventricular myocytes isolated from guinea pig heart. Circ. Res. 1990, 66, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Dorado, D.; Theroux, P.; Desco, M.; Solares, J.; Elizaga, J.; Fernandez-Aviles, F.; Alonso, J.; Soriano, J. Cell-to-cell interaction: A mechanism to explain wave-front progression of myocardial necrosis. Am. J. Physiol. 1989, 256, H1266–H1273. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Inserte, J.; Ruiz-Meana, M.; Gonzalez, M.A.; Solares, J.; Julia, M.; Barrabes, J.A.; Soler-Soler, J. Gap junction uncoupler heptanol prevents cell-to-cell progression of hypercontracture and limits necrosis during myocardial reperfusion. Circulation 1997, 96, 3579–3586. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Garcia-Dorado, D.; Lane, S.; Pina, P.; Inserte, J.; Mirabet, M.; Soler-Soler, J. Persistence of gap junction communication during myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2563–H2571. [Google Scholar] [CrossRef]
- Saltman, A.E.; Aksehirli, T.O.; Valiunas, V.; Gaudette, G.R.; Matsuyama, N.; Brink, P.; Krukenkamp, I.B. Gap junction uncoupling protects the heart against ischemia. J. Thorac. Cardiovasc. Surg. 2002, 124, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Gysembergh, A.; Kloner, R.A.; Przyklenk, K. Pretreatment with the gap junction uncoupler heptanol does not limit infarct size in rabbit heart. Cardiovasc. Pathol. 2001, 10, 13–17. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A.; Garcia-Dorado, D.; Ruiz-Meana, M.; Soler-Soler, J. Protective effect of gap junction uncouplers given during hypoxia against reoxygenation injury in isolated rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H648–H656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, D.; Sanden, E.; Hagve, M.; Chu, X.; Sundset, R.; Ytrehus, K. Heptanol triggers cardioprotection via mitochondrial mechanisms and mitochondrial potassium channel opening in rat hearts. Acta Physiol. 2011, 201, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, V.; Caprini, M.; Nicchia, G.P.; Rossi, A.; Dovizio, M.; Cervetto, C.; Nobile, M.; Ferroni, S. Carbenoxolone inhibits volume-regulated anion conductance in cultured rat cortical astroglia. Channels 2009, 3, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Liang, C.; Sun, W.; Chen, J.; Chen, X. Glycyrrhizic acid ameliorates myocardial ischemic injury by the regulation of inflammation and oxidative state. Drug Des. Dev. Ther. 2018, 12, 1311–1319. [Google Scholar] [CrossRef] [Green Version]
- Nelson, W.L.; Makielski, J.C. Block of sodium current by heptanol in voltage-clamped canine cardiac Purkinje cells. Circ. Res. 1991, 68, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.M.; Rice, R.T.; duBell, W.H.; Spurgeon, H.A. Initial contractile response of isolated rat heart cells to halothane, enflurane, and isoflurane. Anesthesiology 1997, 86, 137–146. [Google Scholar] [CrossRef]
- Shimada, T.; Somlyo, A.P. Modulation of voltage-dependent Ca channel current by arachidonic acid and other long-chain fatty acids in rabbit intestinal smooth muscle. J. Gen. Physiol. 1992, 100, 27–44. [Google Scholar] [CrossRef]
- Kanno, S.; Kovacs, A.; Yamada, K.A.; Saffitz, J.E. Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J. Am. Coll. Cardiol. 2003, 41, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Schwanke, U.; Konietzka, I.; Duschin, A.; Li, X.; Schulz, R.; Heusch, G. No ischemic preconditioning in heterozygous connexin43-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1740–H1742. [Google Scholar] [CrossRef] [PubMed]
- Schwanke, U.; Li, X.; Schulz, R.; Heusch, G. No ischemic preconditioning in heterozygous connexin 43-deficient mice—A further in vivo study. Basic Res. Cardiol. 2003, 98, 181–182. [Google Scholar] [CrossRef]
- Maass, K.; Chase, S.E.; Lin, X.; Delmar, M. Cx43 CT domain influences infarct size and susceptibility to ventricular tachyarrhythmias in acute myocardial infarction. Cardiovasc. Res. 2009, 84, 361–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prestia, K.A.; Sosunov, E.A.; Anyukhovsky, E.P.; Dolmatova, E.; Kelly, C.W.; Brink, P.R.; Robinson, R.B.; Rosen, M.R.; Duffy, H.S. Increased Cell-Cell Coupling Increases Infarct Size and Does not Decrease Incidence of Ventricular Tachycardia in Mice. Front. Physiol. 2011, 2, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasui, K.; Kada, K.; Hojo, M.; Lee, J.K.; Kamiya, K.; Toyama, J.; Opthof, T.; Kodama, I. Cell-to-cell interaction prevents cell death in cultured neonatal rat ventricular myocytes. Cardiovasc. Res. 2000, 48, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Blanc, E.M.; Bruce-Keller, A.J.; Mattson, M.P. Astrocytic gap junctional communication decreases neuronal vulnerability to oxidative stress-induced disruption of Ca2+ homeostasis and cell death. J. Neurochem. 1998, 70, 958–970. [Google Scholar] [CrossRef]
- Li, G.; Whittaker, P.; Yao, M.; Kloner, R.A.; Przyklenk, K. The gap junction uncoupler heptanol abrogates infarct size reduction with preconditioning in mouse hearts. Cardiovasc. Pathol. 2002, 11, 158–165. [Google Scholar] [CrossRef]
- Hund, T.J.; Lerner, D.L.; Yamada, K.A.; Schuessler, R.B.; Saffitz, J.E. Protein kinase Cepsilon mediates salutary effects on electrical coupling induced by ischemic preconditioning. Heart Rhythm 2007, 4, 1183–1193. [Google Scholar] [CrossRef] [Green Version]
- Papp, R.; Gonczi, M.; Kovacs, M.; Seprenyi, G.; Vegh, A. Gap junctional uncoupling plays a trigger role in the antiarrhythmic effect of ischaemic preconditioning. Cardiovasc. Res. 2007, 74, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Haugan, K.; Marcussen, N.; Kjolbye, A.L.; Nielsen, M.S.; Hennan, J.K.; Petersen, J.S. Treatment with the Gap Junction Modifier Rotigaptide (ZP123) Reduces Infarct Size in Rats with Chronic Myocardial Infarction. J. Cardiovasc. Pharmacol. 2006, 47, 236–242. [Google Scholar] [CrossRef]
- Pedersen, C.M.; Venkatasubramanian, S.; Vase, H.; Hyldebrandt, J.A.; Contractor, H.; Schmidt, M.R.; Botker, H.E.; Cruden, N.L.; Newby, D.E.; Kharbanda, R.K.; et al. Rotigaptide protects the myocardium and arterial vasculature from ischaemia reperfusion injury. Br. J. Clin. Pharmacol. 2016, 81, 1037–1045. [Google Scholar] [CrossRef]
- Diez, E.R.; Sanchez, J.A.; Prado, N.J.; Ponce Zumino, A.Z.; Garcia-Dorado, D.; Miatello, R.M.; Rodriguez-Sinovas, A. Ischemic Postconditioning Reduces Reperfusion Arrhythmias by Adenosine Receptors and Protein Kinase C Activation but Is Independent of K(ATP) Channels or Connexin 43. Int. J. Mol. Sci. 2019, 20, 5927. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Buchert, A.; Feldhaus, S.; Schulz, R. No loss of cardioprotection by postconditioning in connexin 43-deficient mice. Basic Res. Cardiol. 2006, 101, 354–356. [Google Scholar] [CrossRef]
- Morel, S.; Braunersreuther, V.; Chanson, M.; Bouis, D.; Rochemont, V.; Foglia, B.; Pelli, G.; Sutter, E.; Pinsky, D.J.; Mach, F.; et al. Endothelial Cx40 limits myocardial ischaemia/reperfusion injury in mice. Cardiovasc. Res. 2014, 102, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Sinovas, A.; Cabestrero, A.; Lopez, D.; Torre, I.; Morente, M.; Abellan, A.; Miro, E.; Ruiz-Meana, M.; Garcia-Dorado, D. The modulatory effects of connexin 43 on cell death/survival beyond cell coupling. Prog. Biophys. Mol. Biol. 2007, 94, 219–232. [Google Scholar] [CrossRef]
- Kardami, E.; Dang, X.; Iacobas, D.A.; Nickel, B.E.; Jeyaraman, M.; Srisakuldee, W.; Makazan, J.; Tanguy, S.; Spray, D.C. The role of connexins in controlling cell growth and gene expression. Prog. Biophys. Mol. Biol. 2007, 94, 245–264. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and the control of tissue growth: Lack of communication between cancer cells. Nature 1966, 209, 1248–1249. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Wang, H.; McLachlan, E.; Veitch, G.I.; Laird, D.W. Down-regulation of Cx43 by retroviral delivery of small interfering RNA promotes an aggressive breast cancer cell phenotype. Cancer Res. 2005, 65, 2705–2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschi, K.K.; Xu, C.E.; Tsukamoto, T.; Sager, R. Gap junction genes Cx26 and Cx43 individually suppress the cancer phenotype of human mammary carcinoma cells and restore differentiation potential. Cell Growth Differ. 1996, 7, 861–870. [Google Scholar]
- Stains, J.P.; Lecanda, F.; Screen, J.; Towler, D.A.; Civitelli, R. Gap junctional communication modulates gene transcription by altering the recruitment of Sp1 and Sp3 to connexin-response elements in osteoblast promoters. J. Biol. Chem. 2003, 278, 24377–24387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stains, J.P.; Civitelli, R. Gap junctions regulate extracellular signal-regulated kinase signaling to affect gene transcription. Mol. Biol. Cell 2005, 16, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Saez, J.C.; Schalper, K.A.; Retamal, M.A.; Orellana, J.A.; Shoji, K.F.; Bennett, M.V. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp. Cell Res. 2010, 316, 2377–2389. [Google Scholar] [CrossRef]
- Ponsaerts, R.; Wang, N.; Himpens, B.; Leybaert, L.; Bultynck, G. The contractile system as a negative regulator of the connexin 43 hemichannel. Biol. Cell 2012, 104, 367–377. [Google Scholar] [CrossRef]
- Rana, S.; Dringen, R. Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci. Lett. 2007, 415, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Quist, A.P.; Rhee, S.K.; Lin, H.; Lal, R. Physiological role of gap-junctional hemichannels. Extracellular calcium-dependent isosmotic volume regulation. J. Cell Biol. 2000, 148, 1063–1074. [Google Scholar] [CrossRef]
- Bukauskas, F.F.; Verselis, V.K. Gap junction channel gating. Biochim. Biophys. Acta 2004, 1662, 42–60. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Sinovas, A.; Sanchez, J.A.; Fernandez-Sanz, C.; Ruiz-Meana, M.; Garcia-Dorado, D. Connexin and pannexin as modulators of myocardial injury. Biochim. Biophys. Acta 2012, 1818, 1962–1970. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef] [Green Version]
- Ebihara, L. New roles for connexons. News Physiol. Sci. 2003, 18, 100–103. [Google Scholar] [CrossRef]
- Goodenough, D.A.; Paul, D.L. Beyond the gap: Functions of unpaired connexon channels. Nat. Rev. Mol. Cell. Biol. 2003, 4, 285–294. [Google Scholar] [CrossRef]
- Evans, W.H.; De Vuyst, E.; Leybaert, L. The gap junction cellular internet: Connexin hemichannels enter the signalling limelight. Biochem. J. 2006, 397, 1–14. [Google Scholar] [CrossRef]
- Bruzzone, S.; Guida, L.; Zocchi, E.; Franco, L.; De Flora, A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001, 15, 10–12. [Google Scholar] [CrossRef]
- Karagiannis, A.; Sylantyev, S.; Hadjihambi, A.; Hosford, P.S.; Kasparov, S.; Gourine, A.V. Hemichannel-mediated release of lactate. J. Cereb. Blood Flow Metab. 2016, 36, 1202–1211. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Vinken, M.; Rogiers, V.; Bukauskas, F.F.; Bultynck, G.; Leybaert, L. Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta 2013, 1828, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, P.B.; Knappenberger, J.; Segal, M.; Bennett, M.V.; Charles, A.C.; Kater, S.B. ATP released from astrocytes mediates glial calcium waves. J. Neurosci. 1999, 19, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Cotrina, M.L.; Lin, J.H.; Lopez-Garcia, J.C.; Naus, C.C.; Nedergaard, M. ATP-mediated glia signaling. J. Neurosci. 2000, 20, 2835–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, N.R.; Henriksen, Z.; Sorensen, O.H.; Eriksen, E.F.; Civitelli, R.; Steinberg, T.H. Intercellular calcium signaling occurs between human osteoblasts and osteoclasts and requires activation of osteoclast P2X7 receptors. J. Biol. Chem. 2002, 277, 7574–7580. [Google Scholar] [CrossRef] [Green Version]
- Davidson, J.O.; Green, C.R.; Bennet, L.; Gunn, A.J. Battle of the hemichannels—Connexins and Pannexins in ischemic brain injury. Int. J. Dev. Neurosci. 2015, 45, 66–74. [Google Scholar] [CrossRef]
- Braet, K.; Vandamme, W.; Martin, P.E.; Evans, W.H.; Leybaert, L. Photoliberating inositol-1,4,5-trisphosphate triggers ATP release that is blocked by the connexin mimetic peptide gap 26. Cell Calcium 2003, 33, 37–48. [Google Scholar] [CrossRef]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.; Charles, A.C. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef] [Green Version]
- Ai, Z.; Fischer, A.; Spray, D.C.; Brown, A.M.; Fishman, G.I. Wnt-1 regulation of connexin43 in cardiac myocytes. J. Clin. Investig. 2000, 105, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukimoto, M.; Harada, H.; Ikari, A.; Takagi, K. Involvement of chloride in apoptotic cell death induced by activation of ATP-sensitive P2X7 purinoceptor. J. Biol. Chem. 2005, 280, 2653–2658. [Google Scholar] [CrossRef] [Green Version]
- Schrier, S.M.; Florea, B.I.; Mulder, G.J.; Nagelkerke, J.F.; IJzerman, A.P. Apoptosis induced by extracellular ATP in the mouse neuroblastoma cell line N1E-115: Studies on involvement of P2 receptors and adenosine. Biochem. Pharmacol. 2002, 63, 1119–1126. [Google Scholar] [CrossRef]
- Zipfel, G.J.; Babcock, D.J.; Lee, J.M.; Choi, D.W. Neuronal apoptosis after CNS injury: The roles of glutamate and calcium. J. Neurotrauma 2000, 17, 857–869. [Google Scholar] [CrossRef]
- Schalper, K.A.; Sanchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Saez, J.C. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 2010, 299, C1504–C1515. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Piper, H.M.; Garcia-Dorado, D.; Ovize, M. A fresh look at reperfusion injury. Cardiovasc. Res. 1998, 38, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Inserte, J.; Rodriguez-Sinovas, A.; Piper, H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012, 94, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.L.; Ebihara, L.; Takemoto, L.J.; Swenson, K.I.; Goodenough, D.A. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J. Cell Biol. 1991, 115, 1077–1089. [Google Scholar] [CrossRef] [Green Version]
- Clarke, T.C.; Williams, O.J.; Martin, P.E.; Evans, W.H. ATP release by cardiac myocytes in a simulated ischaemia model: Inhibition by a connexin mimetic and enhancement by an antiarrhythmic peptide. Eur. J. Pharmacol. 2009, 605, 9–14. [Google Scholar] [CrossRef]
- Shintani-Ishida, K.; Uemura, K.; Yoshida, K. Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1714–H1720. [Google Scholar] [CrossRef] [Green Version]
- John, S.; Cesario, D.; Weiss, J.N. Gap junctional hemichannels in the heart. Acta Physiol. Scand. 2003, 179, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Hawat, G.; Benderdour, M.; Rousseau, G.; Baroudi, G. Connexin 43 mimetic peptide Gap26 confers protection to intact heart against myocardial ischemia injury. Pflugers Arch. 2010, 460, 583–592. [Google Scholar] [CrossRef]
- Hawat, G.; Helie, P.; Baroudi, G. Single intravenous low-dose injections of connexin 43 mimetic peptides protect ischemic heart in vivo against myocardial infarction. J. Mol. Cell Cardiol. 2012, 53, 559–566. [Google Scholar] [CrossRef]
- Iyyathurai, J.; D’hondt, C.; Wang, N.; De Bock, M.; Himpens, B.; Retamal, M.A.; Stehberg, J.; Leybaert, L.; Bultynck, G. Peptides and peptide-derived molecules targeting the intracellular domains of Cx43: Gap junctions versus hemichannels. Neuropharmacology 2013, 75, 491–505. [Google Scholar] [CrossRef]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef] [Green Version]
- Braet, K.; Aspeslagh, S.; Vandamme, W.; Willecke, K.; Martin, P.E.; Evans, W.H.; Leybaert, L. Pharmacological sensitivity of ATP release triggered by photoliberation of inositol-1,4,5-trisphosphate and zero extracellular calcium in brain endothelial cells. J. Cell Physiol. 2003, 197, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Lou, N.; Kang, N.; Takano, T.; Hu, F.; Han, X.; Xu, Q.; Lovatt, D.; Torres, A.; Willecke, K.; et al. A central role of connexin 43 in hypoxic preconditioning. J. Neurosci. 2008, 28, 681–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schock, S.C.; Leblanc, D.; Hakim, A.M.; Thompson, C.S. ATP release by way of connexin 36 hemichannels mediates ischemic tolerance in vitro. Biochem. Biophys. Res. Commun. 2008, 368, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Miki, T.; Yano, T. Role of the gap junction in ischemic preconditioning in the heart. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1115–H1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girao, H. Role of connexin 43 in different forms of intercellular communication—Gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins-Marques, T.; Hausenloy, D.J.; Sluijter, J.P.G.; Leybaert, L.; Girao, H. Intercellular Communication in the Heart: Therapeutic Opportunities for Cardiac Ischemia. Trends Mol. Med. 2020. [Google Scholar] [CrossRef]
- He, K.; Shi, X.; Zhang, X.; Dang, S.; Ma, X.; Liu, F.; Xu, M.; Lv, Z.; Han, D.; Fang, X.; et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc. Res. 2011, 92, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Borg, T.K.; Ma, Z.; Xu, M.; Wetzel, G.; Saraf, L.V.; Markwald, R.; Runyan, R.B.; Gao, B.Z. Biochip-based study of unidirectional mitochondrial transfer from stem cells to myocytes via tunneling nanotubes. Biofabrication 2016, 8, 015012. [Google Scholar] [CrossRef] [PubMed]
- Batista-Almeida, D.; Ribeiro-Rodrigues, T.; Martins-Marques, T.; Cortes, L.; Antunes, P.; Gonçalves, L.; Brou, C.; Aasen, T.; Zurzolo, C.; Girao, H. Ischaemia impacts TNT-mediated communication between cardiac cells. Curr. Res. Cell Biol. 2020, 1, 100001. [Google Scholar] [CrossRef]
- Abounit, S.; Zurzolo, C. Wiring through tunneling nanotubes—From electrical signals to organelle transfer. J. Cell Sci. 2012, 125, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gerdes, H.H. Long-distance electrical coupling via tunneling nanotubes. Biochim. Biophys. Acta 2012, 1818, 2082–2086. [Google Scholar] [CrossRef] [Green Version]
- Lock, J.T.; Parker, I.; Smith, I.F. Communication of Ca(2+) signals via tunneling membrane nanotubes is mediated by transmission of inositol trisphosphate through gap junctions. Cell Calcium 2016, 60, 266–272. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Veruki, M.L.; Bukoreshtliev, N.V.; Hartveit, E.; Gerdes, H.H. Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc. Natl. Acad. Sci. USA 2010, 107, 17194–17199. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; Riquelme, J.A.; Kearney, J.; Sharma, V.; et al. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1525–1536. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Riquelme, J.A.; Takov, K.; Vicencio, J.M.; Boi-Doku, C.; Khoo, V.; Doreth, C.; Radenkovic, D.; Lavandero, S.; Yellon, D.M. Cardioprotection mediated by exosomes is impaired in the setting of type II diabetes but can be rescued by the use of non-diabetic exosomes in vitro. J. Cell Mol. Med. 2018, 22, 141–151. [Google Scholar] [CrossRef]
- Giricz, Z.; Varga, Z.V.; Baranyai, T.; Sipos, P.; Paloczi, K.; Kittel, A.; Buzas, E.I.; Ferdinandy, P. Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J. Mol. Cell Cardiol. 2014, 68, 75–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minghua, W.; Zhijian, G.; Chahua, H.; Qiang, L.; Minxuan, X.; Luqiao, W.; Weifang, Z.; Peng, L.; Biming, Z.; Lingling, Y.; et al. Plasma exosomes induced by remote ischaemic preconditioning attenuate myocardial ischaemia/reperfusion injury by transferring miR-24. Cell Death Dis. 2018, 9, 320. [Google Scholar] [CrossRef]
- Deddens, J.C.; Vrijsen, K.R.; Colijn, J.M.; Oerlemans, M.I.; Metz, C.H.; van der Vlist, E.J.; Nolte-’t Hoen, E.N.; den Ouden, K.; Jansen Of Lorkeers, S.J.; van der Spoel, T.I.; et al. Circulating Extracellular Vesicles Contain miRNAs and are Released as Early Biomarkers for Cardiac Injury. J. Cardiovasc. Transl. Res. 2016, 9, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Borosch, S.; Dahmen, E.; Beckers, C.; Stoppe, C.; Buhl, E.M.; Denecke, B.; Goetzenich, A.; Kraemer, S. Characterization of extracellular vesicles derived from cardiac cells in an in vitro model of preconditioning. J. Extracell. Vesicles 2017, 6, 1390391. [Google Scholar] [CrossRef] [Green Version]
- Rana, S.; Yue, S.; Stadel, D.; Zöller, M. Toward tailored exosomes: The exosomal tetraspanin web contributes to target cell selection. Int. J. Biochem. Cell Biol. 2012, 44, 1574–1584. [Google Scholar] [CrossRef]
- Soares, A.R.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Ferreira, J.V.; Catarino, S.; Pinho, M.J.; Zuzarte, M.; Isabel, A.S.; Manadas, B.; Sluijter, P.G.; et al. Gap junctional protein Cx43 is involved in the communication between extracellular vesicles and mammalian cells. Sci. Rep. 2015, 5, 13243. [Google Scholar] [CrossRef] [Green Version]
- Martins-Marques, T.; Ribeiro-Rodrigues, T.; de Jager, S.C.; Zuzarte, M.; Ferreira, C.; Cruz, P.; Reis, L.; Baptista, R.; Gonçalves, L.; Sluijter, J.P.; et al. Myocardial infarction affects Cx43 content of extracellular vesicles secreted by cardiomyocytes. Life Sci. Alliance 2020, 3, e202000821. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Brodsky, S.; Kumari, S.; Valiunas, V.; Brink, P.; Kaide, J.; Nasjletti, A.; Goligorsky, M.S. Paradoxical overexpression and translocation of connexin43 in homocysteine-treated endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2124–H2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miro-Casas, E.; Ruiz-Meana, M.; Agullo, E.; Stahlhofen, S.; Rodriguez-Sinovas, A.; Cabestrero, A.; Jorge, I.; Torre, I.; Vazquez, J.; Boengler, K.; et al. Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc. Res. 2009, 83, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Perrelli, M.G.; Raimondo, S.; Tullio, F.; Merlino, A.; Moro, F.; Geuna, S.; Mancardi, D.; Pagliaro, P. Postconditioning induces an anti-apoptotic effect and preserves mitochondrial integrity in isolated rat hearts. Biochim. Biophys. Acta 2009, 1787, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Goubaeva, F.; Mikami, M.; Giardina, S.; Ding, B.; Abe, J.; Yang, J. Cardiac mitochondrial connexin 43 regulates apoptosis. Biochem. Biophys. Res. Commun. 2007, 352, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Clarke, S.J.; Khaliulin, I. The role of mitochondria in protection of the heart by preconditioning. Biochim. Biophys. Acta 2007, 1767, 1007–1031. [Google Scholar] [CrossRef] [Green Version]
- Pecoraro, M.; Sorrentino, R.; Franceschelli, S.; Del Pizzo, M.; Pinto, A.; Popolo, A. Doxorubicin-Mediated Cardiotoxicity: Role of Mitochondrial Connexin 43. Cardiovasc. Toxicol. 2015, 15, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Azarashvili, T.; Baburina, Y.; Grachev, D.; Krestinina, O.; Evtodienko, Y.; Stricker, R.; Reiser, G. Calcium-induced permeability transition in rat brain mitochondria is promoted by carbenoxolone through targeting connexin43. Am. J. Physiol. Cell Physiol. 2011, 300, C707–C720. [Google Scholar] [CrossRef]
- Hou, S.; Shen, P.P.; Zhao, M.M.; Liu, X.P.; Xie, H.Y.; Deng, F.; Feng, J.C. Mechanism of Mitochondrial Connexin43′s Protection of the Neurovascular Unit under Acute Cerebral Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2016, 17, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozoriz, M.G.; Church, J.; Ozog, M.A.; Naus, C.C.; Krebs, C. Temporary sequestration of potassium by mitochondria in astrocytes. J. Biol. Chem. 2010, 285, 31107–31119. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Haider, H.K.; Porollo, A.; Ashraf, M. Mitochondria-specific transgenic overexpression of connexin-43 simulates preconditioning-induced cytoprotection of stem cells. Cardiovasc. Res. 2010, 88, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Dhein, S. New, emerging roles for cardiac connexins. Mitochondrial Cx43 raises new questions. Cardiovasc. Res. 2005, 67, 179–181. [Google Scholar] [CrossRef]
- Truscott, K.N.; Brandner, K.; Pfanner, N. Mechanisms of protein import into mitochondria. Curr. Biol. 2003, 13, R326–R337. [Google Scholar] [CrossRef] [Green Version]
- Boengler, K.; Stahlhofen, S.; van de, S.A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef]
- Bell, C.L.; Shakespeare, T.I.; Smith, A.R.; Murray, S.A. Visualization of Annular Gap Junction Vesicle Processing: The Interplay Between Annular Gap Junctions and Mitochondria. Int. J. Mol. Sci. 2018, 20, 44. [Google Scholar] [CrossRef] [Green Version]
- Boengler, K.; Ungefug, E.; Heusch, G.; Leybaert, L.; Schulz, R. Connexin 43 impacts on mitochondrial potassium uptake. Front. Pharmacol. 2013, 4, 73. [Google Scholar] [CrossRef] [Green Version]
- Gadicherla, A.K.; Wang, N.; Bulic, M.; Agullo-Pascual, E.; Lissoni, A.; De Smet, M.; Delmar, M.; Bultynck, G.; Krysko, D.V.; Camara, A.; et al. Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res. Cardiol. 2017, 112, 27. [Google Scholar] [CrossRef]
- Boengler, K.; Ruiz-Meana, M.; Gent, S.; Ungefug, E.; Soetkamp, D.; Miro-Casas, E.; Cabestrero, A.; Fernandez-Sanz, C.; Semenzato, M.; Di Lisa, F.; et al. Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J. Cell Mol. Med. 2012, 16, 1649–1655. [Google Scholar] [CrossRef]
- Daleau, P.; Boudriau, S.; Michaud, M.; Jolicoeur, C.; Kingma, J.G., Jr. Preconditioning in the absence or presence of sustained ischemia modulates myocardial Cx43 protein levels and gap junction distribution. Can. J. Physiol. Pharmacol. 2001, 79, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Naitoh, K.; Yano, T.; Miura, T.; Itoh, T.; Miki, T.; Tanno, M.; Sato, T.; Hotta, H.; Terashima, Y.; Shimamoto, K. Roles of Cx43-associated protein kinases in suppression of gap junction-mediated chemical coupling by ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H396–H403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, T.; Yano, T.; Naitoh, K.; Nishihara, M.; Miki, T.; Tanno, M.; Shimamoto, K. Delta-opioid receptor activation before ischemia reduces gap junction permeability in ischemic myocardium by PKC-epsilon-mediated phosphorylation of connexin 43. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1425–H1431. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, W.D.; O’Rourke, B.; Marban, E. Cell-type specificity of preconditioning in an in vitro model. Basic Res. Cardiol. 1996, 91, 450–457. [Google Scholar] [CrossRef]
- Li, X.; Heinzel, F.R.; Boengler, K.; Schulz, R.; Heusch, G. Role of connexin 43 in ischemic preconditioning does not involve intercellular communication through gap junctions. J. Mol. Cell Cardiol. 2004, 36, 161–163. [Google Scholar] [CrossRef]
- Heinzel, F.R.; Luo, Y.; Li, X.; Boengler, K.; Buechert, A.; Garcia-Dorado, D.; Di Lisa, F.; Schulz, R.; Heusch, G. Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ. Res. 2005, 97, 583–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.H.; Yang, J.; Liu, S.; Takano, T.; Wang, X.; Gao, Q.; Willecke, K.; Nedergaard, M. Connexin mediates gap junction-independent resistance to cellular injury. J. Neurosci. 2003, 23, 430–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Soetkamp, D.; Nguyen, T.T.; Menazza, S.; Hirschhauser, C.; Hendgen-Cotta, U.B.; Rassaf, T.; Schluter, K.D.; Boengler, K.; Murphy, E.; Schulz, R. S-nitrosation of mitochondrial connexin 43 regulates mitochondrial function. Basic Res. Cardiol. 2014, 109, 433. [Google Scholar] [CrossRef] [Green Version]
- Srisakuldee, W.; Makazan, Z.; Nickel, B.E.; Zhang, F.; Thliveris, J.A.; Pasumarthi, K.B.; Kardami, E. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin43-dependent. Cardiovasc. Res. 2014, 103, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Renu, K.; Abilash, V.G.; Tirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, M.; Pala, B.; Di Marcantonio, M.C.; Muraro, R.; Marzocco, S.; Pinto, A.; Mincione, G.; Popolo, A. Doxorubicin-induced oxidative and nitrosative stress: Mitochondrial connexin 43 is at the crossroads. Int. J. Mol. Med. 2020, 46, 1197–1209. [Google Scholar] [CrossRef]
- Pecoraro, M.; Pinto, A.; Popolo, A. Trastuzumab-induced cardiotoxicity and role of mitochondrial connexin43 in the adaptive response. Toxicol. In Vitro 2020, 67, 104926. [Google Scholar] [CrossRef]
- Pecoraro, M.; Rodriguez-Sinovas, A.; Marzocco, S.; Ciccarelli, M.; Iaccarino, G.; Pinto, A.; Popolo, A. Cardiotoxic Effects of Short-Term Doxorubicin Administration: Involvement of Connexin 43 in Calcium Impairment. Int. J. Mol. Sci. 2017, 18, 2121. [Google Scholar] [CrossRef]
- Pecoraro, M.; Ciccarelli, M.; Fiordelisi, A.; Iaccarino, G.; Pinto, A.; Popolo, A. Diazoxide Improves Mitochondrial Connexin 43 Expression in a Mouse Model of Doxorubicin-Induced Cardiotoxicity. Int. J. Mol. Sci. 2018, 19, 757. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Si, R.; Scott, B.T.; Makino, A. Mitochondrial connexin40 regulates mitochondrial calcium uptake in coronary endothelial cells. Am. J. Physiol. Cell Physiol. 2017, 312, C398–C406. [Google Scholar] [CrossRef] [Green Version]
- De Feijter, A.W.; Matesic, D.F.; Ruch, R.J.; Guan, X.; Chang, C.C.; Trosko, J.E. Localization and function of the connexin 43 gap-junction protein in normal and various oncogene-expressing rat liver epithelial cells. Mol. Carcinog. 1996, 16, 203–212. [Google Scholar] [CrossRef]
- Cesen-Cummings, K.; Fernstrom, M.J.; Malkinson, A.M.; Ruch, R.J. Frequent reduction of gap junctional intercellular communication and connexin43 expression in human and mouse lung carcinoma cells. Carcinogenesis 1998, 19, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.P.; Fan, Y.; Hossain, M.Z.; Peng, A.; Zeng, Z.L.; Boynton, A.L. Reversion of the neoplastic phenotype of human glioblastoma cells by connexin 43 (cx43). Cancer Res. 1998, 58, 5089–5096. [Google Scholar]
- Crespin, S.; Fromont, G.; Wager, M.; Levillain, P.; Cronier, L.; Monvoisin, A.; Defamie, N.; Mesnil, M. Expression of a gap junction protein, connexin43, in a large panel of human gliomas: New insights. Cancer Med. 2016, 5, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Sirnes, S.; Bruun, J.; Kolberg, M.; Kjenseth, A.; Lind, G.E.; Svindland, A.; Brech, A.; Nesbakken, A.; Lothe, R.A.; Leithe, E.; et al. Connexin43 acts as a colorectal cancer tumor suppressor and predicts disease outcome. Int. J. Cancer 2012, 131, 570–581. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, C.; Zhang, D.; Zou, J.; Liu, W.; Cai, L.; Cui, Y.; Lai, W.; Xie, J. The involvement of the ERK-MAPK pathway in TGF-beta1-mediated connexin43-gap junction formation in chondrocytes. Connect. Tissue Res. 2019, 60, 477–486. [Google Scholar] [CrossRef]
- Chen, X.; Kong, X.; Zhuang, W.; Teng, B.; Yu, X.; Hua, S.; Wang, S.; Liang, F.; Ma, D.; Zhang, S.; et al. Dynamic changes in protein interaction between AKAP95 and Cx43 during cell cycle progression of A549 cells. Sci. Rep. 2016, 6, 21224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Shao, Q.; Curtis, H.; Galipeau, J.; Belliveau, D.J.; Wang, T.; Alaoui-Jamali, M.A.; Laird, D.W. Retroviral delivery of connexin genes to human breast tumor cells inhibits in vivo tumor growth by a mechanism that is independent of significant gap junctional intercellular communication. J. Biol. Chem. 2002, 277, 29132–29138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.Z.; Jiang, J.X. Gap junction and hemichannel-independent actions of connexins on cell and tissue functions—An update. FEBS Lett. 2014, 588, 1186–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Tang, X.; Ma, T.; Ding, M.; Bian, L.; Chen, D.; Li, Y.; Wang, L.; Zhuang, Y.; Xie, M.; et al. Levonorgestrel Inhibits Human Endometrial Cell Proliferation through the Upregulation of Gap Junctional Intercellular Communication via the Nuclear Translocation of Ser255 Phosphorylated Cx43. Biomed. Res. Int. 2015, 2015, 758684. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Khan, M.R.A.; Turmaine, M.; Thrasivoulou, C.; Becker, D.L.; Ahmed, A. Wnt signaling regulates cytosolic translocation of connexin 43. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R248–R261. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Sansano, I.; Montero, M.A.; Romagosa, C.; Temprana-Salvador, J.; Martínez-Marti, A.; Moliné, T.; Hernández-Losa, J.; Cajal, S. Insight into the Role and Regulation of Gap Junction Genes in Lung Cancer and Identification of Nuclear Cx43 as a Putative Biomarker of Poor Prognosis. Cancers 2019, 11, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ul-Hussain, M.; Olk, S.; Schoenebeck, B.; Wasielewski, B.; Meier, C.; Prochnow, N.; May, C.; Galozzi, S.; Marcus, K.; Zoidl, G.; et al. Internal Ribosomal Entry Site (IRES) Activity Generates Endogenous Carboxyl-terminal Domains of Cx43 and Is Responsive to Hypoxic Conditions. J. Biol. Chem. 2014, 289, 20979–20990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salat-Canela, C.; Sese, M.; Peula, C.; Cajal, S.; Aasen, T. Internal translation of the connexin 43 transcript. Cell Commun. Signal. 2014, 12, 31. [Google Scholar] [CrossRef] [Green Version]
- Salat-Canela, C.; Muñoz, M.J.; Sese, M.; Cajal, S.; Aasen, T. Post-transcriptional regulation of connexins. Biochem. Soc. Trans. 2015, 43, 465–470. [Google Scholar] [CrossRef]
- Smyth, J.W.; Shaw, R.M. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013, 5, 611–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Zhang, S.S.; Xiao, S.; Basheer, W.A.; Baum, R.; Epifantseva, I.; Hong, T.; Shaw, R.M. Cx43 Isoform GJA1-20k Promotes Microtubule Dependent Mitochondrial Transport. Front. Physiol. 2017, 8, 905. [Google Scholar] [CrossRef]
- Mennecier, G.; Derangeon, M.; Coronas, V.; Herve, J.C.; Mesnil, M. Aberrant expression and localization of connexin43 and connexin30 in a rat glioma cell line. Mol. Carcinog. 2008, 47, 391–401. [Google Scholar] [CrossRef]
- Jiang, J.X.; Gu, S. Gap junction- and hemichannel-independent actions of connexins. Biochim. Biophys. Acta 2005, 1711, 208–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stout, C.; Goodenough, D.A.; Paul, D.L. Connexins: Functions without junctions. Curr. Opin. Cell Biol. 2004, 16, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Yu, X.S.; Yin, X.; Jiang, J.X. Stimulation of lens cell differentiation by gap junction protein connexin 45.6. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2103–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesnil, M.; Krutovskikh, V.; Piccoli, C.; Elfgang, C.; Traub, O.; Willecke, K.; Yamasaki, H. Negative growth control of HeLa cells by connexin genes: Connexin species specificity. Cancer Res. 1995, 55, 629–639. [Google Scholar] [PubMed]
- Moorby, C.; Patel, M. Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp. Cell Res. 2001, 271, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Olbina, G.; Eckhart, W. Mutations in the second extracellular region of connexin 43 prevent localization to the plasma membrane, but do not affect its ability to suppress cell growth. Mol. Cancer Res. 2003, 1, 690–700. [Google Scholar]
- Banks, E.A.; Yu, X.S.; Shi, Q.; Jiang, J.X. Promotion of lens epithelial-fiber differentiation by the C-terminus of connexin 45.6 a role independent of gap junction communication. J. Cell Sci. 2007, 120, 3602–3612. [Google Scholar] [CrossRef] [Green Version]
- Doble, B.W.; Dang, X.; Ping, P.; Fandrich, R.R.; Nickel, B.E.; Jin, Y.; Cattini, P.A.; Kardami, E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J. Cell Sci. 2004, 117, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Shao, Q.; Thomas, T.; Kalra, J.; Alaoui-Jamali, M.A.; Laird, D.W. Connexin26 regulates the expression of angiogenesis-related genes in human breast tumor cells by both GJIC-dependent and -independent mechanisms. Cell Commun. Adhes. 2003, 10, 387–393. [Google Scholar] [CrossRef]
- Vinken, M.; Maes, M.; Cavill, R.; Valkenborg, D.; Ellis, J.K.; Decrock, E.; Leybaert, L.; Staes, A.; Gevaert, K.; Oliveira, A.G.; et al. Proteomic and metabolomic responses to connexin43 silencing in primary hepatocyte cultures. Arch. Toxicol. 2013, 87, 883–894. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Li, W.E.; Zoidl, G.; Dermietzel, R.; Spray, D.C. Genes controlling multiple functional pathways are transcriptionally regulated in connexin43 null mouse heart. Physiol. Genom. 2005, 20, 211–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambon, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [CrossRef] [PubMed]
- Epifantseva, I.; Xiao, S.; Baum, R.E.; Kleber, A.G.; Hong, T.; Shaw, R.M. An Alternatively Translated Connexin 43 Isoform, GJA1-11k, Localizes to the Nucleus and Can Inhibit Cell Cycle Progression. Biomolecules 2020, 10, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, J.A.; van Veen, T.A.; de Jong, S.; van der, N.R.; van Stuijvenberg, L.; Driessen, H.; Labzowski, R.; Oefner, C.M.; Bosch, A.A.; Nguyen, T.Q.; et al. Reduced cx43 expression triggers increased fibrosis due to enhanced fibroblast activity. Circ. Arrhythm. Electrophysiol. 2012, 5, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valls-Lacalle, L.; Negre-Pujol, C.; Rodriguez, C.; Varona, S.; Valera-Canellas, A.; Consegal, M.; Martinez-Gonzalez, J.; Rodriguez-Sinovas, A. Opposite Effects of Moderate and Extreme Cx43 Deficiency in Conditional Cx43-Deficient Mice on Angiotensin II-Induced Cardiac Fibrosis. Cells 2019, 8, 1299. [Google Scholar] [CrossRef] [Green Version]
- Valls-Lacalle, L.; Consegal, M.; Ruiz-Meana, M.; Benito, B.; Inserte, J.; Barba, I.; Ferreira-Gonzalez, I.; Rodriguez-Sinovas, A. Connexin 43 Deficiency Is Associated with Reduced Myocardial Scar Size and Attenuated TGFbeta Signaling after Transient Coronary Occlusion in Conditional Knock-Out Mice. Biomolecules 2020, 10, 651. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, H.; Kovacs, A.; Kanter, E.M.; Yamada, K.A. Reduced expression of Cx43 attenuates ventricular remodeling after myocardial infarction via impaired TGF-beta signaling. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H477–H487. [Google Scholar] [CrossRef] [Green Version]
- Ongstad, E.L.; O’Quinn, M.P.; Ghatnekar, G.S.; Yost, M.J.; Gourdie, R.G. A Connexin43 Mimetic Peptide Promotes Regenerative Healing and Improves Mechanical Properties in Skin and Heart. Adv. Wound Care 2013, 2, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Coutinho, P.; Frank, S.; Franke, S.; Law, L.Y.; Martin, P.; Green, C.R.; Becker, D.L. Targeting connexin43 expression accelerates the rate of wound repair. Curr. Biol. 2003, 13, 1697–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghatnekar, G.S.; O’Quinn, M.P.; Jourdan, L.J.; Gurjarpadhye, A.A.; Draughn, R.L.; Gourdie, R.G. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 2009, 4, 205–223. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, J.; Ghatnekar, G.S.; Grek, C.L.; Moyer, K.E.; Gourdie, R.G. Connexin 43-Based Therapeutics for Dermal Wound Healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef] [Green Version]
- Elbadawy, H.M.; Mirabelli, P.; Xeroudaki, M.; Parekh, M.; Bertolin, M.; Breda, C.; Cagini, C.; Ponzin, D.; Lagali, N.; Ferrari, S. Effect of connexin 43 inhibition by the mimetic peptide Gap27 on corneal wound healing, inflammation and neovascularization. Br. J. Pharmacol. 2016, 173, 2880–2893. [Google Scholar] [CrossRef]
- Cogliati, B.; Vinken, M.; Silva, T.C.; Araujo, C.M.; Aloia, T.P.; Chaible, L.M.; Mori, C.M.; Dagli, M.L. Connexin 43 deficiency accelerates skin wound healing and extracellular matrix remodeling in mice. J. Dermatol. Sci. 2015, 79, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmartin, D.J.; Soon, A.; Thrasivoulou, C.; Phillips, A.R.; Jayasinghe, S.N.; Becker, D.L. Sustained Release of Cx43 Antisense Oligodeoxynucleotides from Coated Collagen Scaffolds Promotes Wound Healing. Adv. Healthc. Mater. 2016, 5, 1786–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghatnekar, G.S.; Grek, C.L.; Armstrong, D.G.; Desai, S.C.; Gourdie, R.G. The effect of a connexin43-based Peptide on the healing of chronic venous leg ulcers: A multicenter, randomized trial. J. Investig. Dermatol. 2015, 135, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Sinovas, A.; Sánchez, J.A.; Valls-Lacalle, L.; Consegal, M.; Ferreira-González, I. Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease. Int. J. Mol. Sci. 2021, 22, 4413. https://doi.org/10.3390/ijms22094413
Rodríguez-Sinovas A, Sánchez JA, Valls-Lacalle L, Consegal M, Ferreira-González I. Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease. International Journal of Molecular Sciences. 2021; 22(9):4413. https://doi.org/10.3390/ijms22094413
Chicago/Turabian StyleRodríguez-Sinovas, Antonio, Jose Antonio Sánchez, Laura Valls-Lacalle, Marta Consegal, and Ignacio Ferreira-González. 2021. "Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease" International Journal of Molecular Sciences 22, no. 9: 4413. https://doi.org/10.3390/ijms22094413
APA StyleRodríguez-Sinovas, A., Sánchez, J. A., Valls-Lacalle, L., Consegal, M., & Ferreira-González, I. (2021). Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease. International Journal of Molecular Sciences, 22(9), 4413. https://doi.org/10.3390/ijms22094413