
ABCC6, Pyrophosphate and Ectopic Calcification: Therapeutic Solutions

 ,
,
Abstract
:1. Introduction
2. The Pathologies Associated with ABCC6 and ENPP1 Deficiencies
2.1. Pseudoxanthoma Elasticum
2.1.1. Dermal Manifestations
2.1.2. Ocular Manifestations
2.1.3. Cardiovascular Manifestations
2.1.4. Renal Manifestations
2.2. Generalized Arterial Calcification of Infancy (GACI)
2.3. PXE and GACI Are Different Clinical Manifestations of a Phenotypic Continuum
2.4. Thalassemia
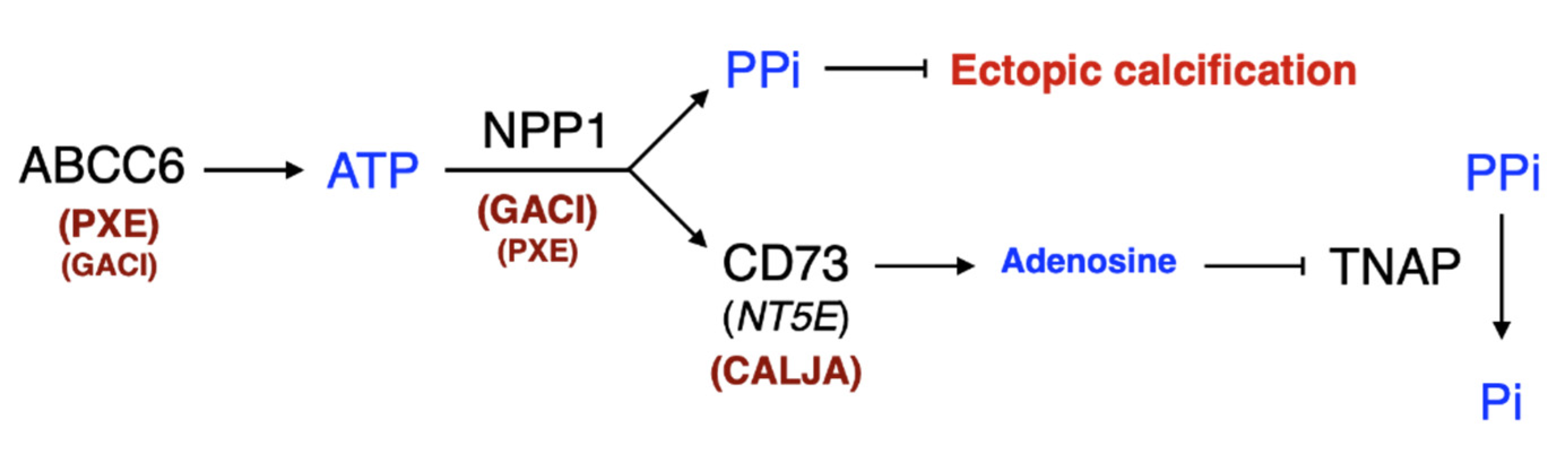
3. The Structure and Molecular Function of ABCC6
3.1. ABCC6 Structure
3.2. ABCC6 Is an Efflux Pump
3.3. Cellular Localization of ABCC6
3.4. NPP1 Structure
4. Mutations in ABCC6 and ENPP1
4.1. ABCC6
4.2. ENPP1
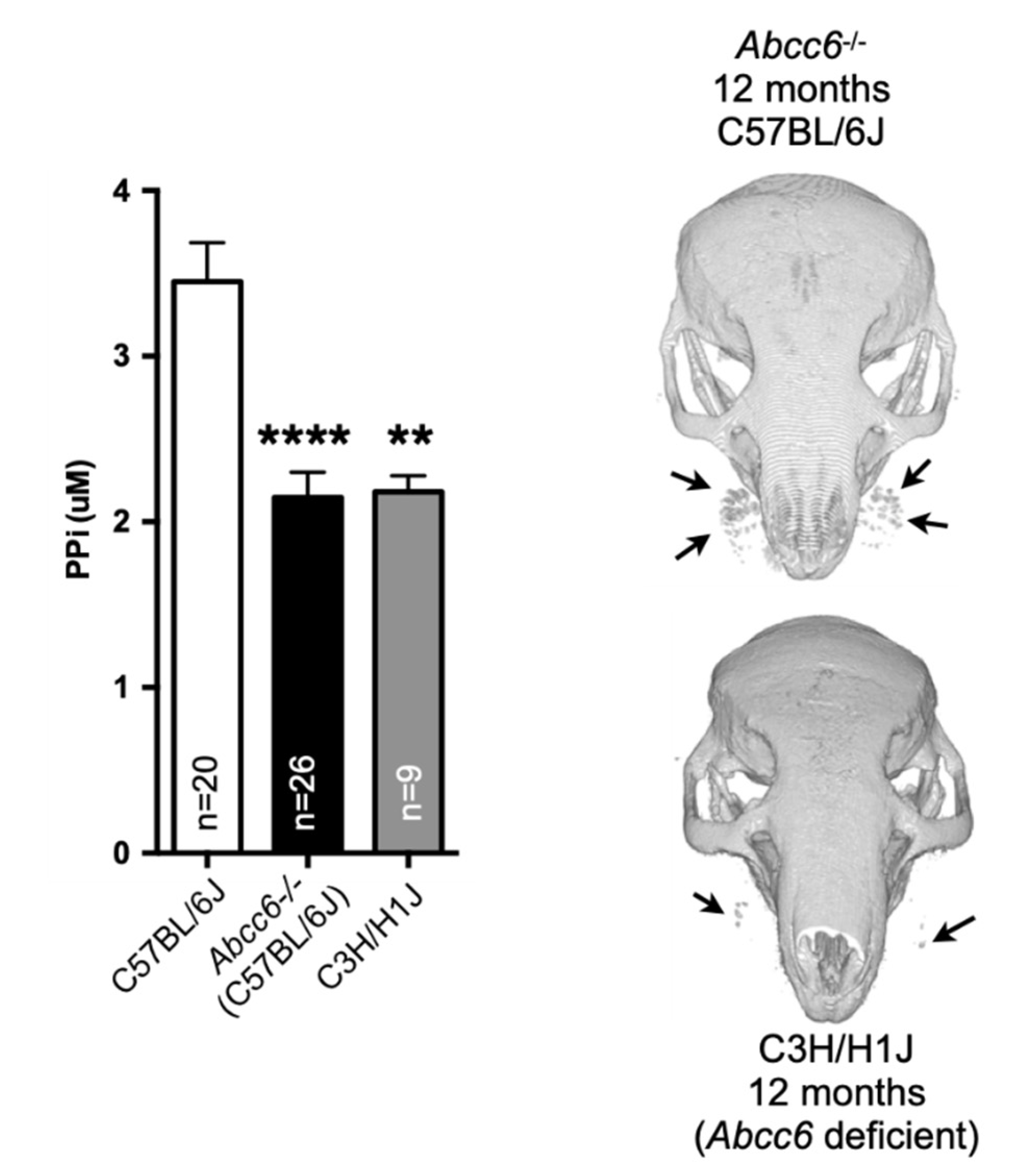
5. Animal Models
5.1. The PXE Mice
5.2. The Murine DCC Phenotype and Other PXE Mouse Models
5.3. The PXE Rat
5.4. The “PXE” Zebrafish
5.5. The GACI Models
5.5.1. GACI Mice
5.5.2. The GACI Zebrafish
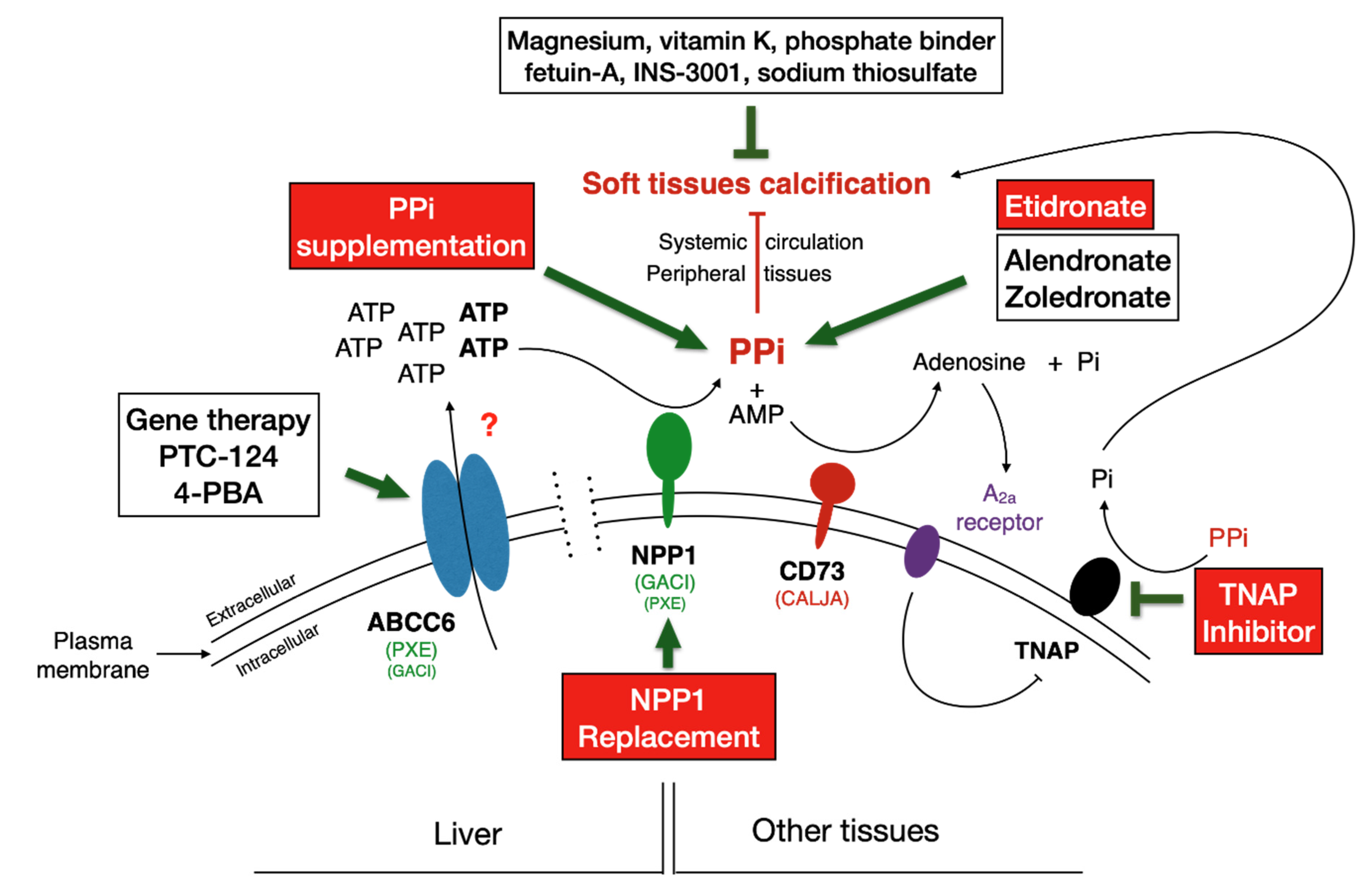
6. Rescue and Therapeutic Solutions
6.1. Treatment Outcome Conundrum
6.2. Palliative Treatment: An Ocular Therapy for PXE
6.3. Targeting ABCC6
6.3.1. PTC-124
6.3.2. 4-Phenylbutyrate (4-PBA)
6.4. Targeting NPP1
6.5. Targeting TNAP
6.6. Gene Therapy
6.7. Supplementation Therapies for Direct Inhibition of Calcification
6.7.1. Calcium and Phosphate
6.7.2. Magnesium
6.7.3. Vitamin K
6.7.4. Bisphosphonate Treatment for PXE and GACI
6.7.5. Pyrophosphate (PPi)
6.7.6. Phytic Acid (IP6)
6.7.7. Sodium Thiosulfate
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, K.; Sawczyk, M.; Liu, C.; Yuan, Y.; Song, B.; Deivanayagam, R.; Nie, A.; Hu, X.; Dravid, V.P.; Lu, J.; et al. Revealing nanoscale mineralization pathways of hydroxyapatite using in situ liquid cell transmission electron microscopy. Sci. Adv. 2020, 6, eaaz7524. [Google Scholar] [CrossRef] [PubMed]
- Atzeni, F.; Sarzi-Puttini, P.; Bevilacqua, M. Calcium deposition and associated chronic diseases (atherosclerosis, diffuse idiopathic skeletal hyperostosis, and others). Rheum. Dis. Clin. N. Am. 2006, 32, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Nitschke, Y.; Terkeltaub, R. Genetics in arterial calcification: Pieces of a puzzle and cogs in a wheel. Circ. Res. 2011, 109, 578–592. [Google Scholar] [CrossRef]
- Ruiz, J.L.; Hutcheson, J.D.; Aikawa, E. Cardiovascular calcification: Current controversies and novel concepts. Cardiovasc. Pathol. 2015, 24, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Bergen, A.A.; Plomp, A.S.; Schuurman, E.J.; Terry, S.F.; Breuning, M.H.; Dauwerse, H.G.; Swart, J.; Kool, M.; van Soest, S.; Baas, F.; et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef]
- Le Saux, O.; Urban, Z.; Tschuch, C.; Csiszar, K.; Bacchelli, B.; Quaglino, D.; Pasquali-Ronchetti, I.; Pope, F.M.; Richards, A.; Terry, S.F.; et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 223–227. [Google Scholar] [CrossRef]
- Ringpfeil, F.; Lebwohl, M.G.; Christiano, A.M.; Uitto, J. Pseudoxanthoma elasticum: Mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc. Natl. Acad. Sci. USA 2000, 97, 6001–6006. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.S.; Küçükosmanoglu, A.; de Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.M.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Borst, P.; van de Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Váradi, A.; Plomp, A.S.; Bergen, A.A.B.; Elferink, R.P.J.O.; Borst, P.; et al. ABCC6–Mediated ATP Secretion by the Liver Is the Main Source of the Mineralization Inhibitor Inorganic Pyrophosphate in the Systemic Circulation—Brief Report. Arter. Thromb. Vasc. Biol. 2014, 34, 1985–1989. [Google Scholar] [CrossRef] [Green Version]
- Markello, T.C.; Pak, L.K.; St. Hilaire, C.; Dorward, H.; Ziegler, S.G.; Chen, M.Y.; Chaganti, K.; Nussbaum, R.L.; Boehm, M.; Gahl, W.A. Vascular pathology of medial arterial calcifications in NT5E deficiency: Implications for the role of adenosine in pseudoxanthoma elasticum. Mol. Genet. Metab. 2011, 103, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Miglionico, R.; Armentano, M.F.; Carmosino, M.; Salvia, A.M.; Cuviello, F.; Bisaccia, F.; Ostuni, A. Dysregulation of gene expression in ABCC6 knockdown HepG2 cells. Cell. Mol. Biol. Lett. 2014, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Aherrahrou, Z.; Doehring, L.C.; Ehlers, E.-M.; Liptau, H.; Depping, R.; Linsel-Nitschke, P.; Kaczmarek, P.M.; Erdmann, J.; Schunkert, H. An Alternative Splice Variant in Abcc6, the Gene Causing Dystrophic Calcification, Leads to Protein Deficiency in C3H/He Mice. J. Biol. Chem. 2008, 283, 7608–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brampton, C.; Aherrahrou, Z.; Chen, L.-H.; Martin, L.; Bergen, A.A.; Gorgels, T.G.; Erdfdi, J.; Schunkert, H.; Szabó, Z.; Váradi, A.; et al. The Level of Hepatic ABCC6 Expression Determines the Severity of Calcification after Cardiac Injury. Am. J. Pathol. 2014, 184, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Vera, I.; Che, N.; Wang, X.; Wang, S.S.; Ingram-Drake, L.; Schadt, E.E.; Drake, T.A.; Lusis, A.J. Identification of Abcc6 as the major causal gene for dystrophic cardiac calcification in mice through inte-grative genomics. Proc. Natl. Acad. Sci. USA 2007, 104, 4530–4535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalal, I.G.; Seetha, D.; Panda, A.; Nitschke, Y.; Rutsch, F. Molecular diagnosis of generalized arterial calcification of infancy (GACI). J. Cardiovasc. Dis. Res. 2012, 3, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Boulanger, G.; Labreze, C.; Croue, A.; Schurgers, L.J.; Chassaing, N.; Wittkampf, T.; Rutsch, F.; Martin, L. An unusual severe vascular case of pseudoxanthoma elasticum presenting as generalized arterial calcifica-tion of infancy. Am. J. Med. Genet. A 2010, 152, 118–123. [Google Scholar] [CrossRef]
- Nitschke, Y.; Baujat, G.; Botschen, U.; Wittkampf, T.; du Moulin, M.; Stella, J.; le Merrer, M.; Guest, G.; Lambot, K.; Tazarourte-Pinturier, M.-F.; et al. Generalized Arterial Calcification of Infancy and Pseudoxanthoma Elasticum Can Be Caused by Mutations in Either ENPP1 or ABCC6. Am. J. Hum. Genet. 2012, 90, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Pomozi, V.; Brampton, C.; van de Wetering, K.; Zoll, J.; Calio, B.; Pham, K.; Owens, J.B.; Marh, J.; Moisyadi, S.; Váradi, A.; et al. Pyrophosphate Supplementation Prevents Chronic and Acute Calcification in ABCC6-Deficient Mice. Am. J. Pathol. 2017, 187, 1258–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomozi, V.; Julian, C.B.; Zoll, J.; Pham, K.; Kuo, S.; Tőkési, N.; Martin, L.; Váradi, A.; le Saux, O. Dietary Pyrophosphate Modulates Calcification in a Mouse Model of Pseudoxanthoma Elasticum: Implication for Treatment of Patients. J. Investig. Dermatol. 2019, 139, 1082–1088. [Google Scholar] [CrossRef]
- Ziegler, S.G.; Ferreira, C.R.; Macfarlane, E.G.; Riddle, R.C.; Tomlinson, R.E.; Chew, E.Y.; Martin, L.; Ma, C.-T.; Sergienko, E.; Pinkerton, A.B.; et al. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci. Transl. Med. 2017, 9, eaal1669. [Google Scholar] [CrossRef] [Green Version]
- Borst, P.; Váradi, A.; van de Wetering, K. PXE, a Mysterious Inborn Error Clarified. Trends Biochem. Sci. 2019, 44, 125–140. [Google Scholar] [CrossRef]
- Ali, S.A.; Ng, C.; Votava-Smith, J.K.; Randolph, L.M.; Pitukcheewanont, P. Bisphosphonate therapy in an infant with generalized arterial calcification with an ABCC6 mutation. Osteoporos. Int. 2018, 29, 2575–2579. [Google Scholar] [CrossRef]
- Edouard, T.; Chabot, G.; Miro, J.; Buhas, D.C.; Nitschke, Y.; Lapierre, C.; Rutsch, F.; Alos, N. Efficacy and safety of 2-year etidronate treatment in a child with generalized arterial calcification of infancy. Eur. J. Nucl. Med. Mol. Imaging 2011, 170, 1585–1590. [Google Scholar] [CrossRef] [PubMed]
- Finger, R.P.; Issa, P.C.; Schmitz-Valckenberg, S.; Holz, F.G.; Scholl, H.N. Long-Term Effectiveness of Intravitreal Bevacizumab for Choroidal Neovascularization Secondary to Angioid Streaks in Pseudoxanthoma Elasticum. Retina 2011, 31, 1268–1278. [Google Scholar] [CrossRef]
- Uitto, J.; Shamban, A. Heritable Skin Diseases with Molecular Defects in Collagen or Elastin. Dermatol. Clin. 1987, 5, 63–84. [Google Scholar] [CrossRef]
- Balzer, F. Recherches sur les caractères anatomiques du xanthelasma. Arch. Physiol. 1884, 4, 65–80. [Google Scholar]
- Chauffard, M.A. Xanthélasma disséminé et symétrique et sans insuffisance hépatique. Bull. Soc. Med. Paris 1889, 6, 412–419. [Google Scholar]
- Rigal, D. Observation pour servir à l’histoire de la chéloide diffuse xanthélasmique. Ann. Dermatol. Syphilol. 1881, 2, 491–501. [Google Scholar]
- Darier, J. Pseudo-Xanthome Elastique; III Congrès International de Dermatoligie de Londres: London, UK, 1896; pp. 289–295. [Google Scholar]
- Neldner, K.H. Pseudoxanthoma Elasticum. Int. J. Dermatol. 1988, 27, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Truter, S.; Rosenbaum-Fiedler, J.; Sapadin, A.; Lebwohl, M. Calcification of elastic fibers in pseudoxan-thoma elasticum. Mt. Sinai. J. Med. 1996, 63, 210–215. [Google Scholar]
- Uitto, J.; Boyd, C.D.; Lebwohl, M.G.; Moshell, A.N.; Rosenbloom, J.; Terry, S. International Centennial Meeting on Pseudoxanthoma Elasticum: Progress in PXE Research. J. Investig. Dermatol. 1998, 110, 840–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weenink, A.C.; Dijkman, G.; de Meijer, P.H. Pseudoxanthoma elasticum and its complications: Two case reports. Neth. J. Med. 1996, 49, 24–29. [Google Scholar] [CrossRef]
- Campens, L.; Vanakker, O.M.; Trachet, B.; Segers, P.; Leroy, B.P.; de Zaeytijd, J.; Voet, D.; de Paepe, A.; de Backer, T.; de Backer, J. Characterization of Cardiovascular Involvement in Pseudoxanthoma Elasticum Families. Arter. Thromb. Vasc. Biol. 2013, 33, 2646–2652. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P.; Boutouyrie, P.; Laloux, B.; Laurent, S. Arterial Remodeling and Stiffness in Patients with Pseudoxanthoma Elasticum. Arter. Thromb. Vasc. Biol. 2003, 23, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Kornet, L.; Bergen, A.A.; Hoeks, A.P.; Cleutjens, J.P.; Oostra, R.-J.; Daemen, M.J.; van Soest, S.; Reneman, R.S. In patients with pseudoxanthoma elasticum a thicker and more elastic carotid artery is associated with elastin fragmentation and proteoglycans accumulation. Ultrasound Med. Biol. 2004, 30, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Bertulezzi, G.; Paris, R.; Moroni, M.; Porta, C.; Nastasi, G.; Amadeo, A. Atrial septal aneurysm in a patient with pseudoxanthoma elasticum. Acta Cardiol. 1998, 53, 223–225. [Google Scholar] [PubMed]
- Prunier, F.; Terrien, G.; le Corre, Y.; Apana, A.L.Y.; Bière, L.; Kauffenstein, G.; Furber, A.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Le Saux, O.; et al. Pseudoxanthoma Elasticum: Cardiac Findings in Patients and Abcc6-Deficient Mouse Model. PLoS ONE 2013, 8, e68700. [Google Scholar] [CrossRef]
- Eddy, D.D.; Farber, E.M. Pseudoxathoam elasticum. Internal manifestatons: A report of cases and a review of the literature. Arch. Dermatol. 1962, 86, 729–740. [Google Scholar] [CrossRef]
- Kauffenstein, G.; Pizard, A.; le Corre, Y.; Vessières, E.; Grimaud, L.; Toutain, B.; Labat, C.; Mauras, Y.; Gorgels, T.G.; Bergen, A.A.; et al. Disseminated arterial calcification and enhanced myogenic response are associated with abcc6 deficiency in a mouse model of pseudoxanthoma elasticum. Arter. Thromb. Vasc. Biol. 2014, 34, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Fabre, B.; Bayle, P.; Bazex, J.; Durand, D.; Lamant, L.; Chassaing, N. Pseudoxanthoma elasticum and nephrolithiasis. J. Eur. Acad. Dermatol. Venereol. 2005, 19, 212–215. [Google Scholar] [CrossRef]
- Mallette, L.E.; Mechanick, J.I. Heritable syndrome of pseudoxanthoma elasticum with abnormal phosphorus and vitamin D metabolism. Am. J. Med. 1987, 83, 1157–1162. [Google Scholar] [CrossRef]
- Seeger, H.; Mohebbi, N. Pseudoxanthoma elasticum and nephrocalcinosis. Kidney Int. 2016, 89, 1407. [Google Scholar] [CrossRef] [PubMed]
- Legrand, A.; Cornez, L.; Samkari, W.; Mazzella, J.-M.; Venisse, A.; Boccio, V.; Auribault, K.; Keren, B.; Benistan, K.; Germain, D.P.; et al. Mutation spectrum in the ABCC6 gene and genotype–phenotype correlations in a French cohort with pseudoxanthoma elasticum. Genet. Med. 2017, 19, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Letavernier, E.; Bouderlique, E.; Zaworski, J.; Martin, L.; Daudon, M. Pseudoxanthoma Elasticum, Kidney Stones and Pyrophosphate: From a Rare Disease to Urolithiasis and Vascular Calcifications. Int. J. Mol. Sci. 2019, 20, 6353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letavernier, E.; Kauffenstein, G.; Huguet, L.; Navasiolava, N.; Bouderlique, E.; Tang, E.; Delaitre, L.; Bazin, D.; de Frutos, M.; Gay, C.; et al. ABCC6 Deficiency Promotes Development of Randall Plaque. J. Am. Soc. Nephrol. 2018, 29, 2337–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheduzzi, D.; Sammarco, R.; Quaglino, D.; Bercovitch, L.; Terry, S.; Taylor, W.; Ronchetti, I.P. Extracutaneous ultrastructural alterations in pseudoxanthoma elasticum. Ultrastruct. Pathol. 2003, 27, 375–384. [Google Scholar] [CrossRef]
- Lebwohl, M.; Halperin, J.; Phelps, R.G. Brief report: Occult pseudoxanthoma elasticum in patients with premature cardiovascular disease. N. Engl. J. Med. 1993, 329, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Vaingankar, S.; Johnson, K.; Goldfine, I.; Maddux, B.; Schauerte, P.; Kalhoff, H.; Sano, K.; Boisvert, W.A.; Superti-Furga, A.; et al. PC-1 Nucleoside Triphosphate Pyrophosphohydrolase Deficiency in Idiopathic Infantile Arterial Calcification. Am. J. Pathol. 2001, 158, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Moran, J.J. Idiopathic arterial calcification of infancy: A clinicopathologic study. Pathol. Annu. 1975, 10, 393–417. [Google Scholar]
- Morton, R. Idiopathic arterial calcification in infancy. Histopathology 1978, 2, 423–432. [Google Scholar] [CrossRef]
- Ramjan, K.A.; Roscioli, T.; Rutsch, F.; Sillence, D.; Munns, C.F. Generalized arterial calcification of infancy: Treatment with bisphosphonates. Nat. Clin. Pract. Endocrinol. Metab. 2009, 5, 167–172. [Google Scholar] [PubMed]
- Rutsch, F.; Boyer, P.; Nitschke, Y.; Ruf, N.; Lorenz-Depierieux, B.; Wittkampf, T.; Weissen-Plenz, G.; Fischer, R.J.; Mughal, Z.; Gregory, J.W.; et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ. Cardiovasc. Genet. 2008, 1, 133–140. [Google Scholar] [CrossRef]
- Lorenz-Depiereux, B.; Schnabel, D.; Tiosano, D.; Hausler, G.; Strom, T.M. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am. J. Hum. Genet. 2010, 86, 267–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutsch, F.; Ruf, N.; Vaingankar, S.; Toliat, M.R.; Suk, A.; Hohne, W.; Schauer, G.; Lehmann, M.; Roscioli, T.; Schnabel, D.; et al. Mutations in ENPP1 are associated with ’idiopathic’ infantile arterial calcification. Nat. Genet. 2003, 34, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Martin, L.; Aherrahrou, Z.; Leftheriotis, G.; Varadi, A.; Brampton, C.N. The molecular and physiological roles of ABCC6: More than meets the eye. Front. Genet. 2012, 3, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitschke, Y.; Rutsch, F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum: Two sides of the same coin. Front. Genet. 2012, 3, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Grange, D.K.; Armstrong, N.L.; Whelan, A.J.; Hurley, M.Y.; Rishavy, M.A.; Hallgren, K.W.; Berkner, K.L.; Schurgers, L.J.; Jiang, Q.; et al. Mutations in the GGCX and ABCC6 Genes in a Family with Pseudoxanthoma Elasticum-Like Phenotypes. J. Investig. Dermatol. 2009, 129, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Omarjee, L.; Nitschke, Y.; Verschuere, S.; Bourrat, E.; Vignon, M.; Navasiolava, N.; Leftheriotis, G.; Kauffenstein, G.; Rutsch, F.; Vanakker, O.; et al. Severe early-onset manifestations of pseudoxanthoma elasticum resulting from the cumulative effects of several deleterious mutations in ENPP1, ABCC6 and HBB: Transient improvement in ectopic calcification with sodium thiosulfate. Br. J. Dermatol. 2019, 183, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Bentley-Phillips, B. Pseudoxanthoma elasticum-like skin changes induced by penicillamine. J. R. Soc. Med. 1985, 78, 787. [Google Scholar]
- Bolognia, J.L.; Braverman, I. Pseudoxanthoma-elasticum-like Skin Changes Induced by Penicillamine. Dermatology 1992, 184, 12–18. [Google Scholar] [CrossRef]
- Coatesworth, A.P.; Darnton, S.J.; Green, R.M.; Cayton, R.M.; Antonakopoulos, G.N. A case of systemic pseudo-pseudoxanthoma elasticum with diverse symptomatology caused by long-term penicillamine use. J. Clin. Pathol. 1998, 51, 169–171. [Google Scholar] [CrossRef] [Green Version]
- Dhurat, R.; Nayak, C.; Pereira, R.; Kagne, R.; Khatu, S. Penicillamine-induced elastosis perforans serpiginosa with abnormal “lumpy-bumpy” elastic fibers in lesional and non-lesional skin. Indian J. Dermatol. Venereol. Leprol. 2011, 77, 55–58. [Google Scholar] [CrossRef]
- Aessopos, A.; Farmakis, D.; Loukopoulos, D. Elastic tissue abnormalities resembling pseudoxanthoma elasticum in beta thalassemia and the sickling syndromes. Blood 2002, 99, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Baccarani-Contri, M.; Bacchelli, B.; Boraldi, F.; Quaglino, D.; Taparelli, F.; Carnevali, E.; Francomano, M.A.; Seidenari, S.; Bettoli, V.; de Sanctis, V.; et al. Characterization of pseudoxanthoma elasticum-like lesions in the skin of patients with beta-thalassemia. J. Am. Acad. Dermatol. 2001, 44, 33–39. [Google Scholar] [CrossRef]
- Cianciulli, P.; Sorrentino, F.; Maffei, L.; Amadori, S.; Cappabianca, M.P.; Foglietta, E.; Carnevali, E.; Pasquali-Ronchetti, I. Cardiovascular involvement in thalassaemic patients with pseudoxanthoma elasticum-like skin lesions: A long-term follow-up study. Eur. J. Clin. Investig. 2002, 32, 700–706. [Google Scholar] [CrossRef]
- Farmakis, D.; Moyssakis, I.; Perakis, A.; Rombos, Y.; Deftereos, S.; Giakoumis, A.; Polymeropoulos, E.; Aessopos, A. Unstable angina associated with coronary arterial calcification in a thalassemia intermedia patient with a pseudoxanthoma elasticum-like syndrome. Eur. J. Haematol. 2003, 70, 64–66. [Google Scholar] [CrossRef]
- Farmakis, D.; Vesleme, V.; Papadogianni, A.; Tsaftaridis, P.; Kapralos, P.; Aessopos, A. Aneurysmatic dilatation of ascending aorta in a patient with beta-thalassemia and a pseudoxanthoma elasticum-like syndrome. Ann. Hematol. 2004, 83, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, N.; Beck, K.; Bacchelli, B.; Cianciulli, P.; Pasquali-Ronchetti, I.; le Saux, O. Acquired Pseudoxanthoma elasticum-like syndrome in beta-thalassaemia patients. Br. J. Haematol. 2003, 122, 852–854. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.; Sarkadi, B.; Váradi, A. An inventory of the human ABC proteins. Biochim. Biophys. Acta Biomembr. 1999, 1461, 237–262. [Google Scholar] [CrossRef] [Green Version]
- Stefková, J.; Poledne, R.; Hubácek, J.A. ATP-binding cassette (ABC) transporters in human metabolism and diseases. Physiol. Res. 2004, 53, 235–243. [Google Scholar] [PubMed]
- Dawson, R.J.; Locher, K.P. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007, 581, 935–938. [Google Scholar] [CrossRef] [Green Version]
- Fülöp, K.; Barna, L.; Symmons, O.; Závodszky, P.; Váradi, A. Clustering of disease-causing mutations on the domain–domain interfaces of ABCC6. Biochem. Biophys. Res. Commun. 2009, 379, 706–709. [Google Scholar] [CrossRef]
- Madon, J.; Hagenbuch, B.; Landmann, L.; Meier, P.J.; Stieger, B. Transport Function and Hepatocellular Localization of mrp6 in Rat Liver. Mol. Pharmacol. 2000, 57, 634–641. [Google Scholar] [CrossRef]
- Cai, J.; Daoud, R.; Alqawi, O.; Georges, E.; Pelletier, J.; Gros, P. Nucleotide binding and nucleotide hydrolysis properties of the ABC transporter MRP6 (ABCC6). Biochemistry 2002, 41, 8058–8067. [Google Scholar] [CrossRef]
- Ilias, A.; Urban, Z.; Seidl, T.L.; le Saux, O.; Sinko, E.; Boyd, C.D.; Sarkadi, B.; Varadi, A. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6). J. Biol. Chem. 2002, 277, 16860–16867. [Google Scholar] [CrossRef] [Green Version]
- Hirohashi, T.; Suzuki, H.; Sugiyama, Y. Characterization of the transport properties of cloned rat multi-drug resistance-associated protein 3 (MRP3). J. Biol. Chem. 1999, 274, 15181–15185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belinsky, M.G.; Chen, Z.-S.; Shchaveleva, I.; Zeng, H.; Kruh, G.D. Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6). Cancer Res. 2002, 62, 6172–6177. [Google Scholar] [PubMed]
- Beck, K.; Hayashi, K.; Nishiguchi, B.; le Saux, O.; Hayashi, M.; Boyd, C.D. The Distribution of Abcc6 in Normal Mouse Tissues Suggests Multiple Functions for this ABC Transporter. J. Histochem. Cytochem. 2003, 51, 887–902. [Google Scholar] [CrossRef] [Green Version]
- Le Saux, O.; Fülöp, K.; Yamaguchi, Y.; Iliás, A.; Szabó, Z.; Brampton, C.N.; Pomozi, V.; Huszár, K.; Arányi, T.; Váradi, A. Expression and In Vivo Rescue of Human ABCC6 Disease-Causing Mutants in Mouse Liver. PLoS ONE 2011, 6, e24738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, K.; Hayashi, K.; Dang, K.; Hayashi, M.; Boyd, C.D. Analysis of ABCC6 (MRP6) in normal human tissues. Histochem. Cell Biol. 2005, 123, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Sinkó, E.; Iliás, A.; Ujhelly, O.; Homolya, L.; Scheffer, G.L.; Bergen, A.A.B.; Sarkadi, B.; Váradi, A. Subcellular localization and N-glycosylation of human ABCC6, expressed in MDCKII cells. Biochem. Biophys. Res. Commun. 2003, 308, 263–269. [Google Scholar] [CrossRef]
- Martin, L.J.; Lau, E.; Singh, H.; Vergnes, L.; Tarling, E.J.; Mehrabian, M.; Mungrue, I.; Xiao, S.; Shih, D.; Castellani, L.; et al. ABCC6 localizes to the mitochondria-associated membrane. Circ. Res. 2012, 111, 516–520. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Nishimasu, H.; Okudaira, S.; Mihara, E.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of Enpp1, an extracellular glycoprotein involved in bone mineralization and insulin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 16876–16881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, F.; Zhu, D.; Farquharson, C.; Macrae, V.E. ENPP1 in the Regulation of Mineralization and Beyond. Trends Biochem. Sci. 2019, 44, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, N.; Martin, L.; Calvas, P.; le Bert, M.; Hovnanian, A. Pseudoxanthoma elasticum: A clinical, pathophysiological and genetic update including 11 novel ABCC6 mutations. J. Med Genet. 2005, 42, 881–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Saux, O.; Beck, K.; Sachsinger, C.; Silvestri, C.; Treiber, C.; Goring, H.H.; Johnson, E.W.; de Paepe, A.; Pope, F.M.; Pasquali-Ronchetti, I.; et al. A spectrum of abcc6 mutations is responsible for pseudoxanthoma elasticum. Am. J. Hum. Genet. 2001, 69, 749–764. [Google Scholar] [CrossRef] [Green Version]
- Pfendner, E.G.; Vanakker, O.M.; Terry, P.F.; Vourthis, S.; McAndrew, E.P.; McClain, M.R.; Fratta, S.; Marais, A.-S.; Hariri, S.; Coucke, P.J.; et al. Mutation detection in the ABCC6 gene and genotype phenotype analysis in a large international case series affected by pseudoxanthoma elasticum. J. Med Genet. 2007, 44, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Nitschke, Y.; Yan, Y.; Buers, I.; Kintziger, K.; Askew, K.; Rutsch, F. ENPP1-Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gorgels, T.G.; Hu, X.; Scheffer, G.L.; van der Wal, A.C.; Toonstra, J.; de Jong, P.T.; van Kuppevelt, T.H.; Levelt, C.N.; de Wolf, A.; Loves, W.J.; et al. Disruption of Abcc6 in the mouse: Novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum. Mol. Genet. 2005, 14, 1763–1773. [Google Scholar] [CrossRef] [Green Version]
- Klement, J.F.; Matsuzaki, Y.; Jiang, Q.-J.; Terlizzi, J.; Choi, H.Y.; Fujimoto, N.; Li, K.; Pulkkinen, L.; Birk, D.E.; Sundberg, J.P.; et al. Targeted Ablation of the Abcc6 Gene Results in Ectopic Mineralization of Connective Tissues. Mol. Cell. Biol. 2005, 25, 8299–8310. [Google Scholar] [CrossRef] [Green Version]
- Brampton, C.; Yamaguchi, Y.; Vanakker, O.; van Laer, L.; Chen, L.-H.; Thakore, M.; de Paepe, A.; Pomozi, V.; Szabó, P.T.; Martin, L.; et al. Vitamin K does not prevent soft tissue mineralization in a mouse model of pseudoxanthoma elasticum. Cell Cycle 2011, 10, 1810–1820. [Google Scholar] [CrossRef] [Green Version]
- Brampton, C.; Pomozi, V.; Chen, L.-H.; Apana, A.; McCurdy, S.; Zoll, J.; Boisvert, W.A.; Lambert, G.; Henrion, D.; Blanchard, S.; et al. ABCC6 deficiency promotes dyslipidemia and atherosclerosis. Sci. Rep. 2021, 11, 3881. [Google Scholar] [CrossRef]
- Ibold, B.; Tiemann, J.; Faust, I.; Ceglarek, U.; Dittrich, J.; Gorgels, T.G.M.F.; Bergen, A.A.B.; Vanakker, O.; van Gils, M.; Knabbe, C.; et al. Genetic deletion of Abcc6 disturbs cholesterol homeostasis in mice. Sci. Rep. 2021, 11, 2137. [Google Scholar] [CrossRef]
- Jiang, Q.; Endo, M.; Dibra, F.; Wang, K.; Uitto, J. Pseudoxanthoma Elasticum Is a Metabolic Disease. J. Investig. Dermatol. 2009, 129, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Oldenburg, R.; Otsuru, S.; Grand-Pierre, A.E.; Horwitz, E.M.; Uitto, J. Parabiotic heterogenetic pairing of Abcc6−/−/Rag1−/− mice and their wild-type counterparts halts ectopic mineralization in a murine model of pseudoxanthoma elasticum. Am. J. Pathol. 2010, 176, 1855–1862. [Google Scholar] [CrossRef]
- Le Saux, O.; Bunda, S.; van Wart, C.M.; Douet, V.; Got, L.; Martin, L.; Hinek, A. Serum Factors from Pseudoxanthoma Elasticum Patients Alter Elastic Fiber Formation In Vitro. J. Investig. Dermatol. 2006, 126, 1497–1505. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Sundberg, J.P.; Levine, M.A.; Terry, S.F.; Uitto, J. The effects of bisphosphonates on ectopic soft tissue mineralization caused by mutations in the ABCC6 gene. Cell Cycle 2015, 14, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomozi, V.; Brampton, C.; Szeri, F.; Dedinszki, D.; Kozak, E.; van de Wetering, K.; Hopkins, H.; Martin, L.; Varadi, A.; le Saux, O. Functional Rescue of ABCC6 Deficiency by 4-Phenylbutyrate Therapy Reduces Dystrophic Calcification in Abcc6(−/−) Mice. J. Investig. Dermatol. 2017, 137, 595–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doehring, L.C.; Kaczmarek, P.M.; Ehlers, E.-M.; Mayer, B.; Erdmann, J.; Schunkert, H.; Aherrahrou, Z. Arterial calcification in mice after freeze-thaw injury. Ann. Anat. Anat. Anz. 2006, 188, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Eaton, G.J.; Custer, R.P.; Johnson, F.N.; Stabenow, K.T. Dystrophic cardiac calcinosis in mice: Genetic, hormonal, and dietary influences. Am. J. Pathol. 1978, 90, 173–186. [Google Scholar]
- Everitt, J.I.; Olson, L.M.; Mangum, J.B.; Visek, W.J. High mortality with severe dystrophic cardiac calcinosis in C3H/OUJ mice fed high fat purified diets. Vet. Pathol. 1988, 25, 113–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aherrahrou, Z.; Axtner, S.B.; Kaczmarek, P.M.; Jurat, A.; Korff, S.; Doehring, L.C.; Weichenhan, D.; Katus, H.A.; Ivandic, B.T. A locus on chromosome 7 determines dramatic up-regulation of osteopontin in dystrophic cardiac calcification in mice. Am. J. Pathol. 2004, 164, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- Ivandic, B.T.; Utz, H.F.; Kaczmarek, P.M.; Aherrahrou, Z.; Axtner, S.B.; Klepsch, C.; Lusis, A.J.; Katus, H.A. New Dyscalc loci for myocardial cell necrosis and calcification (dystrophic cardiac calcinosis) in mice. Physiol. Genom. 2001, 6, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunnert, S.R. Morphologic response of myocardium to freeze-thaw injury in mouse strains with dystrophic cardiac calcification. Lab. Anim. Sci. 1997, 47, 11–18. [Google Scholar]
- Smolen, K.K.; Gelinas, L.; Franzen, L.; Dobson, S.; Dawar, M.; Ogilvie, G.; Krajden, M.; Fortuno, E.S., III; Kollmann, T.R. Age of recipient and number of doses differentially impact human B and T cell immune memory responses to HPV vaccination. Vaccine 2012, 30, 3572–3579. [Google Scholar] [CrossRef]
- Li, Q.; Berndt, A.; Guo, H.; Sundberg, J.P.; Uitto, J. A Novel Animal Model for Pseudoxanthoma Elasticum: The KK/HlJ Mouse. Am. J. Pathol. 2012, 181, 1190–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, A.; Sundberg, B.A.; Silva, K.A.; Kennedy, V.E.; Richardson, M.A.; Li, Q.; Bronson, R.T.; Uitto, J.; Sundberg, J.P. Phenotypic Characterization of the KK/HlJ Inbred Mouse Strain. Veter. Pathol. 2013, 51, 846–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vilder, E.Y.; Hosen, M.J.; Martin, L.; de Zaeytijd, J.; Leroy, B.P.; Ebran, J.; Coucke, P.J.; de Paepe, A.; Vanakker, O.M. VEGFA variants as prognostic markers for the retinopathy in pseudoxanthoma elasticum. Clin. Genet. 2020, 98, 74–79. [Google Scholar] [CrossRef]
- Luo, H.; Faghankhani, M.; Cao, Y.; Uitto, J.; Li, Q. Molecular Genetics and Modifier Genes in Pseudoxanthoma Elasticum, a Heritable Multisystem Ectopic Mineralization Disorder. J. Investig. Dermatol. 2020. [Google Scholar] [CrossRef]
- Le Corre, Y.; le Saux, O.; Froeliger, F.; Libouban, H.; Kauffenstein, G.; Willoteaux, S.; Leftheriotis, G.; Martin, L. Quantification of the calcification phenotype of abcc6-deficient mice with microcomputed tomography. Am. J. Pathol. 2012, 180, 2208–2213. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Kingman, J.; van de Wetering, K.; Tannouri, S.; Sundberg, J.P.; Uitto, J. Abcc6 Knockout Rat Model Highlights the Role of Liver in PPi Homeostasis in Pseudoxanthoma Elasticum. J. Investig. Dermatol. 2017, 137, 1025–1032. [Google Scholar] [CrossRef]
- Li, Q.; Sadowski, S.; Frank, M.M.; Chai, C.; Váradi, A.; Ho, S.-Y.; Lou, H.; Dean, M.; Thisse, C.; Thisse, B.; et al. The abcc6a Gene Expression Is Required for Normal Zebrafish Development. J. Investig. Dermatol. 2010, 130, 2561–2568. [Google Scholar] [CrossRef] [Green Version]
- Van Gils, M.; Willaert, A.; de Vilder, E.; Coucke, P.; Vanakker, O. Generation and Validation of a Complete Knockout Model of abcc6a in Zebrafish. J. Investig. Dermatol. 2018, 138, 2333–2342. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; She, P.; Liu, X.; Gao, B.; Jin, D.; Zhong, T.P. Disruption of Abcc6 Transporter in Zebrafish Causes Ocular Calcification and Cardiac Fibrosis. Int. J. Mol. Sci. 2020, 22, 278. [Google Scholar] [CrossRef]
- Mackay, E.W.; Apschner, A.; Schulte-Merker, S. Vitamin K reduces hypermineralisation in zebrafish models of PXE and GACI. Development 2015, 142, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- Pomozi, V.; Brampton, C.; Fülöp, K.; Chen, L.-H.; Apana, A.; Li, Q.; Uitto, J.; le Saux, O.; Váradi, A. Analysis of Pseudoxanthoma Elasticum–Causing Missense Mutants of ABCC6 In Vivo; Pharmacological Correction of the Mislocalized Proteins. J. Investig. Dermatol. 2014, 134, 946–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okawa, A.; Nakamura, I.; Goto, S.; Moriya, H.; Nakamura, Y.; Ikegawa, S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat. Genet. 1998, 19, 271–273. [Google Scholar] [CrossRef]
- Li, Q.; Guo, H.; Chou, D.W.; Berndt, A.; Sundberg, J.P.; Uitto, J. Mutant Enpp1asj mice as a model for generalized arterial calcification of infancy. Dis. Model. Mech. 2013, 6, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Dedinszki, D.; Szeri, F.; Kozak, E.; Pomozi, V.; Tokesi, N.; Mezei, T.R.; Merczel, K.; Letavernier, E.; Tang, E.; Le Saux, O.; et al. Oral administration of pyrophosphate inhibits con-nective tissue calcification. EMBO Mol. Med. 2017, 9, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Huesa, C.; Zhu, D.; Glover, J.D.; Ferron, M.; Karsenty, G.; Milne, E.M.; Millan, J.L.; Ahmed, S.F.; Farquharson, C.; Morton, N.M.; et al. Deficiency of the bone mineralization inhibitor NPP1 protects mice against obesity and diabetes. Dis. Model. Mech. 2014, 7, 1341–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, N.; Huesa, C.; Rutsch, F.; MacRae, V. New insights into NPP1 function: Lessons from clinical and animal studies. Bone 2012, 51, 961–968. [Google Scholar] [CrossRef] [Green Version]
- Apschner, A.; Huitema, L.F.; Ponsioen, B.; Peterson-Maduro, J.; Schulte-Merker, S. Zebrafish enpp1 mutants exhibit pathological mineralization, mimicking features of generalized arterial calcification of in-fancy (GACI) and pseudoxanthoma elasticum (PXE). Dis. Model Mech. 2014, 7, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Kauffenstein, G.; Yegutkin, G.G.; Khiati, S.; Pomozi, V.; le Saux, O.; Leftheriotis, G.; Lenaers, G.; Henrion, D.; Martin, L. Alteration of Extracellular Nucleotide Metabolism in Pseudoxanthoma Elasticum. J. Investig. Dermatol. 2018, 138, 1862–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uitto, J.; Li, Q.; van de Wetering, K.; Váradi, A.; Terry, S.F. Insights into Pathomechanisms and Treatment Development in Heritable Ectopic Mineralization Disorders: Summary of the PXE International Biennial Research Symposium-2016. J. Investig. Dermatol. 2017, 137, 790–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranenburg, G.; de Jong, P.A.; Bartstra, J.W.; Lagerweij, S.J.; Lam, M.G.; Norel, J.O.-V.; Risseeuw, S.; van Leeuwen, R.; Imhof, S.M.; Verhaar, H.J.; et al. Etidronate for Prevention of Ectopic Mineralization in Patients with Pseudoxanthoma Elasticum. J. Am. Coll. Cardiol. 2018, 71, 1117–1126. [Google Scholar] [CrossRef]
- Li, Q.; Huang, J.; Pinkerton, A.B.; Millan, J.L.; van Zelst, B.D.; Levine, M.A.; Sundberg, J.P.; Uitto, J. Inhibition of Tissue-Nonspecific Alkaline Phosphatase Attenuates Ectopic Mineralization in the Abcc6 (−/−) Mouse Model of PXE but Not in the Enpp1 Mutant Mouse Models of GACI. J. Investig. Dermatol. 2019, 139, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomozi, V.; le Saux, O.; Brampton, C.; Apana, A.; Iliás, A.; Szeri, F.; Martin, L.; Monostory, K.; Paku, S.; Sarkadi, B.; et al. ABCC6 Is a Basolateral Plasma Membrane Protein. Circ. Res. 2013, 112, e148–e151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.; On, S.J.; Fuchs, W.; Chen, C.; Phelps, R.; Kornreich, D.; Haddican, M.; Singer, G.; Wong, V.; Baum, D.; et al. Magnesium Supplementation in the Treatment of Pseudoxanthoma Elasticum (PXE): A randomized trial. J. Am. Acad. Dermatol. 2019, 81, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Blum, R.R.; Singer, G.K.; Stern, D.K.; Emanuel, P.O.; Fuchs, W.; Phelps, R.G.; Terry, S.F.; Lebwohl, M.G. A randomized controlled trial of oral phosphate binders in the treatment of pseudoxanthoma elasticum. J. Am. Acad. Dermatol. 2011, 65, 341–348. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, Q.; Takahagi, S.; Shao, C.; Uitto, J.; Takahagi, S. Premature termination codon read-through in the ABCC6 gene: Potential treatment for pseudoxanthoma elasticum. J. Investig. Dermatol. 2013, 133, 2672–2677. [Google Scholar] [CrossRef] [Green Version]
- Albright, R.A.; Stabach, P.; Cao, W.; Kavanagh, D.; Mullen, I.; Braddock, A.A.; Covo, M.S.; Tehan, M.; Yang, G.; Cheng, Z.; et al. ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat. Commun. 2015, 6, 10006. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Sinkevicius, K.W.; Vong, S.; Avakian, A.; Leavitt, M.C.; Malanson, H.; Marozsan, A.; Askew, K.L. ENPP1 enzyme replacement therapy improves blood pressure and cardiovascular function in a mouse model of generalized arterial calcification of infancy. Dis. Model. Mech. 2018, 11, dmm035691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, C.; Heiss, A.; Schwarz, A.; Westenfeld, R.; Ketteler, M.; Floege, J.; Muller-Esterl, W.; Schinke, T.; Jahnen-Dechent, W. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J. Clin. Investig. 2003, 112, 357–366. [Google Scholar] [CrossRef]
- Jiang, Q.; Dibra, F.; Lee, M.D.; Oldenburg, R.; Uitto, J. Overexpression of fetuin-a counteracts ectopic mineralization in a mouse model of pseudoxanthoma elasticum (abcc6 (−/−)). J. Investig. Dermatol. 2010, 130, 1288–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Snook, A.E.; Uitto, J.; Li, Q. Adenovirus-Mediated ABCC6 Gene Therapy for Heritable Ectopic Mineralization Disorders. J. Investig. Dermatol. 2019, 139, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- La Russo, J.; Jiang, Q.; Li, Q.; Uitto, J. Ectopic mineralization of connective tissue in Abcc6(−/−) mice: Effects of dietary modifications and a phosphate binder—A preliminary study. Exp. Dermatol. 2008, 17, 203–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgels, T.G.M.F.; Waarsing, J.H.; de Wolf, A.; Brink, J.B.T.; Loves, W.J.P.; Bergen, A.A.B. Dietary magnesium, not calcium, prevents vascular calcification in a mouse model for pseudoxanthoma elasticum. J. Mol. Med. 2010, 88, 467–475. [Google Scholar] [CrossRef] [Green Version]
- La Russo, J.; Li, Q.; Jiang, Q.; Uitto, J. Elevated dietary magnesium prevents connective tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6 (−/−)). J. Investig. Dermatol. 2009, 129, 1388–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgels, T.G.M.F.; Waarsing, J.H.; Herfs, M.; Versteeg, D.; Schoensiegel, F.; Sato, T.; Schlingemann, R.O.; Ivandic, B.; Vermeer, C.; Schurgers, L.J.; et al. Vitamin K supplementation increases vitamin K tissue levels but fails to counteract ectopic calcification in a mouse model for pseudoxanthoma elasticum. J. Mol. Med. 2011, 89, 1125–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Li, Q.; Grand-Pierre, A.E.; Schurgers, L.J.; Uitto, J. Administration of vitamin K does not counteract the ectopic mineralization of connective tissues in Abcc6 (−/−) mice, a model for pseudoxanthoma elasticum. Cell Cycle 2011, 10, 701–707. [Google Scholar] [CrossRef]
- Carrillo-Linares, J.L.; García-Fernández, M.I.; Morillo, M.J.; Sánchez, P.; Rioja, J.; Barón, F.J.; Ariza, M.J.; Harrington, D.J.; Card, D.; Boraldi, F.; et al. The Effects of Parenteral K1 Administration in Pseudoxanthoma Elasticum Patients Versus Controls. A Pilot Study. Front. Med. 2018, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Nollet, L.; van Gils, M.; Verschuere, S.; Vanakker, O. The Role of Vitamin K and Its Related Compounds in Mendelian and Acquired Ectopic Mineralization Disorders. Int. J. Mol. Sci. 2019, 20, 2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, C.; le Saux, O.; Pomozi, V.; Aherrahrou, R.; Kriesen, R.; Stölting, S.; Liebers, A.; Kessler, T.S.H.; Erdmann, J.; Aherrahrou, Z. Etidronate prevents but does not completely reverse dystrophic cardiac calcification by.2 inhibiting macrophage aggregation. Sci. Rep. 2018, 8, 5812. [Google Scholar] [CrossRef] [Green Version]
- Otero, J.E.; Gottesman, G.S.; McAlister, W.H.; Mumm, S.; Madson, K.L.; Kiffer-Moreira, T.; Sheen, C.; Millán, J.L.; Ericson, K.L.; Whyte, M.P. Severe skeletal toxicity from protracted etidronate therapy for generalized arterial calcification of infancy. J. Bone Miner. Res. 2013, 28, 419–430. [Google Scholar] [CrossRef]
- Chong, C.R.; Hutchins, G.M. Idiopathic infantile arterial calcification: The spectrum of clinical presentations. Pediatr. Dev. Pathol. 2008, 11, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Dabisch-Ruthe, M.; Kuzaj, P.; Götting, C.; Knabbe, C.; Hendig, D. Pyrophosphates as a major inhibitor of matrix calcification in Pseudoxanthoma elasticum. J. Dermatol. Sci. 2014, 75, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kingman, J.; Sundberg, J.P.; Levine, M.A.; Uitto, J. Etidronate prevents, but does not reverse, ectopic mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−). Oncotarget 2016, 9, 30721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, I.J.; Li, D.; Ivarsson, M.E.; Uitto, J.; Li, Q. A phytic acid analogue INS-3001 prevents ectopic calcification in an Abcc6(−/−) mouse model of pseudoxanthoma elasticum. Exp. Dermatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hosen, M.J.; van Nieuwerburgh, F.; Steyaert, W.; Deforce, D.; Martin, L.; Leftheriotis, G.; de Paepe, A.; Coucke, P.J.; Vanakker, O.M. Efficiency of Exome Sequencing for the Molecular Diagnosis of Pseudoxanthoma Elasticum. J. Investig. Dermatol. 2015, 135, 992–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plomp, A.S.; Bergen, A.A.; Florijn, R.J.; Terry, S.F.; Toonstra, J.; van Dijk, M.R.; de Jong, P.T. Pseudoxanthoma elasticum: Wide phenotypic variation in homozygotes and no signs in heterozygotes for the c.3775delT mutation in ABCC6. Genet. Med. 2009, 11, 852–858. [Google Scholar] [CrossRef] [Green Version]
- Uitto, J.; Jiang, Q.; Varadi, A.; Bercovitch, L.G.; Terry, S.F. Pseudoxanthoma Elasticum: Diagnostic Features, Classification, and Treatment Options. Expert Opin. Orphan Drugs 2014, 2, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Bartstra, J.W.; Risseeuw, S.; de Jong, P.A.; van Os, B.; Kalsbeek, L.; Mol, C.; Baas, A.F.; Verschuere, S.; Vanakker, O.; Florijn, R.J.; et al. Genotype-phenotype correlation in pseudoxanthoma elasticum. Atherosclerosis 2021, 324, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Boraldi, F.; Costa, S.; Rabacchi, C.; Ciani, M.; Vanakker, O.; Quaglino, D. Can APOE and MTHFR poly-morphisms have an influence on the severity of cardiovascular manifestations in Italian Pseudoxanthoma elasticum affected patients? Mol. Genet. Metab. Rep. 2014, 1, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Hendig, D.; Knabbe, C.; Götting, C. New insights into the pathogenesis of pseudoxanthoma elasticum and related soft tissue calcification disorders by identifying genetic interactions and modifiers. Front. Genet. 2013, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Vanakker, O.M.M.; Hosen, M.J.; de Paepe, A. The ABCC6 transporter: What lessons can be learnt from other ATP-binding cassette transporters? Front. Genet. 2013, 4, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navasiolava, N.; Gnanou, M.; Douillard, M.; Saulnier, P.; Aranyi, T.; Ebran, J.-M.; Henni, S.; Humeau, H.; Lefthériotis, G.; Martin, L. The extent of pseudoxanthoma elasticum skin changes is related to cardiovascular complications and visual loss: A cross-sectional study. Br. J. Dermatol. 2019, 180, 207–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Kingman, J.; Sundberg, J.P.; Uitto, J.; Li, Q. Plasma PPi Deficiency Is the Major, but Not the Exclusive, Cause of Ectopic Mineralization in an Abcc6 Mouse Model of PXE. J. Investig. Dermatol. 2017, 137, 2336–2343. [Google Scholar] [CrossRef] [Green Version]
- Mention, P.J.; Lacoeuille, F.; Leftheriotis, G.; Martin, L.; Omarjee, L. 18F-Flurodeoxyglucose and 18F-Sodium Fluoride Positron Emission Tomography/Computed Tomography Imaging of Arterial and Cu-taneous Alterations in Pseudoxanthoma Elasticum. Circ. Cardiovasc. Imaging 2018, 11, e007060. [Google Scholar] [CrossRef] [Green Version]
- Oudkerk, S.F.; de Jong, P.A.; Blomberg, B.A.; Scholtens, A.M.; Mali, W.P.; Spiering, W. Whole-Body Visualization of Ectopic Bone Formation of Arteries and Skin in Pseudoxanthoma Elasticum. JACC Cardiovasc. Imaging 2016, 9, 755–756. [Google Scholar] [CrossRef]
- Humeau-Heurtier, A.; Colominas, M.A.; Schlotthauer, G.; Etienne, M.; Martin, L.; Abraham, P. Bidimensional unconstrained optimization approach to EMD: An algorithm revealing skin perfusion alterations in pseudoxanthoma elasticum patients. Comput. Methods Programs Biomed. 2017, 140, 233–239. [Google Scholar] [CrossRef]
- Gass, J.D. “Comet” lesion: An ocular sign of pseudoxanthoma elasticum. Retina 2003, 23, 729–730. [Google Scholar] [CrossRef]
- Ciulla, T.A.; Rosenfeld, P.J. Anti-vascular endothelial growth factor therapy for neovascular ocular diseases other than age-related macular degeneration. Curr. Opin. Ophthalmol. 2009, 20, 166–174. [Google Scholar] [CrossRef]
- Vasseur, M.; Carsin-Nicol, B.; Ebran, J.; Willoteaux, S.; Martin, L.; Leftheriotis, G. Carotid Rete Mirabile and Pseudoxanthoma Elasticum: An Accidental Association? Eur. J. Vasc. Endovasc. Surg. 2011, 42, 292–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlovic, A.M.; Zidverc-Trajkovic, J.; Milovic, M.M.; Pavlovic, D.M.; Jovanovic, Z.; Mijajlovic, M.; Petrovic, M.; Kostic, V.S.; Sternic, N. Cerebral Small Vessel Disease in Pseudoxanthoma Elasticum: Three Cases. Can. J. Neurol. Sci. 2005, 32, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Miglionico, R.; Ostuni, A.; Armentano, M.F.; Milella, L.; Crescenzi, E.; Carmosino, M.; Bisaccia, F. ABCC6 knockdown in HepG2 cells induces a senescent-like cell phenotype. Cell. Mol. Biol. Lett. 2017, 22, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiemann, J.; Wagner, T.; Lindenkamp, C.; Plumers, R.; Faust, I.; Knabbe, C.; Hendig, D. Linking ABCC6 Deficiency in Primary Human Dermal Fibroblasts of PXE Patients to p21-Mediated Premature Cellular Se-nescence and the Development of a Proinflammatory Secretory Phenotype. Int. J. Mol. Sci. 2020, 21, 24. [Google Scholar] [CrossRef] [PubMed]
- Mungrue, I.N.; Zhao, P.; Yao, Y.; Meng, H.; Rau, C.; Havel, J.V.; Gorgels, T.G.M.F.; Bergen, A.A.; MacLellan, W.R.; Drake, T.A.; et al. Abcc6 Deficiency Causes Increased Infarct Size and Apoptosis in a Mouse Cardiac Ischemia-Reperfusion Model. Arter. Thromb. Vasc. Biol. 2011, 31, 2806–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Peek, R.; Plomp, A.; Brink, J.T.; Scheffer, G.; van Soest, S.; Leys, A.; de Jong, P.T.V.M.; Bergen, A.A.B. Analysis of the Frequent R1141X Mutation in theABCC6Gene in Pseudoxanthoma Elasticum. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1824–1829. [Google Scholar] [CrossRef]
- Pfendner, E.G.; Uitto, J.; Gerard, G.F.; Terry, S.F. Pseudoxanthoma elasticum: Genetic diagnostic markers. Expert Opin. Med. Diagn. 2008, 2, 63–79. [Google Scholar] [CrossRef]
- Keeling, K.M.; Bedwell, D.M. Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. Wiley Interdiscip. Rev. RNA 2011, 2, 837–852. [Google Scholar] [CrossRef] [Green Version]
- Wilschanski, M.; Miller, L.L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatch, T.; et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur. Respir. J. 2011, 38, 59–69. [Google Scholar] [CrossRef]
- Kerem, E.E.; Konstan, M.M.; de Boeck, K.; Accurso, F.F.; Sermet-Gaudelus, I.; Wilschanski, M.M.; Elborn, S.J.; Melotti, P.P.; Bronsveld, I.I.; Fajac, I.; et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Dover, G.J.; Brusilow, S.; Samid, D. Increased Fetal Hemoglobin in Patients Receiving Sodium 4-Phenylbutyrate. N. Engl. J. Med. 1992, 327, 569–570. [Google Scholar] [CrossRef]
- Maestri, N.E.; Brusilow, S.W.; Clissold, D.B.; Bassett, S.S. Long-Term Treatment of Girls with Ornithine Transcarbamylase Deficiency. N. Engl. J. Med. 1996, 335, 855–860. [Google Scholar] [CrossRef]
- Perrine, S.P.; Ginder, G.D.; Faller, D.V.; Dover, G.H.; Ikuta, T.; Witkowska, H.E.; Cai, S.P.; Vichinsky, E.P.; Olivieri, N.F. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N. Engl. J. Med. 1993, 328, 81–86. [Google Scholar] [CrossRef]
- McGuire, B.M.; Zupanets, I.A.; Lowe, M.E.; Xiao, X.; Syplyviy, V.A.; Monteleone, J.; Gargosky, S.; Dickinson, K.; Martinez, A.; Mokhtarani, M.; et al. Pharmacology and safety of glycerol phenyl-butyrate in healthy adults and adults with cirrhosis. Hepatology 2010, 51, 2077–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannitti, T.; Palmieri, B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D 2011, 11, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, E.; Grosse, B.; Schuller, B.; Davit-Spraul, A.; Conti, F.; Guettier, C.; Cassio, D.; Jacquemin, E. Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: Evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatology 2015, 62, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Naoi, S.; Hirose, Y.; Matsuzaka, Y.; Tanikawa, K.; Igarashi, K.; Nagasaka, H.; Kage, M.; Inui, A.; Kusuhara, H. Successful treatment with 4-phenylbutyrate in a patient with benign recurrent intrahepatic cholestasis type 2 refractory to biliary drainage and bilirubin absorption. Hepatol. Res. 2016, 46, 192–200. [Google Scholar] [CrossRef]
- Rubenstein, R.C.; Zeitlin, P.L. Sodium 4-phenylbutyrate downregulates Hsc70: Implications for intra-cellular trafficking of DeltaF508-CFTR. Am. J. Physiol. Cell. Physiol. 2000, 278, C259–C267. [Google Scholar] [CrossRef]
- Sorrenson, B.; Suetani, R.J.; Williams, M.J.A.; Bickley, V.M.; George, P.M.; Jones, G.T.; McCormick, S.P.A. Functional rescue of mutant ABCA1 proteins by sodium 4-phenylbutyrate. J. Lipid Res. 2013, 54, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Millán, J.L.; Whyte, M.P. Alkaline Phosphatase and Hypophosphatasia. Calcif. Tissue Int. 2016, 98, 398–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narisawa, S.; Harmey, D.; Yadav, M.C.; O’Neill, W.C.; Hoylaerts, M.F.; Millán, J.L. Novel Inhibitors of Alkaline Phosphatase Suppress Vascular Smooth Muscle Cell Calcification. J. Bone Miner. Res. 2007, 22, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Sheen, C.R.; Kuss, P.; Narisawa, S.; Yadav, M.C.; Nigro, J.; Wang, W.; Chhea, T.N.; Sergienko, E.A.; Kapoor, K.; Jackson, M.R.; et al. Pathophysiological Role of Vascular Smooth Muscle Alkaline Phosphatase in Medial Artery Calcification. J. Bone Miner. Res. 2015, 30, 824–836. [Google Scholar] [CrossRef] [Green Version]
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.; Millan, J.L.; Savinova, O.V. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J. Am. Heart Assoc. 2015, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St Hilaire, C.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 2011, 364, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Price, T.P.; Sundberg, J.P.; Uitto, J. Juxta-articular joint-capsule mineralization in CD73 deficient mice: Similarities to patients with NT5E mutations. Cell Cycle 2014, 13, 2609–2615. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zheng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef]
- De Baaij, J.H.F.; Hoenderop, J.G.J.; Bindels, R.J.M. Magnesium in Man: Implications for Health and Disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef]
- Ter, B.A.D.; Shanahan, C.M.; de Baaij, J.H.F. Magnesium Counteracts Vascular Calcification: Passive Interference or Active Modulation? Arter. Thromb Vasc. Biol. 2017, 37, 1431–1445. [Google Scholar]
- Alfrey, A.C.; Miller, N.L.; Trow, R. Effect of Age and Magnesium Depletion on Bone Magnesium Pools in Rats. J. Clin. Investig. 1974, 54, 1074–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, Y.; Hamano, T.; Isaka, Y. Magnesium and Progression of Chronic Kidney Disease: Benefits Beyond Cardiovascular Protection? Adv. Chronic Kidney Dis. 2018, 25, 274–280. [Google Scholar] [CrossRef]
- D’Marco, L.; Lima-Martínez, M.; Karohl, C.; Chacín, M.; Bermúdez, V. Pseudoxanthoma Elasticum: An Interesting Model to Evaluate Chronic Kidney Disease-Like Vascular Damage without Renal Disease. Kidney Dis. 2020, 6, 92–97. [Google Scholar] [CrossRef]
- Schinke, T.; McKee, M.D.; Karsenty, G. Extracellular matrix calcification: Where is the action? Nat. Genet. 1999, 21, 150–151. [Google Scholar] [CrossRef] [PubMed]
- Gheduzzi, D.; Boraldi, F.; Annovi, G.; Devincenzi, C.P.; Schurgers, L.J.; Vermeer, C.; Quaglino, D.; Ronchetti, I.P. Matrix Gla protein is involved in elastic fiber calcification in the dermis of pseudoxanthoma elasticum patients. Lab. Investig. 2007, 87, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Vermeer, C. Gamma-carboxyglutamate-containing proteins and the vitamin K-dependent carboxylase. Biochem. J. 1990, 266, 625–636. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, Q.; Schurgers, L.J.; Uitto, J. Pseudoxanthoma elasticum: Reduced gamma-glutamyl carboxylation of matrix gla protein in a mouse model (Abcc6−/−). Biochem. Biophys. Res. Commun. 2007, 364, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Vanakker, O.M.; Martin, L.; Gheduzzi, D.; Leroy, B.P.; Loeys, B.L.; Guerci, V.I.; Matthys, D.; Terry, S.F.; Coucke, P.J.; Pasquali-Ronchetti, I.; et al. Pseudoxanthoma Elasticum-Like Phenotype with Cutis Laxa and Multiple Coagulation Factor Deficiency Represents a Separate Genetic Entity. J. Investig. Dermatol. 2007, 127, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Borst, P.; van de Wetering, K.; Schlingemann, R. Does the absence of ABCC6 (multidrug resistance protein 6) in patients with Pseudoxanthoma elasticum prevent the liver from providing sufficient vitamin K to the periphery? Cell Cycle 2008, 7, 1575–1579. [Google Scholar] [CrossRef] [Green Version]
- Vanakker, O.M.; Martin, L.; Schurgers, L.J.; Quaglino, D.; Costrop, L.; Vermeer, C.; Pasquali-Ronchetti, I.; Coucke, P.J.; de Paepe, A. Low serum vitamin K in PXE results in defective carboxylation of mineralization inhibitors similar to the GGCX mutations in the PXE-like syndrome. Lab. Investig. 2010, 90, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boraldi, F.; Annovi, G.; Guerra, D.; Devincenzi, C.P.; Garcia-Fernandez, M.I.; Panico, F.; de Santis, G.; Tiozzo, R.; Ronchetti, I.; Quaglino, D. Fibroblast protein profile analysis highlights the role of oxidative stress and vitamin K recycling in the pathogenesis of pseudoxanthoma elasticum. Proteom. Clin. Appl. 2009, 3, 1084–1098. [Google Scholar] [CrossRef]
- Huesa, C.; Staines, K.A.; Millan, J.L.; MacRae, V.E. Effects of etidronate on the Enpp1(-)/(-) mouse model of generalized arterial calcification of infancy. Int. J. Mol. Med. 2015, 36, 159–165. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Monier-Faugere, M.C.; Wang, X.; Malluche, H.H.; O’Neill, W.C. Effect of bisphosphonates on vascular calcification and bone metabolism in experimental renal failure. Kidney Int. 2009, 75, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, J.R.M.; Oliveira, M.F. Primary brain calcification in patients undergoing treatment with the biphosphanate alendronate. Sci. Rep. 2016, 6, 22961. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, H.; Bisaz, S. Mechanism of Calcification: Inhibitory Role of Pyrophosphate. Nat. Cell Biol. 1962, 195, 911. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, W.C.; Lomashvili, K.A.; Malluche, H.H.; Faugere, M.C.; Riser, B.L. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int. 2011, 79, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Orriss, I.R.; Arnett, T.R.; Russell, R.G.G. Pyrophosphate: A key inhibitor of mineralisation. Curr. Opin. Pharmacol. 2016, 28, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Grases, F.; Sanchis, P.; Perello, J.; Isern, B.; Prieto, R.M.; Fernandez-Palomeque, C.; Fiol, M.; Bonnin, O.; Torres, J.J. Phytate (Myo-inositol hexakisphosphate) inhibits cardiovascular calcifications in rats. Front. Biosci. 2006, 11, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Van den Berg, C.J.; Hill, L.F.; Stanbury, S.W. Inositol phosphates and phytic acid as inhibitors of biological calcification in the rat. Clin. Sci. 1972, 43, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Schantl, A.E.; Verhulst, A.; Neven, E.; Behets, G.J.; D’Haese, P.C.; Maillard, M.; Mordasini, D.; Phan, O.; Burnier, M.; Spaggiari, D.; et al. Inhibition of vascular calcification by inositol phosphates derivatized with ethylene glycol oligomers. Nat. Commun. 2020, 11, 721. [Google Scholar] [CrossRef]
- Dennie, W.; McBride, C. Treatment of arsphenamine dermatitis and certain other metallic poisonings. Arch. Dermatol. Syphilol. 1923, 11, 63. [Google Scholar]
- Halliday, H.M.; Sutherland, C.E. Arsenical Poisoning Treated by Sodium Thiosulphate. BMJ 1925, 1, 407. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R.; Goldsmith, D.J.A. Sodium Thiosulfate: New Hope for the Treatment of Calciphylaxis. Semin. Dial. 2010, 23, 258–262. [Google Scholar] [CrossRef]
- Arányi, T.; Bacquet, C.; de Boussac, H.; Ratajewski, M.; Pomozi, V.; Fülöp, K.; Brampton, C.N.; Pulaski, L.; le Saux, O.; Váradi, A. Transcriptional regulation of the ABCC6 gene and the background of impaired function of missense disease-causing mutations. Front. Genet. 2013, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Douet, V.; Heller, M.B.; le Saux, O. DNA methylation and Sp1 binding determine the tissue-specific transcriptional activity of the mouse Abcc6 promoter. Biochem. Biophys. Res. Commun. 2007, 354, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douet, V.; van Wart, C.M.; Heller, M.B.; Reinhard, S.; le Saux, O. HNF4alpha and NF-E2 are key transcriptional regulators of the murine Abcc6 gene expression. Biochim. Biophys. Acta 2006, 1759, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Väärämäki, S.; Pelttari, S.; Uusitalo, H.; Tokesi, N.; Varadi, A.; Nevalainen, P. Pyrophosphate Treatment in Pseudoxanthoma Elasticum (PXE)-Preventing ReOcclusion After Surgery for Critical Limb Ischaemia. Surg. Case Rep. 2019, 2, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Leopold, J.A. Vascular calcification: An age-old problem of old age. Circulation 2013, 127, 2380–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennenberg, R.J.M.W.; Kessels, A.G.H.; Schurgers, L.J.; Van Engelshoven, J.M.A.; de Leeuw, P.; Kroon, A.A. Vascular calcifications as a marker of increased cardiovascular risk: A meta-analysis. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Kranenburg, G.; Baas, A.F.; de Jong, P.A.; Asselbergs, F.W.; Visseren, F.L.J.; Spiering, W. The prevalence of pseudoxanthoma elasticum: Revised estimations based on genotyping in a high vascular risk cohort. Eur. J. Med. Genet. 2019, 62, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Buchheiser, A.; Ebner, A.; Burghoff, S.; Ding, Z.; Romio, M.; Viethen, C.; Lindecke, A.; Köhrer, K.; Fischer, J.W.; Schrader, J. Inactivation of CD73 promotes atherogenesis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2011, 92, 338–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalkanen, J.; Hollmén, M.; Jalkanen, S.; Hakovirta, H. Regulation of CD73 in the development of lower limb atherosclerosis. Purinergic Signal. 2016, 13, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitschke, Y.; Weissen-Plenz, G.; Terkeltaub, R.; Rutsch, F. Npp1 promotes atherosclerosis in ApoE knockout mice. J. Cell. Mol. Med. 2011, 15, 2273–2283. [Google Scholar] [CrossRef]
- Sutton, N.R.; Bouïs, D.; Mann, K.M.; Rashid, I.M.; McCubbrey, A.L.; Hyman, M.C.; Goldstein, D.R.; Mei, A.; Pinsky, D.J. CD73 Promotes Age-Dependent Accretion of Atherosclerosis. Arter. Thromb. Vasc. Biol. 2020, 40, 61–71. [Google Scholar] [CrossRef]
- Li, Q.; Guo, H.; Chou, D.W.; Berndt, A.; Sundberg, J.P.; Uitto, J. Mouse Models for Pseudoxanthoma Elasticum: Genetic and Dietary Modulation of the Ectopic Mineralization Phenotypes. PLoS ONE 2014, 9, e89268. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Treatment/Therapy | Rationale/Target | Diagnosis | References |
|---|---|---|---|
| Correction, replacement, or inhibition of dysfunctional genes/proteins | |||
| PTC-124 (Ataluren or Translarna) | Allows read-through of PTC codons, targets nonsense mutations | PXE | [131] |
| 4-Phenylbutyrate (4-PBA) | Corrects missense mutations allowing for correct cellular localization | PXE | [80,99,117] |
| Rh-NPP1-Fc | Replacement for ENPP1 | GACI | [89,132,133] |
| SBI-425 (TNAP inhibitor) | Inhibits the enzymatic activity of TNAP | PXE | [20,127] |
| Fetuin-A | Glycoprotein that forms complexes with calcium and phosphate ions, acts as an inhibitor of ectopic calcification | PXE | [134,135] |
| Adenovirus with ABCC6 cDNA | Transiently express ABCC6 in the liver | PXE | [136] |
| Bevacizumab (Anti-VEGF) † | Anti-angiogenic therapy; Preserves ocular function in advanced and early disease stages | PXE | [24] |
| Supplementation therapies for direct inhibition of calcification | |||
| Sevelamer hydrochloride (Renagel) | Phosphate binder | PXE | [130,137] |
| Magnesium | Inhibits the formation of apatite | PXE | [129,138,139] |
| Vitamin K (phylloquinone/menaquinone) | Correct for insufficient carboxylation of matrix gla protein (MGP) | PXE | [92,115,116,140,141,142,143] |
| Bisphosphonate (etidronate *, zoledronate) | Non-hydrolyzable analog of PPi, inhibits enzymes that utilize pyrophosphate | PXE and GACI | [18,55,126,144,145,146,147,148] |
| Pyrophosphate * | Potent inhibitor of calcification, ABCC6 modulates PPi production | PXE | [18,19,120], PROPHECI |
| INS-3001 (IP6 derivative) | Known inhibitor of calcification | PXE | [149] |
| Sodium thiosulfate | Approved for calciphlaxis | PXE | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimada, B.K.; Pomozi, V.; Zoll, J.; Kuo, S.; Martin, L.; Le Saux, O. ABCC6, Pyrophosphate and Ectopic Calcification: Therapeutic Solutions. Int. J. Mol. Sci. 2021, 22, 4555. https://doi.org/10.3390/ijms22094555
Shimada BK, Pomozi V, Zoll J, Kuo S, Martin L, Le Saux O. ABCC6, Pyrophosphate and Ectopic Calcification: Therapeutic Solutions. International Journal of Molecular Sciences. 2021; 22(9):4555. https://doi.org/10.3390/ijms22094555
Chicago/Turabian StyleShimada, Briana K., Viola Pomozi, Janna Zoll, Sheree Kuo, Ludovic Martin, and Olivier Le Saux. 2021. "ABCC6, Pyrophosphate and Ectopic Calcification: Therapeutic Solutions" International Journal of Molecular Sciences 22, no. 9: 4555. https://doi.org/10.3390/ijms22094555
APA StyleShimada, B. K., Pomozi, V., Zoll, J., Kuo, S., Martin, L., & Le Saux, O. (2021). ABCC6, Pyrophosphate and Ectopic Calcification: Therapeutic Solutions. International Journal of Molecular Sciences, 22(9), 4555. https://doi.org/10.3390/ijms22094555