
S-Nitrosylation in Tumor Microenvironment

 , ,
, , 
Abstract
:1. Introduction
2. Nitric Oxide (NO) Signaling
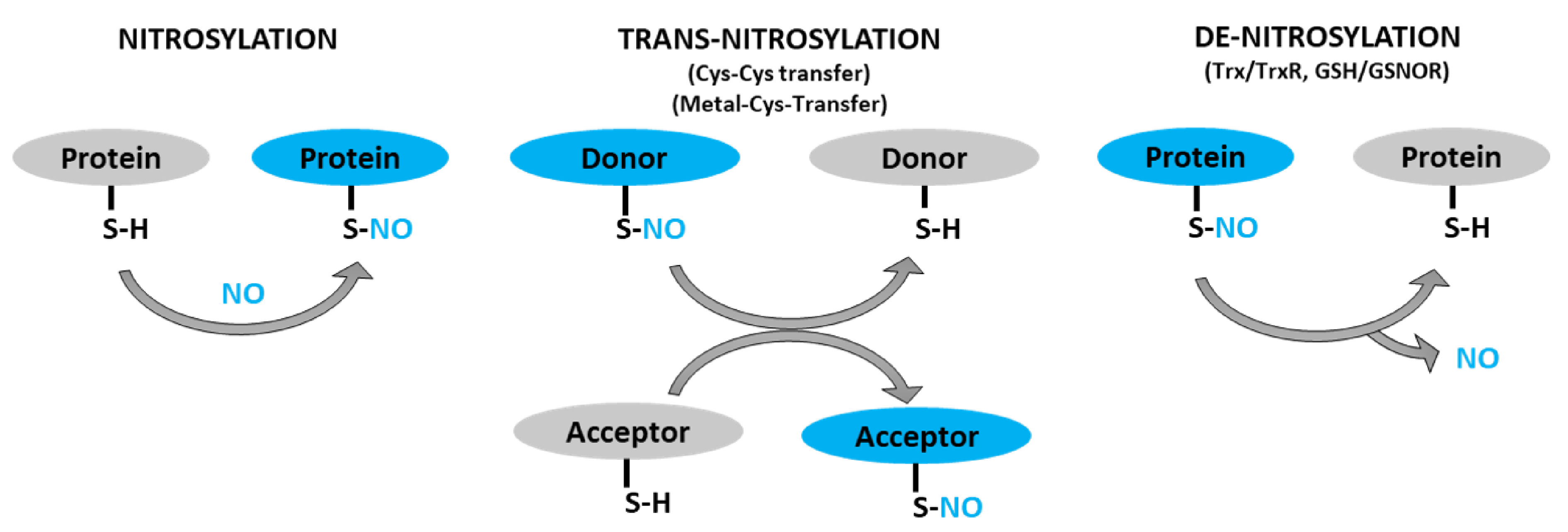
3. What Is S-Nitrosylation?
4. S-Nitrosylation in Diseases
4.1. S-Nitrosylation in Cancer
4.1.1. S-Nitrosylation Influenced by Altered Expression of NOSs and Denitrosylases as Well as Oxidative Stress
4.1.2. S-Nitrosylation Influenced by Hypoxia
4.1.3. S-Nitrosylation Influenced by Oncogenic Mutations
4.1.4. Dichotomous Effects of S-Nitrosylation on Cancer
4.2. S-Nitrosylation in Other Diseases
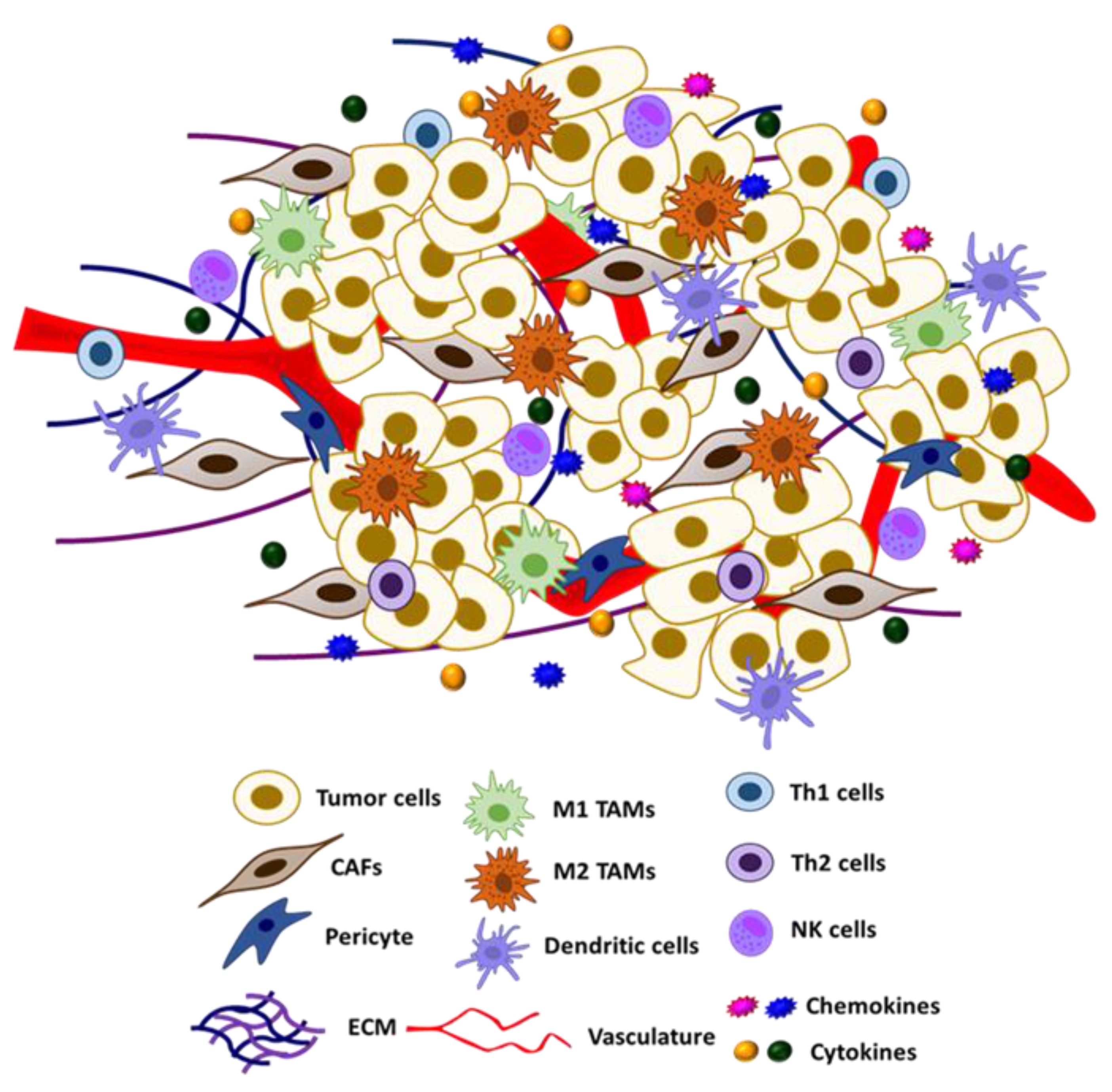
5. S-Nitrosylation in the Tumor Micro-Environment
5.1. S-Nitrosylation in Tumor-Associated Immune Cells
5.1.1. Tumor Associated Macrophages (TAMs)
5.1.2. T Cells
5.1.3. Natural Killer (NK) Cells
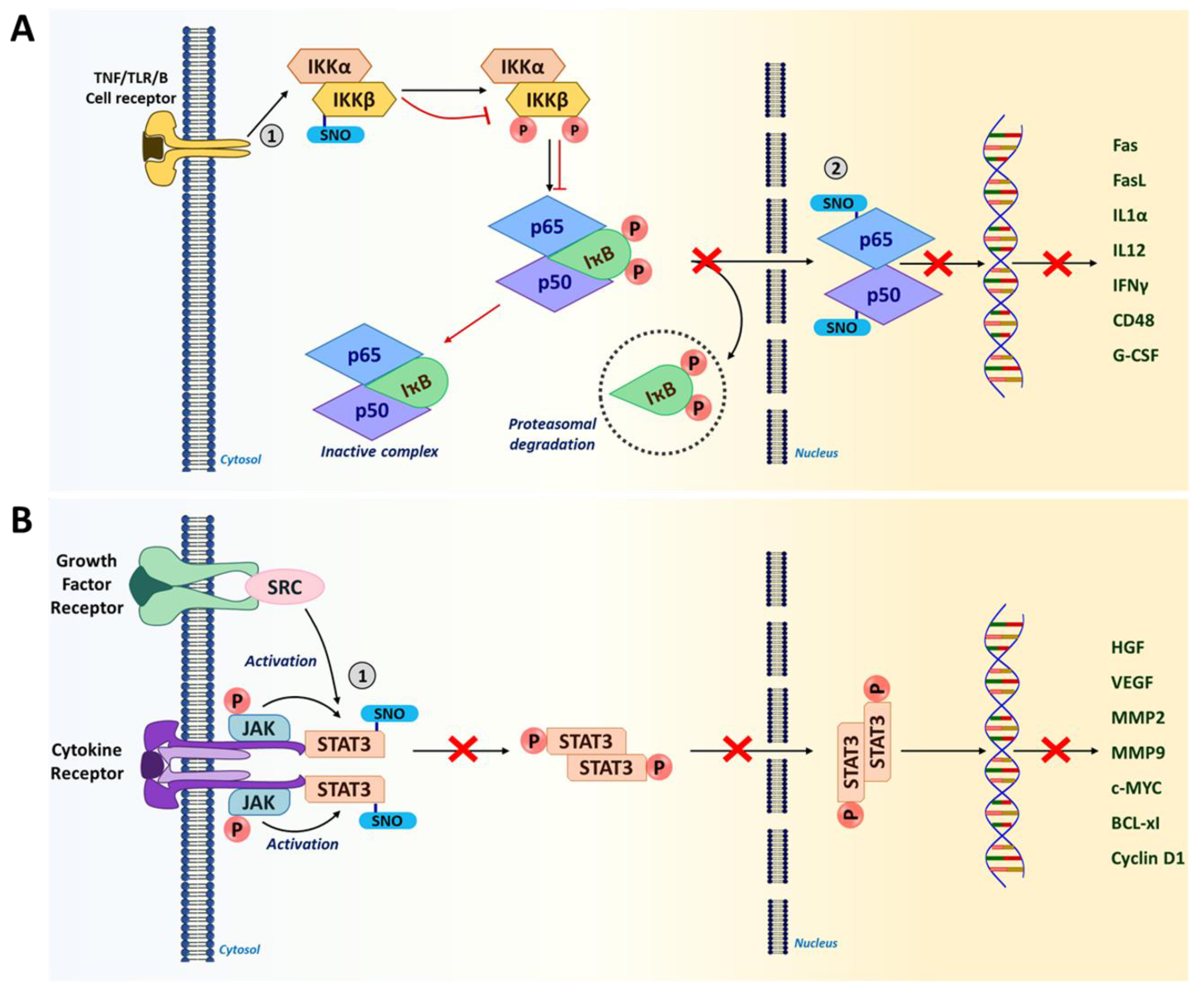
5.1.4. Nuclear Factor Kappa B (NF-κB)
5.1.5. Signal Transducer and Activator of Transcription 3 (STAT3)
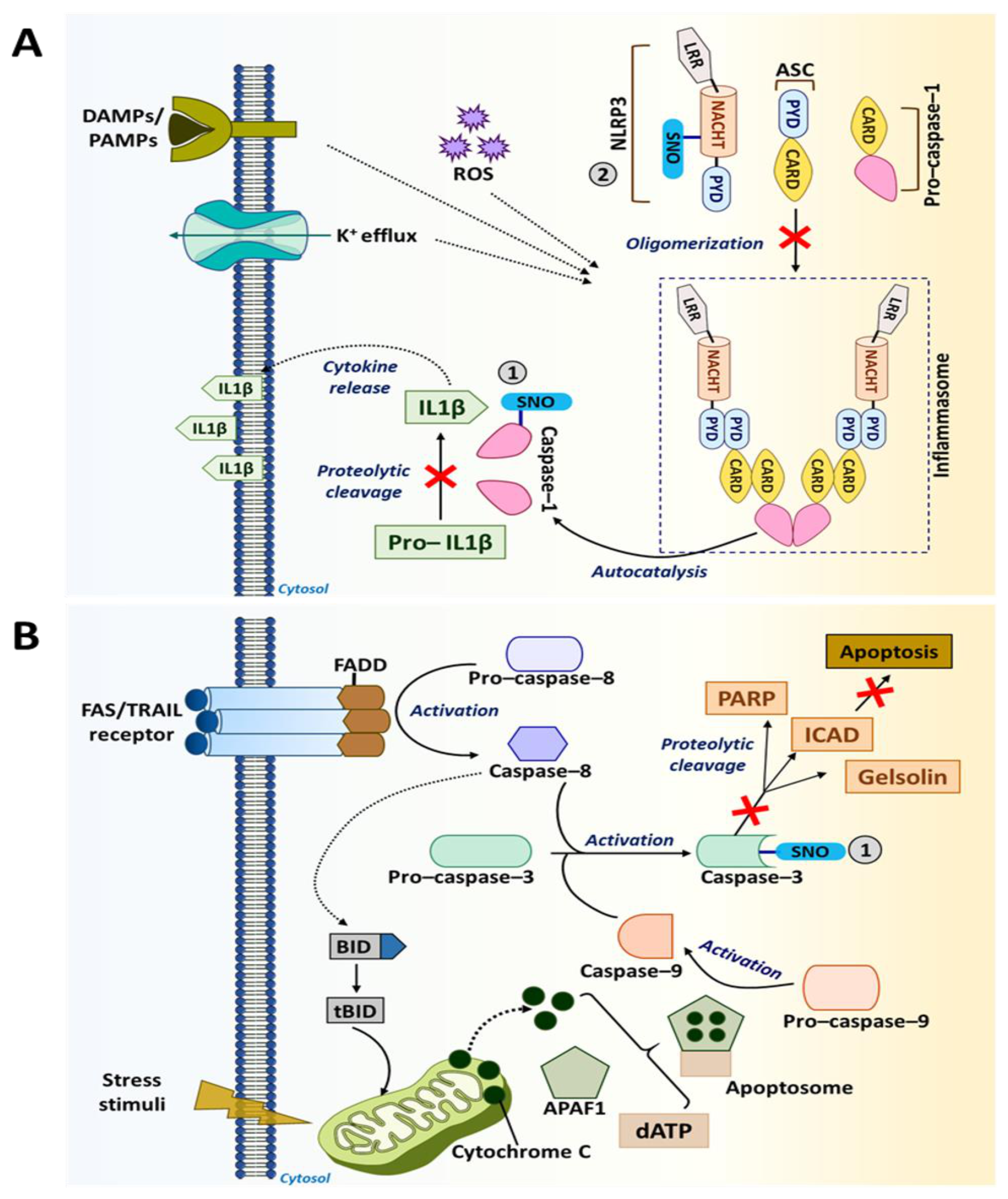
5.1.6. Caspases
5.2. S-Nitrosylation in Endothelial Cells
5.3. S-Nitrosylation in the Extracellular Matrix (ECM)
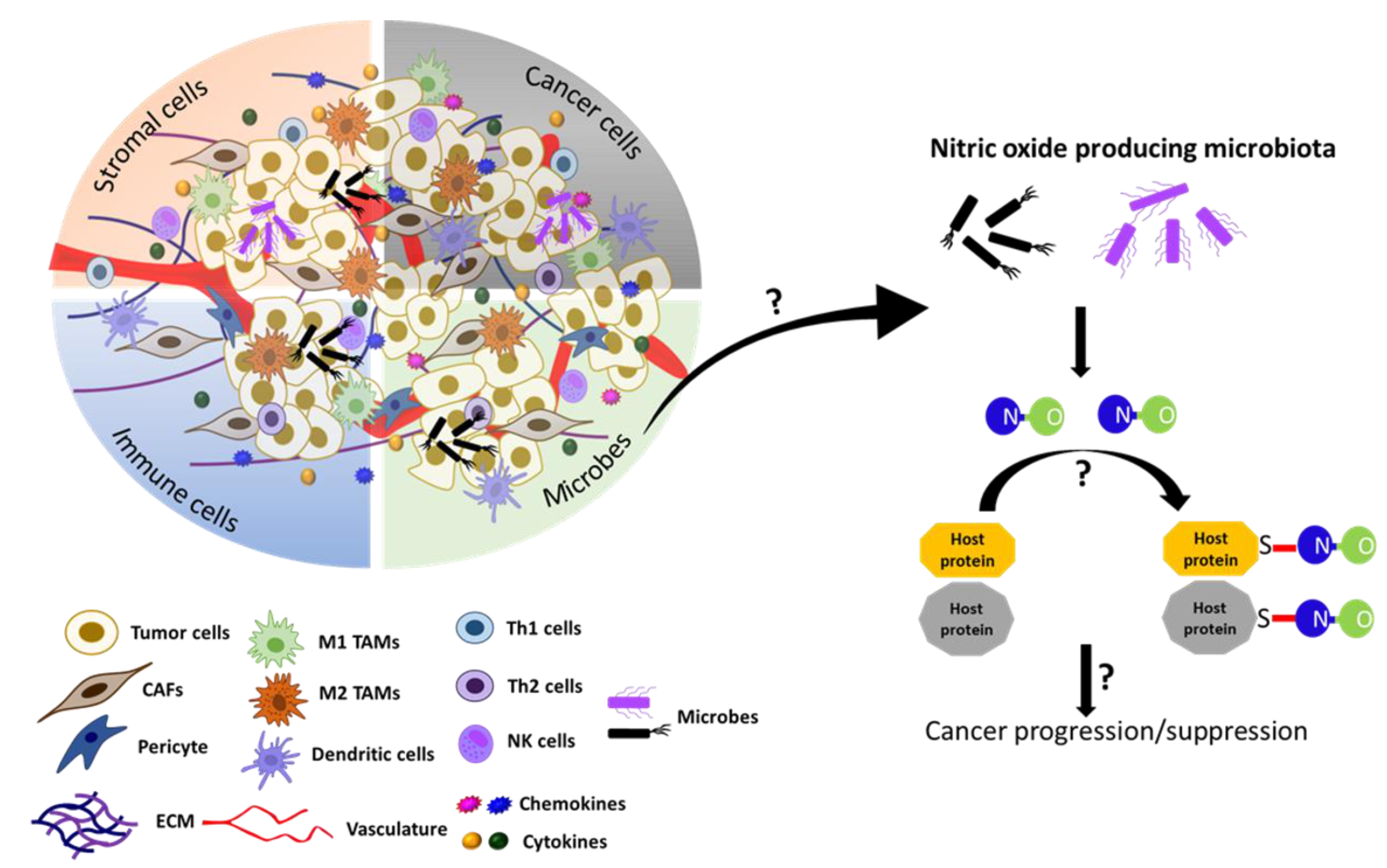
5.4. S-Nitrosylation in Tumor Microbiome
6. S-Nitrosylation in Anti-Cancer Therapy
6.1. Reducing S-Nitrosylation
6.2. Increasing S-Nitrosylation
6.3. Challenges in S-Nitrosylation-Based Anti-Cancer Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| AGT | O(6)-alkylguanine-DNA alkyltransferase |
| AKT | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| APC | Antigen presenting cell |
| ARG1 | Arginase 1 |
| ASK1 | Activation of apoptosis signal regulating kinase |
| BCL2 | B-cell lymphoma 2 |
| BH4 | Tetrahydrobiopterin |
| CAFs | Cancer-associated fibroblasts |
| CAV1 | Caveolin-1 |
| CDK5 | Cyclin-dependent kinase 5 |
| cGMP | Cyclic guanosine monophosphate |
| Cys | Cysteine |
| DETA-NO | Diethylenetriamine/nitric oxide |
| ECM | Extracellular matrix |
| eNOS/NOS3 | Endothelial NOS |
| ERK | Extracellular signal-regulated Kinase |
| ETC | Electron transport chain |
| ETS1 | ETS Proto-Oncogene 1 |
| FAK1 | Focal adhesion kinase |
| FOXO1 | Forkhead Box O1 |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| G-CSF | Granulocyte-colony-stimulating factor |
| M-CSF | Macrophage-colony-stimulating factor |
| GLUT1 | Glucose transporter 1 |
| GSH | Gluthatione |
| GSNO | S-Nitrosoglutathione |
| GSNOR | Glyceryl trinitrate |
| GTN | Guanosine-5′-triphosphate |
| GTP | S-Nitrosoglutathione reductase |
| H2O2 | Hydrogen peroxide |
| HCC | Hepatocellular carcinoma |
| HHcy | Hyperhomocysteinemia |
| HGF | Hepatocyte growth factor |
| HIF1-α | Hypoxia-inducible factor 1α |
| HNSCC | Head and neck squamous cell carcinoma |
| IFNγ | Interferon gamma |
| IL-2 | Interleukin-2 |
| iNOS/NOS2 | Inducible NOS |
| IκB | Inhibitor of NF-κB |
| IKKβ | IκB kinase |
| L-NAME | L-NG-Nitro arginine methyl ester |
| L-NMMA | NG-Monomethyl-L-Arginine |
| LTBP1 | Latent TGF-β binding protein |
| MAPK | Mitogen-activated protein kinases |
| MDSCs | Myeloid-derived suppressor cells |
| MHC I | Major histocompatibility complex I |
| MMP2/9 | Matrix metalloproteinase-2/-9 |
| mtNOS | Mitochondrial NOS |
| MTOC | Microtubule-organizing center |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kappa B |
| NK cells | Natural killer cells |
| nNOS/NOS1 | Neuronal NOS |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NSAID | Non-steroidal anti-inflammatory drugs |
| NSCLC | Non-small cell lung carcinoma |
| ODD | Oxygen-dependent degradation domain |
| PARP | poly [adenosine diphosphate (ADP-ribose)] polymerase |
| PDAC | Pancreatic ductal adenocarcinoma |
| PKG | cGMP-dependent protein kinase |
| PRDX2 | Peroxiredoxin-2 |
| PTM | Post-translational modification |
| PTPS | 6-pyruvoyl-tetrahydropterin synthase |
| RNS | Reactive nitrogen species |
| sGC | Soluble gualylyl cyclase |
| SNO | S-nitrosotyiol |
| SIRT1 | Sirtuin 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| SNAP | S-nitroso-N-acetylpenicillamine |
| SNO | S-nitrosothiols |
| TAMs | Tumor-associated macrophages |
| TCA cycle | Tricarboxylic acid cycle |
| TCR | T cell receptor |
| TG2 | Transglutaminase 2 |
| TGF-β | Transforming growth factor beta |
| Th1 | T helper 1 |
| Th2 | T helper 2 |
| TIE2 | TEK tyrosine kinase |
| TLR | Toll-like receptor |
| TNBC | Triple-negative breast cancer |
| TNF | Tumor necrosis factor |
| Trx | Thioredoxin |
| Trx/R | Thioredoxin reductase |
| TME | Tumor micro-environment |
| TRAP1 | TNF receptor associated protein 1 |
| TWIST1 | Twist-related protein 1 |
| VEGF | Vascular endothelial growth factor |
References
- Stamler, J.S.; Simon, D.I.; Osborne, J.A.; Mullins, M.E.; Jaraki, O.; Michel, T.; Singel, D.J.; Loscalzo, J. S-nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA 1992, 89, 444–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mnatsakanyan, R.; Markoutsa, S.; Walbrunn, K.; Roos, A.; Verhelst, S.H.L.; Zahedi, R.P. Proteome-wide detection of S-nitrosylation targets and motifs using bioorthogonal cleavable-linker-based enrichment and switch technique. Nat. Commun. 2019, 10, 2195. [Google Scholar] [CrossRef] [PubMed]
- Furuta, S. Basal S-Nitrosylation Is the Guardian of Tissue Homeostasis. Trends Cancer 2017, 3, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cocheme, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasinska, I.M.; Sumbayev, V.V. S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003, 549, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.; Nakamura, T.; Cieplak, P.; Chan, S.F.; Kalashnikova, E.; Liao, L.; Saleem, S.; Han, X.; Clemente, A.; Nutter, A.; et al. S-nitrosylation-mediated redox transcriptional switch modulates neurogenesis and neuronal cell death. Cell Rep. 2014, 8, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; James, A.M.; Methner, C.; Pell, V.R.; Prime, T.A.; Erickson, B.K.; Forkink, M.; Lau, G.Y.; Bright, T.P.; Menger, K.E.; et al. Identification and quantification of protein S-nitrosation by nitrite in the mouse heart during ischemia. J. Biol. Chem. 2017, 292, 14486–14495. [Google Scholar] [CrossRef] [Green Version]
- Raju, K.; Doulias, P.T.; Evans, P.; Krizman, E.N.; Jackson, J.G.; Horyn, O.; Daikhin, Y.; Nissim, I.; Yudkoff, M.; Nissim, I.; et al. Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci. Signal 2015, 8, ra68. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.; Lipton, S.A. S-Nitrosylation in neurogenesis and neuronal development. Biochim. Biophys. Acta 2015, 1850, 1588–1593. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Sun, J.; Picht, E.; Ginsburg, K.S.; Bers, D.M.; Steenbergen, C.; Murphy, E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ. Res. 2006, 98, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, I.; Lindermayr, C. Nitric oxide-based protein modification: Formation and site-specificity of protein S-nitrosylation. Front. Plant. Sci. 2013, 4, 137. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. Protein S-nitrosylation and cancer. Cancer Lett. 2012, 320, 123–129. [Google Scholar] [CrossRef]
- Plenchette, S.; Romagny, S.; Laurens, V.; Bettaieb, A. S-Nitrosylation in TNF superfamily signaling pathway: Implication in cancer. Redox Biol. 2015, 6, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351. [Google Scholar] [CrossRef]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef] [Green Version]
- Rizza, S.; Filomeni, G. Role, Targets and Regulation of (de)nitrosylation in Malignancy. Front. Oncol. 2018, 8, 334. [Google Scholar] [CrossRef]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Karlenius, T.C.; Tonissen, K.F. Thioredoxin and Cancer: A Role for Thioredoxin in all States of Tumor Oxygenation. Cancers 2010, 2, 209–232. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Zheng, X.; Bommarito, M.; Metzger, S.; Walia, Y.; Letson, J.; Schroering, A.; Kalinoski, A.; Weaver, D.; Figy, C.; et al. Reduced Basal Nitric Oxide Production Induces Precancerous Mammary Lesions via ERBB2 and TGFβ. Sci. Rep. 2019, 9, 6688. [Google Scholar] [CrossRef]
- Rizza, S.; Filomeni, G. Exploiting S-nitrosylation for cancer therapy: Facts and perspectives. Biochem. J. 2020, 477, 3649–3672. [Google Scholar] [CrossRef]
- Sciacca, M.; Belgorosky, D.; Zambrano, M.; Gomez Escalante, J.I.; Roca, F.; Langle, Y.V.; Sandes, E.O.; Lodillinsky, C.; Eijan, A.M. Inhibition of breast tumor growth by N(G)-nitro-l-arginine methyl ester (l-NAME) is accompanied by activation of fibroblasts. Nitric Oxide 2019, 93, 34–43. [Google Scholar] [CrossRef]
- Christmann, M.; Verbeek, B.; Roos, W.P.; Kaina, B. O(6)-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: Enzyme activity, promoter methylation and immunohistochemistry. Biochim. Biophys. Acta 2011, 1816, 179–190. [Google Scholar] [CrossRef]
- Jaiswal, M.; LaRusso, N.F.; Nishioka, N.; Nakabeppu, Y.; Gores, G.J. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001, 61, 6388–6393. [Google Scholar]
- Tan, C.; Li, Y.; Huang, X.; Wei, M.; Huang, Y.; Tang, Z.; Huang, H.; Zhou, W.; Wang, Y.; Hu, J. Extensive protein S-nitrosylation associated with human pancreatic ductal adenocarcinoma pathogenesis. Cell Death Dis. 2019, 10, 914. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, R.L.; Intabli, H.; Falcinelli, M.; Bucca, G.; Hesketh, A.; Patel, B.A.; Allen, M.C.; Smith, C.P.; Flint, M.S. Stress hormone-mediated acceleration of breast cancer metastasis is halted by inhibition of nitric oxide synthase. Cancer Lett. 2019, 459, 59–71. [Google Scholar] [CrossRef]
- Yarlagadda, K.; Hassani, J.; Foote, I.P.; Markowitz, J. The role of nitric oxide in melanoma. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 500–509. [Google Scholar] [CrossRef]
- Qin, Y.; Dey, A.; Purayil, H.T.; Daaka, Y. Maintenance of androgen receptor inactivation by S-nitrosylation. Cancer Res. 2013, 73, 6690–6699. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.L.; Ji, P.; Ouyang, N.; Kopelovich, L.; Rigas, B. Protein nitration and nitrosylation by NO-donating aspirin in colon cancer cells: Relevance to its mechanism of action. Exp. Cell Res. 2011, 317, 1359–1367. [Google Scholar] [CrossRef] [Green Version]
- Song, J.M.; Upadhyaya, P.; Kassie, F. Nitric oxide-donating aspirin (NO-Aspirin) suppresses lung tumorigenesis in vitro and in vivo and these effects are associated with modulation of the EGFR signaling pathway. Carcinogenesis 2018, 39, 911–920. [Google Scholar] [CrossRef] [Green Version]
- Aranda, E.; Lopez-Pedrera, C.; De La Haba-Rodriguez, J.R.; Rodriguez-Ariza, A. Nitric Oxide and Cancer: The Emerging Role of S-Nitrosylation. Curr. Mol. Med. 2012, 12, 50–67. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M. Emerging Roles of Protein S-Nitrosylation in Macrophages and Cancer Cells. Curr. Med. Chem. 2016, 23, 2602–2617. [Google Scholar] [CrossRef] [PubMed]
- Bignon, E.; Allega, M.F.; Lucchetta, M.; Tiberti, M.; Papaleo, E. Computational Structural Biology of S-nitrosylation of Cancer Targets. Front. Oncol. 2018, 8, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Prajapati, P.; Lambert, D.W. Cancer-associated fibroblasts—Not-so-innocent bystanders in metastasis to bone? J. Bone Oncol. 2016, 5, 128–131. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Bronte, V. Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 2016, 39, 1–6. [Google Scholar] [CrossRef] [Green Version]
- McAllister, S.S.; Weinberg, R.A. Tumor-host interactions: A far-reaching relationship. J. Clin. Oncol. 2010, 28, 4022–4028. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Izquierdo-Alvarez, A.; Garcia-Ortiz, A.; Ibiza, S.; Serrador, J.M.; Martinez-Ruiz, A. Nitrosothiols in the immune system: Signaling and protection. Antioxid. Redox Signal. 2013, 18, 288–308. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R.J. The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA 2001, 98, 355–360. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharm. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Lacza, Z.; Puskar, M.; Figueroa, J.P.; Zhang, J.; Rajapakse, N.; Busija, D.W. Mitochondrial nitric oxide synthase is constitutively active and is functionally upregulated in hypoxia. Free Radic. Biol. Med. 2001, 31, 1609–1615. [Google Scholar] [CrossRef]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharm. Sci. 2005, 26, 190–195. [Google Scholar] [CrossRef]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, Q.; Zhang, L.; Fang, Z.; Zhao, F.; Lv, Z.; Gu, Z.; Zhang, J.; Wang, J.; Zen, K.; et al. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 2010, 20, 676–687. [Google Scholar] [CrossRef]
- Victor, V.M.; Nun˜ez, C.; D’Oco´n, P.; Taylor, C.T.; Esplugues, J.V.; Moncada, S. Regulation of oxygen distribution in tissues by endothelial nitric oxide. Circ. Res. 2009, 104, 1178–1183. [Google Scholar] [CrossRef] [Green Version]
- Moens, A.L.; Kass, D.A. Tetrahydrobiopterin and cardiovascular disease. Arter. Thromb. Vasc. Biol. 2006, 26, 2439–2444. [Google Scholar] [CrossRef]
- Edgar, K.S.; Matesanz, N.; Gardiner, T.A.; Katusic, Z.S.; McDonald, D.M. Hyperoxia depletes (6R)-5,6,7,8-tetrahydrobiopterin levels in the neonatal retina: Implications for nitric oxide synthase function in retinopathy. Am. J. Pathol. 2015, 185, 1769–1782. [Google Scholar] [CrossRef]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [Green Version]
- Veron, D.; Aggarwal, P.K.; Velazquez, H.; Kashgarian, M.; Moeckel, G.; Tufro, A. Podocyte-specific VEGF-a gain of function induces nodular glomerulosclerosis in eNOS null mice. J. Am. Soc. Nephrol. 2014, 25, 1814–1824. [Google Scholar] [CrossRef] [Green Version]
- Rabender, C.S.; Alam, A.; Sundaresan, G.; Cardnell, R.J.; Yakovlev, V.A.; Mukhopadhyay, N.D.; Graves, P.; Zweit, J.; Mikkelsen, R.B. The Role of Nitric Oxide Synthase Uncoupling in Tumor Progression. Mol. Cancer Res. 2015, 13, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Hoang, H.H.; Padgham, S.V.; Meininger, C.J. L-arginine, tetrahydrobiopterin, nitric oxide and diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 76–82. [Google Scholar] [CrossRef]
- Gámez-Méndez, A.M.; Vargas-Robles, H.; Arellano-Mendoza, M.; Cruz-Laguna, E.; Rios, A.; Escalante, B. Early stage of obesity potentiates nitric oxide reduction during the development of renal failure. J. Nephrol. 2014, 27, 281–287. [Google Scholar] [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Antosova, M.; Plevkova, J.; Strapkova, A.; Buday, T. Nitric oxide—Important messenger in human body. OJMIP 2012, 2, 98–106. [Google Scholar] [CrossRef]
- Peunova, N.; Scheinker, V.; Ravi, K.; Enikolopov, G. Nitric oxide coordinates cell proliferation and cell movements during early development of Xenopus. Cell Cycle 2007, 6, 3132–3144. [Google Scholar] [CrossRef]
- Young, S.L.; Evans, K.; Eu, J.P. Nitric oxide modulates branching morphogenesis in fetal rat lung explants. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L379–L385. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.G.; Mashkournia, A.; Parries, S.C.; Goldberg, J.I. Regulation of early embryonic behavior by nitric oxide in the pond snail Helisoma trivolvis. J. Exp. Biol. 2002, 205, 3143–3152. [Google Scholar] [CrossRef]
- Slezinger, M.S.; Kuzin, B.A. Nitric oxide synthase mediates regulation of cell polarity and movement during Drosophila melanogaster morphogenesis. Ontogenez 2009, 40, 40–47. [Google Scholar] [CrossRef]
- Bradley, S.; Tossell, K.; Lockley, R.; McDearmid, J.R. Nitric oxide synthase regulates morphogenesis of zebrafish spinal cord motoneurons. J. Neurosci. 2010, 30, 16816–16831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.P.; Cheng, C.Y. Nitric oxide/nitric oxide synthase, spermatogenesis, and tight junction dynamics. Biol. Reprod. 2004, 70, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Ruiz, A.; Araújo, I.M.; Izquierdo-Álvarez, A.; Hernansanz-Agustín, P.; Lamas, S.; Serrador, J. Specificity in S-nitrosylation: A short-range mechanism for NO signaling? Antioxid. Redox Signal. 2013, 19, 1220–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scicinski, J.; Oronsky, B.; Ning, S.; Knox, S.; Peehl, D.; Kim, M.M.; Langecker, P.; Fanger, G. NO to cancer: The complex and multifaceted role of nitric oxide and the epigenetic nitric oxide donor, RRx-001. Redox Biol. 2015, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Choudhari, S.K.; Chaudhary, M.; Sachin, B.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.D.; Espey, M.G.; Ridnour, L.A.; Hofseth, L.J.; Mancardi, D.; Harris, C.C.; Wink, D.A. Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc. Natl. Acad. Sci. USA 2004, 101, 8894–8899. [Google Scholar] [CrossRef] [Green Version]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharm. Rev. 1991, 43, 109–142. [Google Scholar]
- Thomsen, L.L.; Miles, D.W.; Happerfield, L.; Bobrow, L.G.; Knowles, R.G.; Moncada, S. Nitric oxide synthase activity in human breast cancer. Br. J. Cancer 2020, 72, 41–44. [Google Scholar] [CrossRef]
- Sangle, V.A.; Chaware, S.J.; Kulkarni, M.A.; Ingle, Y.C.; Singh, P.; Pooja, V.K. Elevated tissue nitric oxide in oral squamous cell carcinoma. J. Oral Maxillofac. Pathol. 2018, 22, 35–39. [Google Scholar]
- Abdel-Salam, O.; Youness, E.; Hafez, H. The antioxidant status of the plasma in patients with breast cancer undergoing chemotherapy. Open J. Mol. Integr. Physiol. 2011, 1, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Simeone, A.M.; Broemeling, L.D.; Rosenblum, J.; Tari, A.M. HER2/neu reduces the apoptotic effects of N-(4-hydroxyphenyl)retinamide (4-HPR) in breast cancer cells by decreasing nitric oxide production. Oncogenet 2003, 22, 6739–6747. [Google Scholar] [CrossRef] [Green Version]
- Furuta, S.; Ren, G.; Mao, J.; Bissell, M.J. Laminin signals initiate the reciprocal loop that informs breast-specific gene expression and homeostasis by activating NO, p53 and microRNA. eLife 2018, 7, e26148. [Google Scholar] [CrossRef]
- Hickok, J.R.; Thomas, D.D. Nitric oxide and cancer therapy: The emperor has NO clothes. Curr. Pharm. Des. 2010, 16, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Burke, A.J.; Sullivan, F.J.; Giles, F.J.; Glynn, S.A. The yin and yang of nitric oxide in cancer progression. Carcinogenesis 2013, 34, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Ridnour, L.A.; Thomas, D.D.; Switzer, C.; Flores-Santana, W.; Isenberg, J.S.; Ambs, S.; Roberts, D.D.; Wink, D.A. Molecular mechanisms for discrete nitric oxide levels in cancer. Nitric Oxide 2000, 19, 73–76. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Turkowski, K.; Mora, J.; Brüne, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef] [Green Version]
- Gould, N.; Doulias, P.T.; Tenopoulou, M.; Raju, K.; Ischiropoulos, H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J. Biol. Chem. 2013, 288, 26473–26479. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.S.; Wang, S.B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine oxidative posttranslational modifications: Emerging regulation in the cardiovascular system. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, G.; Koning, T.; Ehrenfeld, P.; Sanchez, F.A. Role of NO and S-nitrosylation in the Expression of Endothelial Adhesion Proteins That Regulate Leukocyte and Tumor Cell Adhesion. Front. Physiol. 2020, 11, 595526. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends. Pharm. Sci. 2016, 37, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Bartberger, M.D.; Liu, W.; Ford, E.; Miranda, K.M.; Switzer, C.; Fukuto, J.M.; Farmer, P.J.; Wink, D.A.; Houk, K.N. The reduction potential of nitric oxide (NO) and its importance to NO biochemistry. Proc. Natl. Acad. Sci. USA 2002, 99, 10958–10963. [Google Scholar] [CrossRef] [Green Version]
- Moller, M.N.; Li, Q.; Vitturi, D.A.; Robinson, J.M.; Lancaster, J.R., Jr.; Denicola, A. Membrane “lens” effect: Focusing the formation of reactive nitrogen oxides from the *NO/O2 reaction. Chem. Res. Toxicol. 2007, 20, 709–714. [Google Scholar] [CrossRef]
- Broniowska, K.A.; Hogg, N. The chemical biology of S-nitrosothiols. Antioxid. Redox Signal. 2012, 17, 969–980. [Google Scholar] [CrossRef]
- Jia, J.; Arif, A.; Terenzi, F.; Willard, B.; Plow, E.F.; Hazen, S.L.; Fox, P.L. Target-selective protein S-nitrosylation by sequence motif recognition. Cell 2014, 159, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Lipton, S.A. Emerging role of protein-protein transnitrosylation in cell signaling pathways. Antioxid. Redox Signal. 2013, 18, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.T.; Stamler, J.S. Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem. 2012, 287, 4411–4418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Stamler, J.S. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J. Mol. Med. 2012, 90, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Hausladen, A.; Wang, Y.J.; Zhang, G.F.; Stomberski, C.; Brunengraber, H.; Hess, D.T.; Stamler, J.S. Identification of S-nitroso-CoA reductases that regulate protein S-nitrosylation. Proc. Natl. Acad. Sci. USA 2014, 111, 18572–18577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Deng, Y.; Yang, X.; Xue, H.; Lang, Y. The Relationship Between Protein S-Nitrosylation and Human Diseases: A Review. Neurochem. Res. 2020, 45, 2815–2827. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, P.; Cordova, F.; Duran, W.N.; Sanchez, F.A. S-nitrosylation and its role in breast cancer angiogenesis and metastasis. Nitric Oxide 2019, 87, 52–59. [Google Scholar] [CrossRef]
- Basudhar, D.; Somasundaram, V.; de Oliveira, G.A.; Kesarwala, A.; Heinecke, J.L.; Cheng, R.Y.; Glynn, S.A.; Ambs, S.; Wink, D.A.; Ridnour, L.A. Nitric Oxide Synthase-2-Derived Nitric Oxide Drives Multiple Pathways of Breast Cancer Progression. Antioxid. Redox Signal. 2017, 26, 1044–1058. [Google Scholar] [CrossRef]
- Thompson, L.; Dong, Y.; Evans, L. Chronic hypoxia increases inducible NOS-derived nitric oxide in fetal guinea pig hearts. Pediatr. Res. 2009, 65, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Senga, T.; Ito, S.; Hyodo, T.; Hasegawa, H.; Hamaguchi, M. S-nitrosylation at cysteine 498 of c-Src tyrosine kinase regulates nitric oxide-mediated cell invasion. J. Biol. Chem. 2010, 285, 3806–3814. [Google Scholar] [CrossRef] [Green Version]
- Marshall, H.E.; Foster, M.W. S-nitrosylation of Ras in breast cancer. Breast Cancer Res. 2012, 14, 113. [Google Scholar] [CrossRef] [Green Version]
- Jindal, S.; Pennock, N.D.; Klug, A.; Narasimhan, J.; Calhoun, A.; Roberts, M.R.; Tamimi, R.M.; Eliassen, A.H.; Weinmann, S.; Borges, V.F.; et al. S-nitrosylated and non-nitrosylated COX2 have differential expression and distinct subcellular localization in normal and breast cancer tissue. NPJ Breast Cancer 2020, 6, 62. [Google Scholar] [CrossRef]
- Thomas, D.D.; Espey, M.G.; Pociask, D.A.; Ridnour, L.A.; Donzelli, S.; Wink, D.A. Asbestos redirects nitric oxide signaling through rapid catalytic conversion to nitrite. Cancer Res. 2006, 66, 11600–11604. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Hou, J.; Huang, L.; Huang, X.; Heibeck, T.H.; Zhao, R.; Pasa-Tolic, L.; Smith, R.D.; Li, Y.; Fu, K.; et al. Site-specific proteomics approach for study protein S-nitrosylation. Anal. Chem. 2010, 82, 7160–7168. [Google Scholar] [CrossRef]
- Ben-Lulu, S.; Ziv, T.; Weisman-Shomer, P.; Benhar, M. Nitrosothiol-Trapping-Based Proteomic Analysis of S-Nitrosylation in Human Lung Carcinoma Cells. PLoS ONE 2017, 12, e0169862. [Google Scholar] [CrossRef]
- Zhang, X.; Li, G.; Guo, Y.; Song, Y.; Chen, L.; Ruan, Q.; Wang, Y.; Sun, L.; Hu, Y.; Zhou, J.; et al. Regulation of ezrin tension by S-nitrosylation mediates non-small cell lung cancer invasion and metastasis. Theranostics 2019, 9, 2555–2571. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Nimmannit, U.; Stehlik, C.; Wang, L.; Jiang, B.H.; Ongpipatanakul, B.; Rojanasakul, Y. Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Res. 2006, 66, 6353–6360. [Google Scholar] [CrossRef] [Green Version]
- Chanvorachote, P.; Nimmannit, U.; Lu, Y.; Talbott, S.; Jiang, B.H.; Rojanasakul, Y. Nitric oxide regulates lung carcinoma cell anoikis through inhibition of ubiquitin-proteasomal degradation of caveolin-1. J. Biol. Chem. 2009, 284, 28476–28484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sun, C.; Xiao, G.; Shan, H.; Tang, L.; Yi, Y.; Yu, W.; Gu, Y. S-nitrosylation of the Peroxiredoxin-2 promotes S-nitrosoglutathione-mediated lung cancer cells apoptosis via AMPK-SIRT1 pathway. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Huang, M.; Chen, X.; Chen, J.; Zou, Z.; Li, L.; Ji, K.; Nie, Z.; Yang, B.; Wei, Z.; et al. The role of S-nitrosylation of PFKM in regulation of glycolysis in ovarian cancer cells. Cell Death Dis. 2021, 12, 408. [Google Scholar] [CrossRef]
- Saed, G.M.; Ali-Fehmi, R.; Jiang, Z.L.; Fletcher, N.M.; Diamond, M.P.; Abu-Soud, H.M.; Munkarah, A.R. Myeloperoxidase serves as a redox switch that regulates apoptosis in epithelial ovarian cancer. Gynecol. Oncol. 2010, 116, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, S.; Rattan, R.; Deshpande, M.; Maguire, J.L.; Johnson, Z.; Graham, R.P.; Shridhar, V. Preclinical Therapeutic Potential of a Nitrosylating Agent in the Treatment of Ovarian Cancer. PLoS ONE 2014, 9, e97897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaac, J.; Tarapore, P.; Zhang, X.; Lam, Y.W.; Ho, S.M. Site-specific S-nitrosylation of integrin alpha6 increases the extent of prostate cancer cell migration by enhancing integrin beta1 association and weakening adherence to laminin-1. Biochemistry 2012, 51, 9689–9697. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Cao, Y.; Zhang, T.; Wang, P.; Ji, D.; Liu, X.; Shi, H.; Hua, L.; Yu, R.; Gao, S. Effects of ERK1/2 S-nitrosylation on ERK1/2 phosphorylation and cell survival in glioma cells. Int. J. Mol. Med. 2018, 41, 1339–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.Q.; Kim, M.Y.; Godoy, L.C.; Thiantanawat, A.; Trudel, L.J.; Wogan, G.N. Nitric oxide activation of Keap1/Nrf2 signaling in human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14547–14551. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zheng, K.; Ma, C.; Li, J.; Zhuo, L.; Huang, W.; Chen, T.; Jiang, Y. PTPS facilitates compartmentalized LTBP1 S-nitrosylation and promotes tumor growth under hypoxia. Mol. Cell 2020, 77, 95–107. [Google Scholar] [CrossRef]
- Zhao, Q.-F.; Yu, J.-T.; Tan, L. S-Nitrosylation in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 268–280. [Google Scholar] [CrossRef]
- Gu, Z.; Kaul, M.; Yan, B.; Kridel, S.J.; Cui, J.; Strongin, A.; Smith, J.W.; Liddington, R.C.; Lipton, S.A. S-nitrosylation of matrix metalloproteinases: Signaling pathway to neuronal cell death. Science 2002, 297, 1186–1190. [Google Scholar] [CrossRef]
- Lipton, S.A.; Stamler, J.S. Actions of redox-related congeners of nitric oxide at the NMDA receptor. Neuropharmacology 1994, 33, 1229–1233. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Lee, J.; Sehgal, P.B. Depletion of the ATPase NSF from Golgi membranes with hypo-S-nitrosylation of vasorelevant proteins in endothelial cells exposed to monocrotaline pyrrole. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1943–H1955. [Google Scholar] [CrossRef]
- Tao, L.; Gao, E.; Bryan, N.S.; Qu, Y.; Liu, H.R.; Hu, A.; Christopher, T.A.; Lopez, B.L.; Yodoi, J.; Koch, W.J.; et al. Cardioprotective effects of thioredoxin in myocardial ischemia and reperfusion: Role of S-nitrosation [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 11471–11476. [Google Scholar] [CrossRef] [Green Version]
- Ambs, S.; Ogunfusika, M.O.; Merriam, W.G.; Bennett, W.P.; Billiar, T.R.; Harris, C.C. Up-regulation of inducible nitric oxide synthase expression in cancer-prone p53 knockout mice. Proc. Natl. Acad. Sci. USA 1998, 95, 8823–8828. [Google Scholar] [CrossRef] [Green Version]
- Vakkala, M.; Kahlos, K.; Lakari, E.; Pääkkö, P.; Kinnula, V.; Soini, Y. Inducible Nitric Oxide Synthase Expression, Apoptosis, and Angiogenesis in in Situ and Invasive Breast Carcinomas. Clin. Cancer Res. 2000, 6, 2408–2416. [Google Scholar]
- Switzer, C.H.; Cheng, R.Y.S.; Ridnour, L.A.; Glynn, S.A.; Ambs, S.; Wink, D.A. Ets-1 is a transcriptional mediator of oncogenic nitric oxide signaling in estrogen receptor-negative breast cancer. Breast Cancer Res. 2012, 14, R125. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.; Khoo, J.; Channon, K.M. Augmented BH4 by gene transfer restores nitric oxide synthase function in hyperglycemic human endothelial cells. Cardiovasc. Res. 2005, 65, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Santhanam, A.V.; d’Uscio, L.V.; Smith, L.A.; Katusic, Z.S. Uncoupling of eNOS causes superoxide anion production and impairs NO signaling in the cerebral microvessels of hph-1 mice. J. Neurochem. 2012, 122, v. [Google Scholar] [CrossRef] [Green Version]
- Melo, F.H.; Molognoni, F.; Morais, A.S.; Toricelli, M.; Mouro, M.G.; Higa, E.M.; Lopes, J.D.; Jasiulionis, M.G. Endothelial nitric oxide synthase uncoupling as a key mediator of melanocyte malignant transformation associated with sustained stress conditions. Free Radic. Biol. Med. 2011, 50, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Jo, H.; Otani, H.; Jo, F.; Shimazu, T.; Okazaki, T.; Yoshioka, K.; Fujita, M.; Kosaki, A.; Iwasaka, T. Inhibition of nitric oxide synthase uncoupling by sepiapterin improves left ventricular function in streptozotocin-induced diabetic mice. Clin. Exp. Pharm. Phys. 2011, 38, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Li, B.; Hanes, M.A.; Kakar, S.; Chen, X.; Liu, L. S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Sci. Transl. Med. 2010, 2, 19ra13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messina, S.; De Simone, G.; Ascenzi, P. Cysteine-based regulation of redox-sensitive Ras small GTPases. Redox Biol. 2019, 26, 101282. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, T.; Cho, D.H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.; Iyer, A.K.V.; Kulkarni, Y.; Azad, N. S-Nitrosylation of Bcl-2 Negatively Affects Autophagy in Lung Epithelial Cells. J. Cell Biochem. 2016, 117, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Hetman, M.; Gozdz, A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur. J. Biochem. 2004, 271, 2050–2055. [Google Scholar] [CrossRef]
- Santos, A.I.; Carreira, B.P.; Izquierdo-Álvarez, A.; Ramos, E.; Lourenço, A.S.; Filipa Santos, D.; Morte, M.I.; Ribeiro, L.F.; Marreiros, A.; Sánchez-López, N.; et al. S-Nitrosylation of Ras Mediates Nitric Oxide-Dependent Post-Injury Neurogenesis in a Seizure Model. Antioxid. Redox Signal. 2018, 28, 15–30. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Burguillos, M.A.; Osman, A.M.; Frijhoff, J.; Carrillo-Jiménez, A.; Kanatani, S.; Augsten, M.; Saidi, D.; Rodhe, J.; Kavanagh, E. Glioma-induced inhibition of caspase-3 in microglia promotes a tumor-supportive phenotype. Nat. Immunol. 2016, 17, 1282–1290. [Google Scholar] [CrossRef]
- Huang, J.J.; Blobe, G.C. Dichotomous roles of TGF-β in human cancer. Biochem. Soc. Trans. 2016, 44, 1441–1454. [Google Scholar] [CrossRef] [Green Version]
- Zahid, S.; Khan, R.; Oellerich, M.; Ahmed, N.; Asif, A. Differential S-nitrosylation of proteins in Alzheimer’s disease. Neuroscience 2014, 256, 126–136. [Google Scholar] [CrossRef]
- Sun, J.; Murphy, E. Protein S-nitrosylation and cardioprotection. Circ. Res. 2010, 106, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.L.; Zhang, R.; Anand, P.; Stomberski, C.T.; Qian, Z.; Hausladen, A.; Wang, L.; Rhee, E.P.; Parikh, S.M.; Karumanchi, S.A.; et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 2019, 565, 96–100. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogenet 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Frisch, J.; Angenendt, A.; Hoth, M.; Prates Roma, L.; Lis, A. STIM-Orai Channels and Reactive Oxygen Species in the Tumor Microenvironment. Cancers 2019, 11, 457. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Chen, C. S-nitrosylation Denitrosylation and Apoptosis of Immune Cells. Cell Mol. Immunol. 2007, 4, 353–358. [Google Scholar]
- Ibanez-Vea, M.; Huang, H.; Martinez de Morentin, X.; Perez, E.; Gato, M.; Zuazo, M.; Arasanz, H.; Fernandez-Irigoyen, J.; Santamaria, E.; Fernandez-Hinojal, G.; et al. Characterization of Macrophage Endogenous S-Nitrosoproteome Using a Cysteine-Specific Phosphonate Adaptable Tag in Combination with TiO2 Chromatography. J. Proteome Res. 2018, 17, 1172–1182. [Google Scholar] [CrossRef] [Green Version]
- Jaffrey, S.R.; Snyder, S.H. The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE 2001, 2001, pl1. [Google Scholar] [CrossRef]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Fernando, V.; Sharma, V.; Walia, Y.; Letson, J.; Furuta, S. Correction of arginine metabolism with sepiapterin-the precursor of nitric oxide synthase cofactor BH4-induces immunostimulatory-shift of breast cancer. Biochem. Pharm. 2020, 176, 113887. [Google Scholar] [CrossRef]
- Virág, L.; Jaén, R.I.; Regdon, Z.; Boscá, L.; Prieto, P. Self-defense of macrophages against oxidative injury: Fighting for their own survival. Redox Biol. 2019, 26, 101261. [Google Scholar] [CrossRef]
- Ibiza, S.; Víctor, V.M.; Boscá, I.; Ortega, A.; Urzainqui, A.; O’Connor, J.E.; Sánchez-Madrid, F.; Esplugues, J.V.; Serrador, J.M. Endothelial Nitric Oxide Synthase Regulates T Cell Receptor Signaling at the Immunological Synapse. Immunity. 2006, 24, 753–765. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ortiz, A.; Serrador, J.M. Nitric Oxide Signaling in T Cell-Mediated Immunity. Trends Mol. Med. 2018, 24, 412–427. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Satoh, A.; Chatterjee, S.; Toomre, D.K.; Chalouni, C.M.; Fulton, D.; Groszmann, R.J.; Shah, V.H.; Sessa, W.C. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc. Natl. Acad. Sci. USA 2006, 103, 19777–19782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Wang, Z.E.; Doulias, P.T.; Wei, W.; Ischiropoulos, H.; Locksley, R.M.; Liu, L. Lymphocyte development requires S-nitrosoglutathione reductase. J. Immunol. 2010, 185, 6664–6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, Y.; Zhang, Y.; Lü, S.; Miao, Y.; Yang, J.; Huang, S.; Ma, X.; Han, L.; Deng, J.; et al. GSNOR modulates hyperhomocysteinemia-induced T cell activation and atherosclerosis by switching Akt S-nitrosylation to phosphorylation. Redox Biol. 2018, 17, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Guequen, A.; Zamorano, P.; Cordova, F.; Koning, T.; Torres, A.; Ehrenfeld, P.; Boric, M.P.; Salazar-Onfray, F.; Gavard, J.; Duran, W.N.; et al. Interleukin-8 Secreted by Glioblastoma Cells Induces Microvascular Hyperpermeability Through NO Signaling Involving S-Nitrosylation of VE-Cadherin and p120 in Endothelial Cells. Front. Physiol. 2019, 10, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, N.; Zamorano, P.; Carrasco, R.; Mujica, P.; Gonzalez, F.G.; Quezada, C.; Meininger, C.J.; Boric, M.P.; Duran, W.N.; Sanchez, F.A. S-Nitrosation of beta-Catenin and p120 Catenin A Novel Regulatory Mechanism in Endothelial Hyperpermeability. Circ. Res. 2012, 111, U248–U553. [Google Scholar] [CrossRef] [Green Version]
- Thibeault, S.; Rautureau, Y.; Oubaha, M.; Faubert, D.; Wilkes, B.C.; Delisle, C.; Gratton, J.P. S-Nitrosylation of beta-Catenin by eNOS-Derived NO Promotes VEGF-Induced Endothelial Cell Permeability. Mol. Cell 2010, 39, 468–476. [Google Scholar] [CrossRef]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.Y. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Kang-Decker, N.; Cao, S.; Chatterjee, S.; Yao, J.; Egan, L.J.; Semela, D.; Mukhopadhyay, D.; Shah, V. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J. Cell Sci. 2007, 120, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Moniri, N.H.; Ozawa, K.; Stamler, J.S.; Daaka, Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc. Natl. Acad. Sci. USA 2006, 103, 1295–1300. [Google Scholar] [CrossRef] [Green Version]
- Pi, X.; Wu, Y.; Ferguson, J.E., 3rd; Portbury, A.L.; Patterson, C. SDF-1alpha stimulates JNK3 activity via eNOS-dependent nitrosylation of MKP7 to enhance endothelial migration. Proc. Natl. Acad. Sci. USA 2009, 106, 5675–5680. [Google Scholar] [CrossRef] [Green Version]
- Parker, K.H.; Beury, D.W.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. Adv. Cancer Res. 2015, 128, 95–139. [Google Scholar] [CrossRef] [Green Version]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef]
- Marshall, H.E.; Hess, D.T.; Stamler, J.S. S-nitrosylation—Physiological regulation of NF-κB. Proc. Natl. Acad. Sci. USA 2004, 101, 8841–8842. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Won, J.S.; Singh, A.K.; Sharma, A.K.; Singh, I. STAT3 regulation by S-nitrosylation: Implication for inflammatory disease. Antioxid. Redox Signal. 2014, 20, 2514–2527. [Google Scholar] [CrossRef]
- Mannick, J.B.; Schonhoff, C.; Papeta, N.; Ghafourifar, P.; Szibor, M.; Fang, K.; Gaston, B. S-Nitrosylation of mitochondrial caspases. J. Biol. Chem. 2001, 154, 1111–1116. [Google Scholar] [CrossRef] [Green Version]
- Mao, K.; Chen, S.; Chen, M.; Ma, Y.; Wang, Y.; Huang, B.; He, Z.; Zeng, Y.; Hu, Y.; Sun, S.; et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013, 23, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.B.; Rathinam, V.A.; Martens, G.W.; Martinot, A.J.; Kornfeld, H.; Fitzgerald, K.A.; Sassetti, C.M. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat. Immunol. 2013, 14, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Cuellar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting Edge: Nitric Oxide Inhibits the NLRP3 Inflammasome. J. Immunol. 2012, 189, 5113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhanam, L.; Lim, H.K.; Lim, H.K.; Miriel, V.; Brown, T.; Patel, M.; Balanson, S.; Ryoo, S.; Anderson, M.; Irani, K.; et al. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ. Res. 2007, 101, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Batista, W.L.; Ogata, F.T.; Curcio, M.F.; Miguel, R.B.; Arai, R.J.; Matsuo, A.L.; Moraes, M.S.; Stern, A.; Monteiro, H.P. S-nitrosoglutathione and endothelial nitric oxide synthase-derived nitric oxide regulate compartmentalized ras S-nitrosylation and stimulate cell proliferation. Antioxid. Redox Signal. 2013, 18, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.F.; Huri, D.A.; Snyder, S.H. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science 2005, 310, 1966–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Switzer, C.H.; Glynn, S.A.; Cheng, R.Y.; Ridnour, L.A.; Green, J.E.; Ambs, S.; Wink, D.A. S-nitrosylation of EGFR and Src activates an oncogenic signaling network in human basal-like breast cancer. Mol. Cancer Res. 2012, 10, 1203–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.H.; Wei, W.; Liu, L. Regulation of DNA repair by S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 730–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Xu-Welliver, M.; Kanugula, S.; Pegg, A.E. Inactivation and degradation of O(6)-alkylguanine-DNA alkyltransferase after reaction with nitric oxide. Cancer Res. 2002, 62, 3037–3043. [Google Scholar]
- Tang, Z.; Bauer, J.A.; Morrison, B.; Lindner, D.J. Nitrosylcobalamin promotes cell death via S nitrosylation of Apo2L/TRAIL receptor DR4. Mol. Cell Biol. 2006, 26, 5588–5594. [Google Scholar] [CrossRef] [Green Version]
- Azad, N.; Vallyathan, V.; Wang, L.; Tantishaiyakul, V.; Stehlik, C.; Leonard, S.S.; Rojanasakul, Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 2006, 281, 34124–34134. [Google Scholar] [CrossRef] [Green Version]
- Iyer, A.K.; Azad, N.; Wang, L.; Rojanasakul, Y. Role of S-nitrosylation in apoptosis resistance and carcinogenesis. Nitric Oxide 2008, 19, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Sun, T.; Bei, Y.; Ding, S.; Zheng, W.; Lu, Y.; Shen, P. S-nitrosylation of ERK inhibits ERK phosphorylation and induces apoptosis. Sci. Rep. 2013, 3, 1814. [Google Scholar] [CrossRef] [Green Version]
- Nott, A.; Nitarska, J.; Veenvliet, J.V.; Schacke, S.; Derijck, A.A.; Sirko, P.; Muchardt, C.; Pasterkamp, R.J.; Smidt, M.P.; Riccio, A. S-nitrosylation of HDAC2 regulates the expression of the chromatin-remodeling factor Brm during radial neuron migration. Proc. Natl. Acad. Sci. USA 2013, 110, 3113–3118. [Google Scholar] [CrossRef] [Green Version]
- Nott, A.; Watson, P.M.; Robinson, J.D.; Crepaldi, L.; Riccio, A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 2008, 455, 411–415. [Google Scholar] [CrossRef]
- Yu, C.X.; Li, S.; Whorton, A.R. Redox regulation of PTEN by S-nitrosothiols. Mol. Pharm. 2005, 68, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Calmels, S.; Hainaut, P.; Ohshima, H. Nitric oxide induces conformational and functional modifications of wild-type p53 tumor suppressor protein. Cancer Res. 1997, 57, 3365–3369. [Google Scholar]
- Schonhoff, C.M.; Daou, M.C.; Jones, S.N.; Schiffer, C.A.; Ross, A.H. Nitric oxide-mediated inhibition of Hdm2-p53 binding. Biochemistry 2002, 41, 13570–13574. [Google Scholar] [CrossRef]
- Leon-Bollotte, L.; Subramaniam, S.; Cauvard, O.; Plenchette-Colas, S.; Paul, C.; Godard, C.; Martinez-Ruiz, A.; Legembre, P.; Jeannin, J.F.; Bettaieb, A. S-nitrosylation of the death receptor fas promotes fas ligand-mediated apoptosis in cancer cells. Gastroenterology 2011, 140, 2009–2018.e2. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Rizza, S.; Montagna, C.; Cardaci, S.; Maiani, E.; Di Giacomo, G.; Sanchez-Quiles, V.; Blagoev, B.; Rasola, A.; De Zio, D.; Stamler, J.S.; et al. S-nitrosylation of the Mitochondrial Chaperone TRAP1 Sensitizes Hepatocellular Carcinoma Cells to Inhibitors of Succinate Dehydrogenase. Cancer Res. 2016, 76, 4170–4182. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Cifone, M.G.; Ulisse, S.; Santoni, A. Natural killer cells and nitric oxide. Int. Immunopharmacol. 2001, 1, 1513–1524. [Google Scholar] [CrossRef]
- Jyothi, M.D.; Khar, A. Induction of nitric oxide production by natural killer cells: Its role in tumor cell death. Nitric Oxide 1999, 3, 409–418. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Huxford, T.; Ghosh, G. A structural guide to proteins of the NF-kappaB signaling module. Cold Spring Harb. Perspect. Biol. 2009, 1, a000075. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Yin, Q.; Wu, H. Structural studies of NF-kappaB signaling. Cell Res. 2011, 21, 183–195. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogenet 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [Green Version]
- Caviedes, A.; Maturana, B.; Corvalán, K.; Engler, A.; Gordillo, F.; Varas-Godoy, M.; Smalla, K.-H.; Batiz, L.F.; Lafourcade, C.; Kaehne, T.; et al. eNOS-dependent S-nitrosylation of the NF-κB subunit p65 has neuroprotective effects. bioRxiv 2020. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.-W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Burdelya, L.; Kujawski, M.; Niu, G.; Zhong, B.; Wang, T.; Zhang, S.; Kortylewski, M.; Shain, K.; Kay, H.; Djeu, J.; et al. Stat3 Activity in Melanoma Cells Affects Migration of Immune Effector Cells and Nitric Oxide-Mediated Antitumor Effects. J. Immunol. 2005, 174, 3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butturini, E.; Carcereri de Prati, A.; Mariotto, S. Redox Regulation of STAT1 and STAT3 Signaling. Int. J. Mol. Sci. 2020, 21, 7034. [Google Scholar] [CrossRef] [PubMed]
- Hillmer, E.J.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016, 31, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Pal, S.K.; Reckamp, K.; Figlin, R.A.; Yu, H. STAT3: A target to enhance antitumor immune response. Curr. Top. Microbiol. Immunol. 2011, 344, 41–59. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]
- Niu, Z.; Tang, J.; Zhang, W.; Chen, Y.; Huang, Y.; Chen, B.; Li, J.; Shen, P. Caspase-1 promotes monocyte–macrophage differentiation by repressing PPARγ. FEBS J. 2017, 284, 568–585. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death. Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.C.; Arthurton, L.; Baena-Lopez, L.A. Learning on the Fly: The Interplay between Caspases and Cancer. BioMed Res. Int. 2018, 2018, 5473180. [Google Scholar] [CrossRef]
- Vande Walle, L.; Lamkanfi, M. Inflammasomes: Caspase-1-Activating Platforms with Critical Roles in Host Defense. Front. Microbiol. 2011, 2. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, E.; Sahin, A.A.; Chen, J.S.; Krishnamurthy, R.R.; Aggarwal, N.; Brun, A.M.; Sapino, A.; Zhang, F.; Sharma, D.; Yang, X.H.; et al. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogenet 2002, 21, 8843–8851. [Google Scholar] [CrossRef] [Green Version]
- Ponder, K.G.; Boise, L.H. The prodomain of caspase-3 regulates its own removal and caspase activation. Cell Death Discov. 2019, 5, 56. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.-F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.-C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.R.; Bedford, J.S.; Li, C.-Y. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal 2010, 3, ra13-ra13. [Google Scholar] [CrossRef] [Green Version]
- Sessa, W.C. eNOS at a glance. J. Cell Sci. 2004, 117, 2427–2429. [Google Scholar] [CrossRef] [Green Version]
- Tonini, T.; Rossi, F.; Claudio, P.P. Molecular basis of angiogenesis and cancer. OncoGene 2003, 22, 6549–6556. [Google Scholar] [CrossRef] [Green Version]
- Kostourou, V.; Cartwright, J.E.; Johnstone, A.P.; Boult, J.K.; Cullis, E.R.; Whitley, G.; Robinson, S.P. The role of tumour-derived iNOS in tumour progression and angiogenesis. Br. J. Cancer 2011, 104, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-Inducible Factor-1 Is a Basic-Helix-Loop-Helix-Pas Heterodimer Regulated by Cellular O-2 Tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 2013, 9, 1623–1636. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Mimeault, M.; Batra, S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell Mol. Med. 2013, 17, 30–54. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhou, S.; Xu, Y.; Sheng, S.; Qian, S.Y.; Huo, X. Nitric oxide synthase inhibitors 1400W and L-NIO inhibit angiogenesis pathway of colorectal cancer. Nitric Oxide 2019, 83, 33–39. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, S.; Pang, L.; Yang, J.; Li, H.J.; Huo, X.; Qian, S.Y. Celastrol suppresses nitric oxide synthases and the angiogenesis pathway in colorectal cancer. Free Radic. Res. 2019, 53, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Aebi, U. Intermediate filaments: Molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu. Rev. Biochem. 2004, 73, 749–789. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome–an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchheit, C.L.; Weigel, K.J.; Schafer, Z.T. Cancer cell survival during detachment from the ECM: Multiple barriers to tumour progression. Nat. Rev. Cancer 2014, 14, 632–641. [Google Scholar] [CrossRef]
- Chiarugi, P.; Giannoni, E. Anoikis: A necessary death program for anchorage-dependent cells. Biochem. Pharmacol. 2008, 76, 1352–1364. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. BBA Mol. Cell Res. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Berrier, A.L.; Yamada, K.M. Cell-matrix adhesion. J. Cell Physiol. 2007, 213, 565–573. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Miranti, C.K.; Brugge, J.S. Sensing the environment: A historical perspective on integrin signal transduction. Nat. Cell Biol. 2002, 4, E83–E90. [Google Scholar] [CrossRef]
- Parsons, J.T.; Martin, K.H.; Slack, J.K.; Taylor, J.M.; Weed, S.A. Focal adhesion kinase: A regulator of focal adhesion dynamics and cell movement. Oncogenet 2000, 19, 5606–5613. [Google Scholar] [CrossRef] [Green Version]
- da Costa, P.E.; Batista, W.L.; Moraes, M.S.; Stern, A.; Monteiro, H.P. Src kinase activation by nitric oxide promotes resistance to anoikis in tumour cell lines. Free Radic. Res. 2018, 52, 592–604. [Google Scholar] [CrossRef]
- Bernatchez, P.N.; Bauer, P.M.; Yu, J.; Prendergast, J.S.; He, P.; Sessa, W.C. Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc. Natl. Acad. Sci. USA 2005, 102, 761–766. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.-Y.; Zhao, Y.D.; Mirza, M.K.; Huang, J.H.; Potula, H.-H.S.K.; Vogel, S.M.; Brovkovych, V.; Yuan, J.X.J.; Wharton, J.; Malik, A.B. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J. Clin. Investig. 2009, 119, 2009–2018. [Google Scholar] [CrossRef] [Green Version]
- Bakhshi, F.R.; Bonini, M.; Chen, Z.; NieuwAmerongen, G.; Comhair, S.; Erzurum, S.; Minshall, R. Role of Caveolin-1 S-Nitrosylation, Ubiquitination, and Degradation in Idiopathic Pulmonary Arterial Hypertension. FASEB J. 2013, 27, 878.877–878.877. [Google Scholar] [CrossRef]
- Chen, Z.; D S Oliveira, S.; Zimnicka, A.M.; Jiang, Y.; Sharma, T.; Chen, S.; Lazarov, O.; Bonini, M.G.; Haus, J.M.; Minshall, R.D. Reciprocal regulation of eNOS and caveolin-1 functions in endothelial cells. Mol. Biol. Cell 2018, 29, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.M.; Liu, J. Caveolin-1 up-regulation during epithelial to mesenchymal transition is mediated by focal adhesion kinase. J. Biol. Chem. 2008, 283, 13714–13724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamaze, C.; Torrino, S. Caveolae and cancer: A new mechanical perspective. Biomed. J. 2015, 38, 367–379. [Google Scholar] [CrossRef]
- Bakker, E.N.; Pistea, A.; VanBavel, E. Transglutaminases in vascular biology: Relevance for vascular remodeling and atherosclerosis. J. Vasc. Res. 2008, 45, 271–278. [Google Scholar] [CrossRef]
- Santhanam, A.V.; Smith, L.A.; Katusic, Z.S. Brain-derived neurotrophic factor stimulates production of prostacyclin in cerebral arteries. Stroke 2010, 41, 350–356. [Google Scholar] [CrossRef] [Green Version]
- Tabolacci, C.; De Martino, A.; Mischiati, C.; Feriotto, G.; Beninati, S. The Role of Tissue Transglutaminase in Cancer Cell Initiation, Survival and Progression. Med. Sci. 2019, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, A.; et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 2019, 178, 795–806. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Laniewski, P.; Cui, H.; Roe, D.J.; Chase, D.M.; Herbst-Kralovetz, M.M. Vaginal microbiota, genital inflammation, and neoplasia impact immune checkpoint protein profiles in the cervicovaginal microenvironment. NPJ Precis. Oncol. 2020, 4, 22. [Google Scholar] [CrossRef]
- Bashiardes, S.; Tuganbaev, T.; Federici, S.; Elinav, E. The microbiome in anti-cancer therapy. Semin. Immunol. 2017, 32, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Seth, P.; Hsieh, P.N.; Jamal, S.; Wang, L.; Gygi, S.P.; Jain, M.K.; Coller, J.; Stamler, J.S. Regulation of MicroRNA Machinery and Development by Interspecies S-Nitrosylation. Cell 2019, 176, 1014–1025.e1012. [Google Scholar] [CrossRef] [Green Version]
- Urbaniak, C.; Cummins, J.; Brackstone, M.; Macklaim, J.M.; Gloor, G.B.; Baban, C.K.; Scott, L.; O’Hanlon, D.M.; Burton, J.P.; Francis, K.P.; et al. Microbiota of human breast tissue. Appl. Environ. Microbiol. 2014, 80, 3007–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbaniak, C.; Burton, J.P.; Reid, G. Breast, milk and microbes: A complex relationship that does not end with lactation. Women Health 2012, 8, 385–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannaraj, P.S.; Li, F.; Cerini, C.; Bender, J.M.; Yang, S.; Rollie, A.; Adisetiyo, H.; Zabih, S.; Lincez, P.J.; Bittinger, K.; et al. Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatr. 2017, 171, 647–654. [Google Scholar] [CrossRef]
- Urbaniak, C.; Gloor, G.B.; Brackstone, M.; Scott, L.; Tangney, M.; Reid, G. The Microbiota of Breast Tissue and Its Association with Breast Cancer. Appl. Environ. Microbiol. 2016, 82, 5039–5048. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- Jadeski, L.C.; Chakraborty, C.; Lala, P.K. Role of nitric oxide in tumour progression with special reference to a murine breast cancer model. Can. J. Physiol. Pharm. 2002, 80, 125–135. [Google Scholar] [CrossRef]
- Lala, P.K.; Chakraborty, C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001, 2, 149–156. [Google Scholar] [CrossRef]
- Thomsen, L.L.; Miles, D.W. Role of nitric oxide in tumour progression: Lessons from human tumours. Cancer Metastasis Rev. 1998, 17, 107–118. [Google Scholar] [CrossRef]
- Lim, S.Y.; Raftery, M.; Cai, H.; Hsu, K.; Yan, W.X.; Hseih, H.L.; Watts, R.N.; Richardson, D.; Thomas, S.; Perry, M.; et al. S-nitrosylated S100A8: Novel anti-inflammatory properties. J. Immunol. 2008, 181, 5627–5636. [Google Scholar] [CrossRef] [Green Version]
- Cianchi, F.; Cortesini, C.; Fantappie, O.; Messerini, L.; Schiavone, N.; Vannacci, A.; Nistri, S.; Sardi, I.; Baroni, G.; Marzocca, C.; et al. Inducible nitric oxide synthase expression in human colorectal cancer: Correlation with tumor angiogenesis. Am. J. Pathol. 2003, 162, 793–801. [Google Scholar] [CrossRef]
- Thomas, D.D.; Wink, D.A. NOS2 as an Emergent Player in Progression of Cancer. Antioxid. Redox Signal. 2017, 26, 963–965. [Google Scholar] [CrossRef]
- Lopez-Sanchez, L.M.; Corrales, F.J.; Lopez-Pedrera, C.; Aranda, E.; Rodriguez-Ariza, A. Pharmacological impairment of s-nitrosoglutathione or thioredoxin reductases augments protein S-Nitrosation in human hepatocarcinoma cells. AntiCancer Res. 2010, 30, 415–421. [Google Scholar]
- Granados-Principal, S.; Liu, Y.; Guevara, M.L.; Blanco, E.; Choi, D.S.; Qian, W.; Patel, T.; Rodriguez, A.A.; Cusimano, J.; Weiss, H.L.; et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 2015, 17, 25. [Google Scholar] [CrossRef]
- Davila-Gonzalez, D.; Choi, D.S.; Rosato, R.R.; Granados-Principal, S.M.; Kuhn, J.G.; Li, W.F.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.; et al. Pharmacological Inhibition of NOS Activates ASK1/JNK Pathway Augmenting Docetaxel-Mediated Apoptosis in Triple-Negative Breast Cancer. Clin. Cancer Res. 2018, 24, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.H.; Wei, W.; Hanes, M.A.; Liu, L. Hepatocarcinogenesis driven by GSNOR deficiency is prevented by iNOS inhibition. Cancer Res. 2013, 73, 2897–2904. [Google Scholar] [CrossRef] [Green Version]
- Tong, A.W.; Nemunaitis, J.; Su, D.; Zhang, Y.; Cunningham, C.; Senzer, N.; Netto, G.; Rich, D.; Mhashilkar, A.; Parker, K.; et al. Intratumoral injection of INGN 241, a nonreplicating adenovector expressing the melanoma-differentiation associated gene-7 (mda-7/IL24): Biologic outcome in advanced cancer patients. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 11, 160–172. [Google Scholar] [CrossRef]
- Tian, H.; Wang, J.; Zhang, B.; Di, J.; Chen, F.; Li, H.; Li, L.; Pei, D.; Zheng, J. MDA-7/IL-24 induces Bcl-2 denitrosylation and ubiquitin-degradation involved in cancer cell apoptosis. PLoS ONE 2012, 7, e37200. [Google Scholar] [CrossRef] [Green Version]
- Kaliyaperumal, K.; Sharma, A.K.; McDonald, D.G.; Dhindsa, J.S.; Yount, C.; Singh, A.K.; Won, J.S.; Singh, I. S-Nitrosoglutathione-mediated STAT3 regulation in efficacy of radiotherapy and cisplatin therapy in head and neck squamous cell carcinoma. Redox Biol. 2015, 6, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Romagny, S.; Bouaouiche, S.; Lucchi, G.; Ducoroy, P.; Bertoldo, J.B.; Terenzi, H.; Bettaieb, A.; Plenchette, S. S-Nitrosylation of cIAP1 Switches Cancer Cell Fate from TNFalpha/TNFR1-Mediated Cell Survival to Cell Death. Cancer Res. 2018, 78, 1948–1957. [Google Scholar] [CrossRef] [Green Version]
- Tan, G.; Qiu, M.; Chen, L.; Zhang, S.; Ke, L.; Liu, J. JS-K, a nitric oxide pro-drug, regulates growth and apoptosis through the ubiquitin-proteasome pathway in prostate cancer cells. BMC Cancer 2017, 17, 376. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Goswami, S.; Rodes, D.B.; Kodela, R.; Velazquez, C.A.; Boring, D.; Crowell, J.A.; Kashfi, K. NO-releasing NSAIDs suppress NF-κB signaling in vitro and in vivo through S-nitrosylation. Cancer Lett. 2010, 298, 204–211. [Google Scholar] [CrossRef]
- Kashfi, K. Nitric Oxide-Releasing Hybrid Drugs Target Cellular Processes Through S-Nitrosylation. Immunopathol. Dis. Ther. 2012, 3, 97–108. [Google Scholar] [CrossRef]
- Rigas, B.; Williams, J.L. NO-donating NSAIDs and cancer: An overview with a note on whether NO is required for their action. Nitric Oxide Biol. Chem. 2008, 19, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonavida, B.; Garban, H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol. 2015, 6, 486–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caneba, C.A.; Yang, L.; Baddour, J.; Curtis, R.; Win, J.; Hartig, S.; Marini, J.; Nagrath, D. Nitric oxide is a positive regulator of the Warburg effect in ovarian cancer cells. Cell Death Dis. 2014, 5, e1302. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanchez, L.M.; Aranda, E.; Rodriguez-Ariza, A. Nitric oxide and tumor metabolic reprogramming. Biochem. Pharm. 2020, 176, 113769. [Google Scholar] [CrossRef]
- Drapier, J.C.; Hibbs, J.B., Jr. Aconitases: A class of metalloproteins highly sensitive to nitric oxide synthesis. Methods Enzymol. 1996, 269, 26–36. [Google Scholar] [CrossRef]
- Piantadosi, C.A. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 712–721. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.S.; Richter, C.; Chun, J.S.; Huh, T.L.; Kang, S.S.; Park, J.W. Inactivation of NADP(+)-dependent isocitrate dehydrogenase by nitric oxide. Free Radic. Biol. Med. 2002, 33, 927–937. [Google Scholar] [CrossRef]
- Boveris, A.; Costa, L.E.; Poderoso, J.J.; Carreras, M.C.; Cadenas, E. Regulation of mitochondrial respiration by oxygen and nitric oxide. Ann. N. Y. Acad. Sci. 2000, 899, 121–135. [Google Scholar] [CrossRef]
- Dahm, C.C.; Moore, K.; Murphy, M.P. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: Implications for the interaction of nitric oxide with mitochondria. J. Biol. Chem. 2006, 281, 10056–10065. [Google Scholar] [CrossRef] [Green Version]
- Montagna, C.; Cirotti, C.; Rizza, S.; Filomeni, G. When S-Nitrosylation Gets to Mitochondria: From Signaling to Age-Related Diseases. Antioxid. Redox Signal. 2020, 32, 884–905. [Google Scholar] [CrossRef]
- Rasola, A.; Neckers, L.; Picard, D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. 2014, 24, 455–463. [Google Scholar] [CrossRef]
- Yoshida, S.; Tsutsumi, S.; Muhlebach, G.; Sourbier, C.; Lee, M.J.; Lee, S.; Vartholomaiou, E.; Tatokoro, M.; Beebe, K.; Miyajima, N.; et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc. Natl. Acad. Sci. USA 2013, 110, E1604–E1612. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wang, F.; Trachootham, D.; Huang, P. Preferential killing of cancer cells with mitochondrial dysfunction by natural compounds. Mitochondrion 2010, 10, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Neuzil, J.; Dong, L.F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef]
- Kashfi, K.; Rigas, B. Molecular targets of nitric-oxide-donating aspirin in cancer. Biochem. Soc. Trans. 2005, 33, 701–704. [Google Scholar] [CrossRef]
- Alimoradi, H.; Greish, K.; Gamble, A.B.; Giles, G.I. Controlled Delivery of Nitric Oxide for Cancer Therapy. Pharm. Nanotechnol. 2019, 7, 279–303. [Google Scholar] [CrossRef] [PubMed]
- Chegaev, K.; Riganti, C.; Lazzarato, L.; Rolando, B.; Guglielmo, S.; Campia, I.; Fruttero, R.; Bosia, A.; Gasco, A. Nitric Oxide Donor Doxorubicins Accumulate into Doxorubicin-Resistant Human Colon Cancer Cells Inducing Cytotoxicity. ACS Med. Chem. Lett. 2011, 2, 494–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murad, F. Cyclic guanosine monophosphate as a mediator of vasodilation. J. Clin. Investig. 1986, 78, 1–5. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Associated Disease | Status of S-Nitrosylation in Disease | Reference |
|---|---|---|---|
| Cancer | |||
| C-Src | Breast cancer | Increased | [93] |
| H-Ras | Increased | [94] | |
| COX2 | Increased | [95] | |
| HIF1α | Breast cancer | Decreased | [90,96] |
| Galectin-1 | Lung cancer | Increased | [97,98] |
| Ezrin | Lung cancer | Increased | [99] |
| BCL-2 | Increased | [100] | |
| Caveolin-1 | Increased | [101] | |
| Peroxiredoxin-2 | Decreased | [102] | |
| Rac1 | Pancreatic cancer | Increased | [25] |
| Rac2 | Increased | ||
| STAT1 | - | ||
| PGK1 | - | ||
| RB | - | ||
| PFKM | Ovarian cancer | Increased | [103] |
| Caspase-3 | Decreased | [104] | |
| STAT3 | Ovarian cancer Pancreatic cancer Head and neck cancer | Increased | [25,105] |
| Androgen receptor | Prostate cancer | Increased | [28] |
| Integrin α6 | Increased | [106] | |
| ERK1/2 | Glioma | Decreased | [107] |
| Keap1 | Colon cancer | Increased | [108] |
| LTBP1 | Colorectal cancer | Increased | [109] |
| Neurodegenerative Disease | |||
| PTEN | Alzheimer’s disease | Increased | [110] |
| CDK5 | Increased | ||
| APOE | Increased | ||
| DNM1L | Increased | ||
| Tubulin | Increased | ||
| SOD2 | - | ||
| MMP9 | Cerebral ischemia | Increased | [111] |
| NMDA Receptor | Dementia | Increased | [112] |
| Cardiovascular Disease | |||
| NSF | Pulmonary arterial hypertension | Decreased | [113] |
| NOS3 | Decreased | ||
| CLTC | Decreased | ||
| Thioredoxin 1 (Trx) | Myocardial ischemia | Increased | [114] |
| Protein | Signaling Pathway | Impact on Protein | Physiological Impact of S–Nitrosylation during Cancer | S-Nitrosylation Site (* Potential Site) | Ref |
|---|---|---|---|---|---|
| Endothelial cells | |||||
| VE–Cadherin | Disassembled adherens junction between endothelial cells | Induced phosphorylation and internalization | Increased cell migration; hyperpermeability | – | [149] |
| p120 | Disassembled adherens junction between endothelial cells | Inhibited binding with β–Catenin | Increased cell migration | Cys579 | [150] |
| β–Catenin | Disassembled adherens junction between endothelial cells | Inhibited binding with p120 | Increased cell migration | Csy619 | [151] |
| HIF1–α | Activated HIF1 signaling pathway | Increased activation and stability | Increased angiogenesis and cancer metastasis | Cys533 | [152] |
| Dynamin | Promoted clathrin–dependent endocytosis of β–Adrenergic receptor | Increased self–assembly and GTPase activity | Increased angiogenesis | Cys86, Cys607 | [153,154] |
| MKP7 | Activated JNK3 signaling pathway | Inhibited phosphatase activity | Increased angiogenesis and migration | Cys244 * | [13,155] |
| Immune cells | |||||
| T cell receptor | – | – | Decreased T cell proliferation and migration; increased T cell apoptosis | – | [156] |
| CCL–2 | Reduced activity of CCR2/CCL2 signaling pathway | Decreased protein expression. | Decreased T cell infiltration | – | [157] |
| NF–kB | Inactivated NF–kB signaling pathway | Inhibited DNA binding activity | Decreased inflammation | Cys179 | [158] |
| STAT3 | Inactivated STAT3 signaling pathway | Inhibited activation | Decreased immune inflammatory response | Cys259 | [159] |
| Caspase–1 | Inhibited activation of NLRP3–Caspase–1 inflammasome | Inhibited activation | Decreased immune inflammatory response | Cys285 | [32] |
| Caspase–3 | Inhibited downstream activation of Caspase–3 signaling | Inhibited activation | Decreased cancer cell apoptosis | Cys163 | [160] |
| JNK1 | Inhibited activation of JNK signaling pathway | Inhibited activation | Decreased inflammation | – | [32] |
| NLRP3 | Inhibited activation of NLRP3–Caspase–1 inflammasome | Inhibited activity | Decreased immune inflammatory response | – | [161,162,163] |
| NOS2 | – | Suppressed activity | Decreased immune inflammatory response | – | [32] |
| ARG1 | – | Increased protein stability | Increased immunosuppressive response | Cys303 | [164] |
| Others (e.g., tumor cells) | |||||
| p21Ras | Promoted Guanine Nucleotide Exchange and activate downstream signaling pathways | Promoted protein activity | Increased Ras induced tumor growth | Cys118 | [94,165] |
| p21Ras (oncogenic) | – | – | Increased tumorigenic growth | Gly12Cy, Gly13Cys | [123] |
| COX2 | – | Stimulated protein activity | Increased inflammation | Cys526 | [95,166] |
| EGFR | Inhibited activation of EGF/EGFR signaling pathway | Impaired tyrosine kinase activity | Decreased tumorigenic growth | – | [167] |
| OGG1 | Reduced activity of BER (Base excision repair) pathway | Inhibited activity | Impaired DNA damage repair response | – | [168] |
| AGT1 | Suppressed activity of direct DNA repair pathway | Promoted protein degradation | Impaired DNA damage repair response | Cys145 | [169] |
| Apo2L/TRAIL receptor DR4 | Inhibited activation of death receptor signaling pathway | Inhibited activity | Decreased cancer cell apoptosis | Cys336 | [170] |
| Bcl–2 | – | Promoted protein stability | Decreased cancer cell apoptosis | Cys158, Cys229 | [100,171,172] |
| ERK | Suppressed activity of ERK/MAPK pathway | Suppressed kinase activity | Increased cancer cell apoptosis | Cys183 | [173] |
| HDAC2 | Induced protein release from chromatin. | Increased acetylation activity. | Increased histone acetylation | Cys262, Cys274 | [174,175] |
| PTEN | Activated downstream Akt signaling pathway | Inhibited enzymatic activity | Increased tumor progression | – | [176] |
| Src | Activated oncogenic signaling pathways (Akt, c–MYC) | Increased kinase activity | Increased tumor growth and proliferation | Cys498 | [93] |
| Androgen receptor | Suppression of androgen receptor signaling | Suppressed DNA binding activity | Increased tumor growth | Cys601 | [28] |
| Integrin α6 | – | Suppressed binding to ECM | Increased cell migration | Cys86 | [106] |
| Caveolin–1 | – | Prevented proteasomal degradation | Increased tumor progression | Cys156 | [33] |
| p53 | – | Induced activation | Increased transactivation of antioxidant genes | – | [177] |
| MDM2 | – | Inhibited activity | Decreased p53 binding and inhibition | Cys77 | [178] |
| Fas | Activated Fas/FasL signaling pathway | Increased sensitivity to Fas ligand | Increased cancer cell apoptosis | Cys304 | [179] |
| MKP1 | – | Increased phosphatase activity | Decreased radiation induced apoptosis | Cys258 | [154] |
| TRAP1 | Increased mitochondrial ROS production & permeability transition pore opening | Promoted proteasomal degradation | Increased cell death in GSNOR deficient cells (HCC) | Cys501 | [33,180,181] |
| Drug | Molecular Signaling Changes | Biological Outcome | Model and Cell Type | Reference |
|---|---|---|---|---|
| Reducing S-Nitrosylation | ||||
| 1400W, L-NAME, L-NMMA | iNOS inhibition, HIF-1α, and IRE1α/XBP1 impairment | Decreased cell growth and motility | TNBC, MDA-MB-231 and SUM159 | [251] |
| L-NMMA+ Docetaxel | iNOS inhibition, ASK1 activation | Increased cytotoxicity in docetaxel-resistant cells | TNBC, SUM-159PT, MDA-MB-436, and MDA-MB-468 | [257] |
| 1400W | Rescues AGT depletion | Reduced DNA mutagenesis | HCC, Diethylnitrosamine (DEN) induced HCC in murine model | [258] |
| MDA-7/IL-24 | Increased BCL-2 denitrosylation | Increased apoptosis | Pan cancer, melanoma A375, and renal carcinoma 7860 | [260] |
| 1400W | Increased OGG1 activity | Increased DNA-repair activity | Cholangiocarcinoma, KMBC | [24] |
| 1400W, L-NIO | Inhibition of angiogenesis related genes | Decreased cell growth, migration, and angiogenesis | CRC; HT 29, and HCT 116 | [217] |
| L-NAME | Inhibition of MAPK signaling | Decreased cell growth and survival | Breast cancer, LM-2, LM-3, LMM3, MDA-MB-231 | [22] |
| Increasing S-nitrosylation | ||||
| SNP, GSNO | Increased ERK1/2 S-nitrosylation | Decreased cell growth | Glioma, U251 cells | |
| GSNO | Increased STAT3 S-nitrosylation | Decreased cell growth of chemo-resistant cells | Ovarian cancer. Ovarian cancer cell lines and HNSCC | [105,261] |
| GTN | cIAP S-nitrosylation | Increased apoptosis and cell death | Colon and breast cancer. SW480, CT26, MDA-MB-231, and EMT6, macrophages | [262] |
| JSK | Inhibition of ubiquitination | Decreased cell growth | Prostate cancer, LNCaP, and C4-2 | [263] |
| NO-ASA and NO-naproxen | Increased NF-κB S-nitrosylation | Decreased cell growth | Colon cancer, HT-29 cells | [264] |
| NO-NSAID | Increased NF-κB and caspase-3 S-nitrosylation | Decreased cell growth | Pan-cancer | [265] |
| SNOC, GSNO, and DETA-NO | Increased Androgen receptor | Decreased cell growth | Prostate cancer, LNCaP, PC3, and 22Rv1 cells | [28] |
| SNP | Increased ERK1/2 S-nitrosylation | Increased apoptosis | Breast cancer, MCF-7 cells | [173] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, V.; Fernando, V.; Letson, J.; Walia, Y.; Zheng, X.; Fackelman, D.; Furuta, S. S-Nitrosylation in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 4600. https://doi.org/10.3390/ijms22094600
Sharma V, Fernando V, Letson J, Walia Y, Zheng X, Fackelman D, Furuta S. S-Nitrosylation in Tumor Microenvironment. International Journal of Molecular Sciences. 2021; 22(9):4600. https://doi.org/10.3390/ijms22094600
Chicago/Turabian StyleSharma, Vandana, Veani Fernando, Joshua Letson, Yashna Walia, Xunzhen Zheng, Daniel Fackelman, and Saori Furuta. 2021. "S-Nitrosylation in Tumor Microenvironment" International Journal of Molecular Sciences 22, no. 9: 4600. https://doi.org/10.3390/ijms22094600
APA StyleSharma, V., Fernando, V., Letson, J., Walia, Y., Zheng, X., Fackelman, D., & Furuta, S. (2021). S-Nitrosylation in Tumor Microenvironment. International Journal of Molecular Sciences, 22(9), 4600. https://doi.org/10.3390/ijms22094600